新型聚合物的制作方法
本申请是分案申请,其原申请的国际申请号为pct/us2015/061036,中国国家申请号为201580062578.2,申请日为2015年11月17日,发明名称为“新型聚合物及其制备方法”。
相关申请的交叉引用
本申请要求于2014年11月18日递交的共同未决的美国临时专利申请第62/081,144号的权益,将其如同完全引述一样并入本文。
背景技术:
碱性交换膜或阴离子交换膜(aem)允许在电化学反应中从阴极至阳极输送阴离子(例如oh-、cl-、br-)。aem是aem燃料电池的关键组件,aem燃料电池中使用氢气和氧气来发电,水作为副产物。aem也用于水电解,其中利用电将水分解为氢气和氧气。在aem燃料电池和水电解中,氢氧根离子(oh-)与水分子一起输送通过aem。aem也可以用于例如蓄电池、传感器和作为致动器(actuator)。
已知的aem通常不适用于aem燃料电池或水电解。许多商业上可获得的aem基于聚苯乙烯,这对于aem燃料电池或水电解通常被认为是差的选择。
其他aem材料包括聚砜、聚(苯醚)、聚(苯撑)、聚(苯并咪唑)、聚(亚芳基醚酮)和聚(亚芳基醚砜)。这些聚合物在中链(mid-chain)中含有亚芳基醚键(-o-),在侧链中含有苄基三甲基铵基团。然而,这种组合已被发现在化学上不稳定,并且在高碱性条件下容易降解。特别地,已知的聚亚芳基(polyarylene)将在聚合物主链中包含醚键,因为它们通常通过二醇单体和二卤化物单体之间的碱性缩合反应合成,该反应产生作为副产物的氯化氢。
此外,涉及制造这些聚合物的氯甲基化反应需要使用有毒试剂、反应时间长和深入的优化,才能达到所需的官能度。在长反应时间内经常发生副反应(例如凝胶化),使得难以实现高于2.5mequiv(毫当量)/g的离子交换容量(iec)。
技术实现要素:
在一个实施方式中,本发明提供了一种形成聚合物的方法,所述方法包括:在强酸的存在下,使芳香族化合物和三氟烷基酮反应以形成溴烷基化前体聚合物;和使所述溴烷基化前体聚合物与三烷基胺和氢氧化钠反应以形成主链不含醚键的聚亚芳基。
在另一个实施方式中,本发明提供了一种式i所示的聚合物,
(式i),其中,ar是芳香族化合物,r为100至1,000,000,r2是
在又一个实施方式中,本发明提供了式iii的聚合物
附图说明
根据对本发明各方面的下述详细描述,并结合描绘本发明各实施方式的附图,更容易理解本发明的上述特征和其他特征,所述附图中:
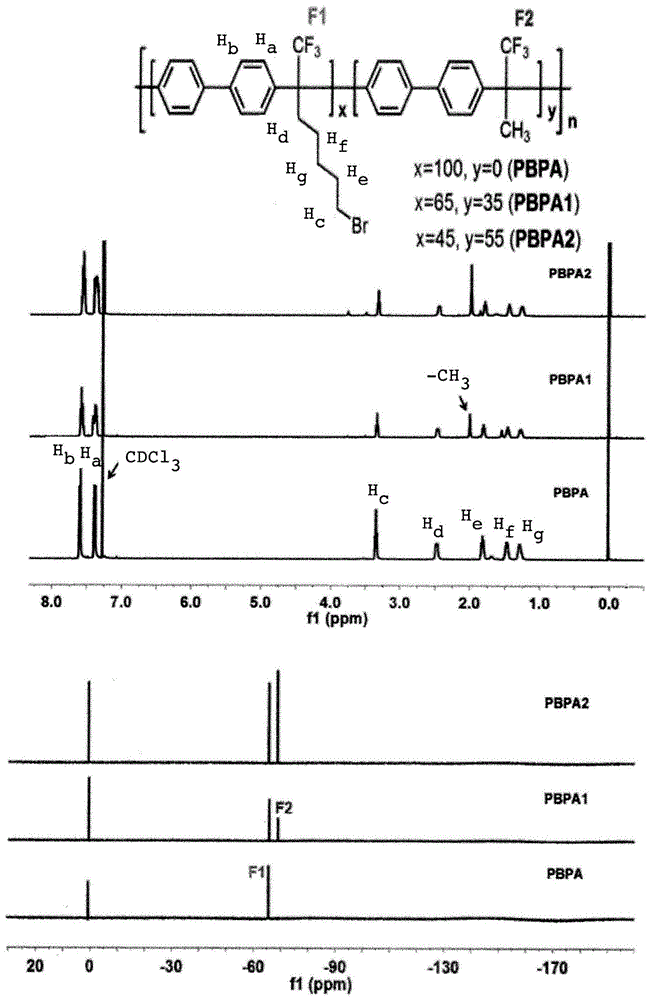
图1显示了根据本发明实施方式的3种示例性溴烷基化前体聚合物的1h和19fnmr谱;
图2显示了根据本发明实施方式的3种示例性聚亚芳基的1h和19fnmr谱;
图3显示了碱稳定性测试前后图2的3种聚亚芳基的1hnmr谱;
图4显示了碱稳定性测试前后图2的3种聚亚芳基之一的1hnmr谱;和
图5显示了(a)本发明的实施方式3种聚亚芳基的应力和应变曲线,和(b)3种聚亚芳基之一的h2/o2极化、高频电阻和功率密度曲线。
注意,本发明的附图不是按比例的。附图仅旨在描绘本发明的典型方面,因此不应被认为是限制本发明的范围。
具体实施方式
本发明的一些实施方式涉及一类新型的聚亚芳基及其制造方法,所述聚亚芳基包含季铵化氢氧化铵。申请人首次使用酸催化缩聚反应生产出高分子量的季铵连接的聚亚芳基(包括聚(联苯基亚烷基)),其不具有碱性不稳定的c-o键。
除了它们在上述燃料电池和水电解方面的aem中的应用之外,申请人还发现本发明的聚合物可用于金属-空气电池技术。令人惊奇的是,申请人还发现这些聚合物表现出抗微生物活性,使其潜在地可用作多种产品的抗微生物涂层。
如下面将更详细说明的,本发明的聚合物通过酮与芳香族化合物之间的酸性缩合合成。结果,副产物是水,而不是如已知的聚亚芳基合成方法中的氯化氢。
申请人开发了一种新的聚合物制造方法,该方法通常包括:使芳香族化合物与三氟烷基酮在强酸的存在下反应(酸催化的friedel-crafts缩聚),以形成溴烷基化前体聚合物;和,在氢氧化钠存在下使溴烷基化前体聚合物与三烷基胺反应,以形成主链不含醚键的聚亚芳基。
根据本发明的一些实施方式,芳香族化合物选自由以下化合物组成的组:
根据本发明的其他实施方式,所述芳香族化合物选自由以下化合物组成的组:
在本发明的一些特定实施方式中,所述芳香族化合物是联苯。
根据本发明的一些实施方式,三氟烷基酮选自由7-溴-1,1,1-三氟庚-2-酮和甲基三氟甲基酮组成的组。
根据本发明的一些实施方式,根据以下反应1制备聚合物,其中ar是聚亚芳基,r为100至1,000,000,r1是
适合用于反应1的强酸包括三氟甲磺酸,不过其他适合的酸对本领域技术人员而言也是清楚的。
如以下反应1a中所示,在本发明的其他实施方式中,芳香基团可以与多个三氟烷基酮结合,其中ar是芳香族化合物,r为100至1,000,000,r1是
下面描述本发明的实施方式的3个示例性聚(联苯基亚烷基)的制造。所述聚(联苯基亚烷基)具有通式ia
其中r2是
实施例1—pbpa+
联苯(0.70g,4.53mmol)、7-溴-1,1,1-三氟庚-2-酮(1.12g,4.53mmol)、二氯甲烷(3.0ml)和三氟甲磺酸(tfsa)在室温下于氮气中利用磁力搅拌棒混合。10小时后,反应混合物溶液变成高粘度,再搅拌2小时。然后将得到的深棕色凝胶状物质用超声波处理粉碎并缓慢倒入甲醇中,形成白色纤维,然后将其过滤并用热甲醇洗涤。真空干燥后,得到1.70g(97%产率)白色纤维状固体,即本文称为pbpa的溴烷基化前体聚合物。图1显示了三种溴烷基化聚(联苯亚烷基)前体(包括pbpa)的1h和19fnmr谱。
将pbpa(200mg)溶于四氢呋喃(thf;2ml)中,在溶液中加入三甲胺水溶液(1ml),在室温下搅拌。聚合物的溶解度逐渐降低,并且6小时后离子聚合物沉淀。向溶液中加入去离子水(1ml),以溶解沉淀物。重复:加入thf,在室温下搅拌6小时,并用去离子水溶解。然后使用旋转蒸发器蒸发挥发性溶剂,并将残余物用少量甲醇(约2ml)再溶解。离子聚合物通过加入乙醚进行沉淀,过滤并真空干燥,得到97%收率(227mg)的聚(联苯亚烷基)pbpa+。图2显示了三种聚(联苯亚烷基)(包括pbpa+)的1h和19fnmr光谱。
实施例2—pbpa1+
联苯(0.70g,4.53mmol)、7-溴-1,1,1-三氟庚-2-酮(0.73g,2.95mmol)、甲基三氟甲基酮(0.18g,1.60mmol)、二氯甲烷(3.0ml)和tfsa(2.0ml)在室温下于氮气中利用磁力搅拌棒混合。5小时后,反应混合物溶液变成高粘度,再搅拌2小时。然后将得到的深棕色凝胶状物质用超声波处理粉碎并缓慢倒入甲醇中。形成白色纤维,然后将其过滤并用热甲醇洗涤。真空干燥后,得到1.4g(96%产率)固体,即本文称为pbpa1的溴烷基化前体聚合物。图1显示了三种溴烷基化聚(联苯亚烷基)前体(包括pbpa1)的1h和19fnmr谱。
将pbpa1(200mg)溶于四氢呋喃(thf;2ml)中,在溶液中加入三甲胺水溶液(1ml),在室温下搅拌。聚合物的溶解度逐渐降低,并且6小时后离子聚合物沉淀。向溶液中加入去离子水(1ml),以溶解沉淀物。重复:加入thf,在室温下搅拌6小时,并用去离子水溶解。然后使用旋转蒸发器蒸发挥发性溶剂,并将残余物用少量甲醇(约2ml)再溶解。离子聚合物通过加入乙醚进行沉淀,过滤并真空干燥,得到98%收率(219mg)的聚(联苯亚烷基)pbpa1+。图2显示了三种聚(联苯亚烷基)(包括pbpa1+)的1h和19fnmr光谱。
实施例3—pbpa2+
联苯(0.50g,3.24mmol)、7-溴-1,1,1-三氟庚-2-酮(0.40g,1.62mmol)、甲基三氟甲基酮(0.19g,1.69mmol)、二氯甲烷(2.5ml)和tfsa(2.3ml)在室温下于氮气中利用磁力搅拌棒混合。3小时后,反应混合物溶液变成高粘度,再搅拌2小时。然后将得到的深棕色凝胶状物质用超声波处理粉碎并缓慢倒入甲醇中。形成白色纤维,然后将其过滤并用热甲醇洗涤。真空干燥后,得到0.94g白色纤维状固体,即本文称为pbpa2的溴烷基化前体聚合物。图1显示了三种溴烷基化聚(联苯亚烷基)前体(包括pbpa2)的1h和19fnmr谱。
将pbpa2(200mg)溶于四氢呋喃(thf;2ml)中,在溶液中加入三甲胺水溶液(1ml),在室温下搅拌。聚合物的溶解度逐渐降低,并且6小时后离子聚合物沉淀。向溶液中加入去离子水(1ml),以溶解沉淀物。重复:加入thf,在室温下搅拌6小时,并用去离子水溶解。然后使用旋转蒸发器蒸发挥发性溶剂,并将残余物用少量甲醇(约2ml)再溶解。离子聚合物通过加入乙醚进行沉淀,过滤并真空干燥,得到98%收率(210mg)的聚(联苯亚烷基)pbpa2+。图2显示了三种聚(联苯亚烷基)(包括pbpa2+)的1h和19fnmr光谱。
下表1显示了pbpa+、pbpa1+和pbpa2+聚合物的吸水量(wu)和阴离子电导率数据。
a在1mnaoh溶液中浸没30天后
所有3种聚合物均展示出优异的wu和导电率性质,特别是pbpa+。尽管有这些wu值,所有3种聚合物展示出低溶胀比(pbpa+为40%,pbpa1+为10%,pbpa2+为5%),这可能是由于存在刚性的芳香族骨架。
下表2显示出在碱稳定性测试前、后pbpa+、pbpa1+和pbpa2+的以mequiv/g表示的离子交换容量(iec)数据。
从表2的数据中可以看出,即使在长时间之后,所有三种聚(联苯亚烷基)在碱性环境(1mnaoh)中也表现出显著的iec稳定性。从表2和上述实施例的结果也可以看出,可以通过调节不同三氟烷基酮的相对比例来控制聚合物的iec。
有趣的是,与具有类似iec(例如,季铵化聚(苯醚)、聚(亚芳基醚酮)和聚(亚芳基醚砜))的其它报道的芳族aem相比,pbpa1+提供了显著更高的氢氧根离子电导率。这可能归因于pbpa1+的相对较高的wu,这有助于水合膜更有效地扩散氢氧根离子。所有三种聚合物随着温度的升高显示出增加的氢氧根离子传导性,这主要是由于随着温度的升高,离子的迁移更快,扩散性更高。
图3显示了上表2中描述的30天碱稳定性测试(1mnaoh,80℃)前、后pbpa+、pbpa1+和pbpa2+的1hnmr谱。图4显示了另一碱测试(1mnaoh,100℃,30天)后pbpa+的1hnmr谱数据。
pbpa+、pbpa1+和pbpa2+不溶于水、四氢呋喃、三氯甲烷(chcl3)和二氯乙烯(ch2cl2),但在室温下可溶于n,n-二甲基甲酰胺、二甲基亚砜和甲醇。这些聚合物的季铵基在270℃分解,热稳定性大于qa聚(亚芳基醚砜)报道的热稳定性。前体聚合物(pbpa、pbpa1、pbpa2)是热稳定的,直至350℃也不分解。
aem的机械性能在燃料电池应用中至关重要。对于pbpa+、pbpa1+和pbpa2+聚合物中每一种,膜的拉伸强度和断裂伸长率分别为20mpa至35mpa和40%至140%,这满足构建aem燃料电池的膜电极组件(mea)的要求。
图5显示了所有三种聚合物的应力-应变曲线(图(a))。在50℃和50%相对湿度下,含有iec最低的聚合物(pbpa2+)的膜的机械强度(35mpa)大于包含pbpa+的膜的机械强度(22mpa),pbpa+是iec最高的聚合物。并且与diels-alder聚(亚苯基)aems(iec=1.7mequiv/g,32mpa最大强度,40%最大应变)相比,包含pbpa1+聚合物的膜显示出相似的拉伸强度(iec=1.9mequiv/g,最大强度33mpa,100%最大应变),但显著更好的断裂伸长率,这可能是由于其具有四级sp3碳的更柔性的主链结构。这些机械强度数据表明,本发明的聚合物具有足以用作燃料电池中的aem材料的韧性和延展性。
图5的图(b)显示80℃时含有pbpa1+的燃料电池的极化曲线。开路电压(ocv)为1.01v,这在供氢的燃料电池中是典型的。80℃时获得155mw/cm2的最大功率密度,并且电池的高频电阻(hfr)<0.1ωcm2。而从hfr获得的膜电导率为19.9s/cm,低于非原位测量值(由于mea的非膜电阻贡献),这里报告的hfr值远小于aem燃料电池的文献报道中发现的典型值。
这些结果清楚地表明:与其他aem材料相比,本发明的qa聚(联苯亚烷基)具有优异的化学稳定性和燃料电池性能。
上述聚(联苯亚烷基)以外的聚亚芳基在本发明的范围内,并且可按照类似的方法制造。例如,可以使用本发明的方法来制造式iii的聚合物
其中,ar是芳香族化合物,r"是
根据本发明的一些实施方式,式iii聚合物可根据以下反应2制备,其中,ar是聚亚芳基,r'是
根据本发明的一些实施方式,芳香族化合物选自由以下化合物组成的组:
其他芳香族化合物当然根据本领域技术人员的认知也可以采用,并且处于本发明的范围内。相似的是,在一些实施方式中,
最后,尽管上述实施方式包括溴化芳香族化合物或三氟烷基酮,但也可以使用其它卤素。例如,可以使用7-氯-1,1,1-三氟庚-2-酮来制备氯代烷基化的前体聚合物,根据本发明由该前体聚合物通过亲核取代制备聚合物。在这样的实施方式中,可以在所得聚合物中包括除铵基以外的基团(例如烷氧基、磺酸酯、羧酸酯、膦酸酯)。
如本文所使用的,单数形式“一个”、“一种”和“该”意图也包括复数形式,除非上下文另有外明确指出。将进一步理解,当在本说明书中使用时,术语“包括”和/或“包含”指示存在所述特征、整数、步骤、操作、元件和/或部件,但不排除存在或添加一个或多个其它特征、整数、步骤、操作、元件、部件和/或其组合。
此书面描述使用示例来公开包括最佳模式的本发明,并且还使得本领域技术人员能够实践本发明,包括制作和使用任何设备或系统并执行任何相关或并入的方法。本发明的可专利范围由权利要求限定,并且可以包括对本领域技术人员想到的其它示例。如果这些其他示例具有与权利要求的文字语言没有不同的结构要素,或者如果它们包括与权利要求的文字语言无实质差异的等同的结构要素,则这些其他示例也在权利要求的范围内。
- 还没有人留言评论。精彩留言会获得点赞!