调控可逆加成-断裂链转移聚合法的产物的分散度的方法与流程
本发明涉及可逆加成-断裂链转移聚合领域,尤其涉及一种调控可逆加成-断裂链转移聚合法的产物的分散度的方法。
背景技术:
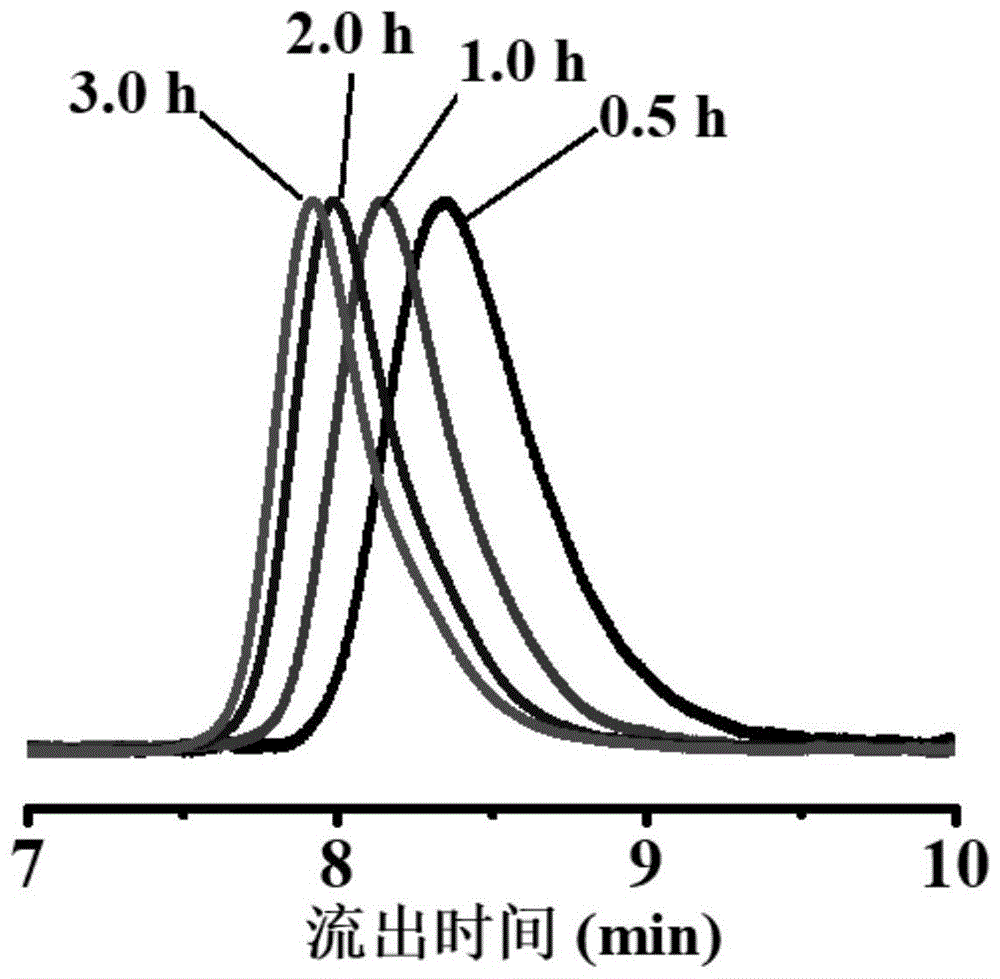
:多分散性对聚合物的玻璃化转变温度,粘度,可加工性以及嵌段共聚物的自组装行为等,都具有深远的影响。目前人们已经证明,中间或两端为多分散的三嵌段聚合物能够形成与单分散聚合物完全不同的相图。在工业生产中,多分散聚乙烯被用来制造轮胎,其高分子量部分保证了足够的弹性,而低分子量部分粘度小,提升了材料的可加工性。目前制备多分散性聚合物的方法主要有以下几种:1.共混法:常见的是混合各种分子量的预聚物,它适用于任何类型和分子量的聚合物。junkers等人对共混方法进行了改善,利用全自动设备,减少人工干预,分离和混合均交给机器完成,更加便捷且达到更高的可靠性(polymerchemistry2019,10(46),6315-6323)。董学会等人利用保护-脱保护的迭代增长方案制备了一系列单分散预聚物,并通过某些分布函数(如舒尔兹-齐姆分布,高斯分布)精确混合它们,从而完全控制分子量分布的宽度、对称性和形状(chemicalscience2019,10(46),10698-10705),虽然该方法可以实现峰型的正态,但合成预聚物的方法属于精密合成,更加复杂耗时。但是共混法通常工艺复杂,耗时长,因为它需要合成和纯化多种材料,然后以精确的比例混合。同时大多数共混物具有多峰,很难实现正态分布。2.使用时间控制的引发:fors课题在氮氧化物介导的聚合(nmp),阴离子聚合,可控配位插入聚合中均实现了这种方法,在这种方案中,通过逐步加入引发剂实现对分散度的调控,先加入的引发剂先引发,链长长;后加入的引发剂后引发,链长短,从而提高分散度(j.am.chem.soc.2016,138(6),1848-1851;acsmacrolett.2016,5(7),796-800;j.am.chem.soc.2020,142(3),1443-1448)。但是使用时间控制的引发法依赖泵的辅助,同时设备复杂,他人难以重复;同时不能应用于聚合物刷等异质结构。3.降低催化剂浓度:anastasaki等人通过降低atrp催化剂铜盐的浓度,在原子转移自由基聚合(atrp)中实现了对分散度的控制。通过降低催化剂浓度,原子转移自由基聚合的控制性降低,分散度升高(angew.chem.int.ed.engl.2019,58(38),13323-13328)。但是,该方法引发剂效率降低,这会得到分子量比目标分子量高的聚合物。单体转化率过低,同时控制分散度最大仅到1.62,此时单体转化率仅19%。若再降低催化剂浓度,聚合将失去活性可控特征,为普通自由基聚合,不能按需求调控分散度。4.加入终止剂:goto等人在可逆络合介导聚合(rcmp)中的丙烯酸甲酯的聚合加入低温下可终止链增长的丙烯酸正丁酯来调控分散度,并通过升温活化再引发聚合从而形成复杂的嵌段,星形和刷状聚合物。随着丙烯酸正丁酯的含量增加,聚丙烯酸甲酯的分散度升高(angew.chem.int.ed.engl.2019,58(17),5598-5603)。该方法仅适用于丙烯酸甲酯的可逆络合介导聚合(rcmp)聚合,其他单体不适用。虽然可以制备嵌段共聚物,但链中掺杂有一定量丙烯酸丁酯,不是纯粹的两嵌段共聚物。5.活性末端修饰终止:robertson和conrad等人通过加入苯肼在原子转移自由基聚合(atrp)中取代链末端br从而使链逐步失去活性实现增加的分散度的目的(polym.chem.2018,9(33),4332-4342)。该方法适用于原子转移自由基聚合(atrp),文中提供的分散度仅有1.08、1.71和1.80。因此,开发底物适用性广泛、操作简单且可根据需求调控聚合物分散度的方法十分必要。技术实现要素:为解决上述技术问题,本发明的目的是提供一种调控可逆加成-断裂链转移聚合法的产物的分散度的方法,仅需在raft聚合中额外添加少量链终止剂即可获得目标分散度的产品,反应操作简单,底物适用性广。本发明的一种调控可逆加成-断裂链转移聚合法的产物的分散度的方法,包括以下步骤:在链终止剂的存在下,将乙烯基类单体在链转移剂和引发剂的作用下,在有机溶剂中发生可逆加成-断裂链转移(raft)聚合反应,其中,链终止剂为2-溴代马来酰亚胺(mbr)或呋喃保护的2-溴代马来酰亚胺(fmbr);链转移剂包括芳香族二硫代酯类化合物;链终止剂为2-溴代马来酰亚胺时,可逆加成-断裂链转移聚合反应在40-110℃的恒温条件下进行;链终止剂为呋喃保护的2-溴代马来酰亚胺时,可逆加成-断裂链转移聚合反应在程序变温条件下进行,程序变温条件为先在低温段反应然后在至少一个高温段反应,所述低温段的反应温度不高于40℃,所述高温段的反应温度高于40℃。进一步地,乙烯基类单体包括甲基丙烯酸甲酯(mma)或苯乙烯(st)。进一步地,芳香族二硫代酯类化合物包括二硫代苯甲酸异丁腈酯(cpdb)和/或α-二硫代萘甲酸异丁腈酯(cpdn)。cpdb和cpdn的结构式依次如下:进一步地,引发剂包括偶氮二异丁腈(aibn)、偶氮二环己基甲腈(accn)和偶氮二异庚腈(abvn)中的一种或几种。进一步地,以摩尔比计,乙烯基类单体:链终止剂:链转移剂:引发剂=(200~800):(0.1~30):1:0.2。可以通过调整链终止剂mbr或fmbr与乙烯基类单体的摩尔比控制聚合物的分散度。进一步地,有机溶剂为二甲亚砜(dmso)和/或n,n’-二甲基甲酰胺(dmf)。进一步地,有机溶剂中,乙烯基类单体的浓度为4-20mol/l。进一步地,聚合时间为2-71h。进一步地,链终止剂为2-溴代马来酰亚胺,可逆加成-断裂链转移聚合反应在100-110℃(优选为110℃)下进行。进一步地,链终止剂为呋喃保护的2-溴代马来酰亚胺,可逆加成-断裂链转移聚合反应首先在40℃下反应4-48h,然后在100-120℃下反应3-5h。优选地,先在40℃下反应4-48h,然后在110℃下反应3-5h。引发剂为偶氮二环己基甲腈和偶氮二异庚腈。如无特殊说明,本发明的反应均在无氧条件下进行。本发明通过加入少量大位阻聚合单体mbr,降低乙烯基类单体在raft聚合过程中产生的部分大分子链的聚合活性,从而达到控制分散度的目的。除此以外,加入链终止剂不可避免地会降低单体转化率,转化率会随着链终止剂添加量增加——分散度升高而降低。此外,本发明还可使用fmbr作为链终止剂,与其他加入链终止剂的方案相比,本发明通过呋喃对2-溴代马来酰亚胺的保护作用,在raft的聚合过程中,由于采用程序变温反应条件,在低温(40℃)下体系中不存在2-溴代马来酰亚胺,乙烯基类单体能够正常活性聚合,而升到高温(高于40℃)时,2-溴代马来酰亚胺被释放,在此时间点开始增加分散度这样的方案实现了同一投料比、不同分子量、相近分散度的产品的获得,同时增加了单体的转化率。本发明中,fmbr在高于40℃的条件下,2-溴代马来酰亚胺随即释放,只是随着温度的升高,其释放速度会加快,在达到110℃时,2-溴代马来酰亚胺会快速释放。借由上述方案,本发明至少具有以下优点:1、本发明使用mbr或fmbr作为链终止剂,首次成功实现了在可逆加成-断裂链转移(raft)聚合中调控分散度的目的。本发明保证了聚合物分散度在较宽的范围内随意调控的同时,单体转化率相对较高。2、本发明使用mbr或fmbr作为链终止剂,对甲基丙烯酸甲酯及其衍生物和苯乙烯及其衍生物这两类典型的单体均可适用,其应用具有很好的前景。3、本发明使用fmbr作为链终止剂,可控制mbr的释放时间,在一定时间释放内可提高单体转化率,且实现了同一投料比、不同分子量、相近分散度的产品的获得。4、本发明的聚合产物的分散度调控范围较为广泛,对于甲基丙烯酸甲酯,可实现的调控范围为1.21-1.95,通过简单的加减链终止剂的使用量,即可获得目标分散度的产品。5、本发明无需外部设备帮助(包括但不限于泵、定制反应装置),操作简单,普通科研人员可轻松使用。上述说明仅是本发明技术方案的概述,为了能够更清楚了解本发明的技术手段,并可依照说明书的内容予以实施,以下以本发明的较佳实施例并配合详细附图说明如后。附图说明图1是实施例一中mma的raft聚合的gpc流出曲线图;图2是实施例一中mma的raft聚合的聚合动力学,以及聚合产物分散度、分子量的变化过程;图3是实施例二中mma的raft聚合的gpc流出曲线图;图4是实施例二中mma的raft聚合的聚合动力学,以及聚合产物分散度、分子量的变化过程;图5是实施例三中mma的raft聚合的gpc流出曲线图;图6是实施例三中mma的raft聚合的聚合动力学,以及聚合产物分散度、分子量的变化过程;图7是实施例四中mma的raft聚合的gpc流出曲线图;图8是实施例四中mma的raft聚合的聚合动力学,以及聚合产物分散度、分子量的变化过程;图9是实施例五中mma的raft聚合的gpc流出曲线图;图10是实施例五中mma的raft聚合的聚合动力学,以及聚合产物分散度、分子量的变化过程;图11是实施例八中mma的raft聚合的gpc流出曲线图;图12是实施例八中呋喃保护的2-溴代马来酰亚胺(fmbr)脱保护动力学、mma的raft聚合的聚合动力学,以及聚合产物分散度、分子量的变化过程。具体实施方式下面结合实施例,对本发明的具体实施方式作进一步详细描述。以下实施例用于说明本发明,但不用来限制本发明的范围。实施例一不添加链终止剂合成聚甲基丙烯酸甲酯(pmmas)平行进行多组实验,每组实验反应不同时间,具体方法如下:按摩尔比,[mma]0:[mbr]0:[cpdb]0:[accn]0=200:0:1:0.2,依次加入cpdb(0.0209g)、accn(0.0046g)、mma(2.0ml)及dmf(2.0ml)于10ml的安瓿瓶中,加入搅拌子,经过3次标准的冷冻-抽气-解冻充气循环后,在无氧氛围下封管。将封管后的安瓿瓶置于磁力搅拌器中在110℃下按预定的时间进行反应,转速600rpm。反应结束后,取出封管,打开封管,用2~5ml的四氢呋喃溶解,倒入250ml的甲醇中,过夜放置后抽滤、烘干即可得到聚甲基丙烯酸甲酯(pmma)。图1为实施例一中不添加链终止剂条件下,mma的raft聚合的gpc流出曲线图;gpc测试结果表明,在聚合反应0.5h时,pmma的分子量(mn)为7.9kda,分散度(mw/mn)为1.19。在聚合反应1h时,pmma的分子量(mn)为11.3kda,分散度(mw/mn)为1.17。在聚合反应2h时,pmma的分子量(mn)为14.6kda,分散度(mw/mn)为1.18。在聚合反应3h时,pmma的分子量(mn)为16.0kda,分散度(mw/mn)为1.21。图2为实施例一中不添加链终止剂条件下,mma的raft聚合的聚合动力学,以及聚合产物分散度、分子量的变化过程。实施例一中数据表明,在不添加链终止剂的条件下,mma的raft聚合具有活性特征,能够保证分子量分布较窄(mw/mn=1.17-1.21)。实施例二加样摩尔比单体:链终止剂:链转移剂=200:5:1合成聚甲基丙烯酸甲酯平行进行多组实验,每组实验反应不同时间,具体方法如下:按摩尔比,[mma]0:[mbr]0:[cpdb]0:[accn]0=200:5:1:0.2,依次加入mbr(0.0830g)、cpdb(0.0209g)、accn(0.0046g)、mma(2.0ml)及dmf(2.0ml)于10ml的安瓿瓶中,加入搅拌子,经过3次标准的冷冻-抽气-解冻充气循环后,在无氧氛围下封管。将封管后的安瓿瓶置于磁力搅拌器中在110℃下按预定的时间进行反应,转速600rpm。反应结束后,取出封管,打开封管,用2~5ml的四氢呋喃溶解,倒入250ml的甲醇中,过夜放置后抽滤、烘干即可得到聚甲基丙烯酸甲酯。图3为实施例二中加样摩尔比链终止剂:链转移剂=5:1条件下,mma的raft聚合的gpc流出曲线图;gpc测试结果表明,在聚合反应0.5h时,pmma的分子量(mn)为7.5kda,分散度(mw/mn)为1.32。在聚合反应1h时,pmma的分子量(mn)为10.7kda,分散度(mw/mn)为1.35。在聚合反应3h时,pmma的分子量(mn)为14.5kda,分散度(mw/mn)为1.48。图4为实施例二中加样摩尔比链终止剂:链转移剂=5:1条件下,mma的raft聚合的聚合动力学,以及聚合产物分散度、分子量的变化过程。实施例二中数据表明,在加样摩尔比链终止剂:链转移剂=5:1条件下,mma的raft聚合会随着聚合反应进行,逐渐失去聚合的活性特征,最终转化率约为67.7%。聚合物的分子量分布mw/mn最大值由未添加链终止剂时的1.21增加至1.48,分散度调控良好。实施例三加样摩尔比单体:链终止剂:链转移剂=200:10:1合成聚甲基丙烯酸甲酯平行进行多组实验,每组实验反应不同时间,具体方法如下:按摩尔比,[mma]0:[mbr]0:[cpdb]0:[accn]0=200:10:1:0.2,依次加入mbr(0.1660g)、cpdb(0.0209g)、accn(0.0046g)、mma(2.0ml)及dmf(2.0ml)于10ml的安瓿瓶中,加入搅拌子,经过3次标准的冷冻-抽气-解冻充气循环后,在无氧氛围下封管。将封管后的安瓿瓶置于磁力搅拌器中在110℃下按预定的时间进行反应,转速600rpm。反应结束后,取出封管,打开封管,用2~5ml的四氢呋喃溶解,倒入250ml的甲醇中,过夜放置后抽滤、烘干即可得到聚甲基丙烯酸甲酯。图5为实施例三中加样摩尔比链终止剂:链转移剂=10:1条件下,mma的raft聚合的gpc流出曲线图;gpc测试结果表明,在聚合反应0.5h时,pmma的分子量(mn)为8.2kda,分散度(mw/mn)为1.42。在聚合反应1h时,pmma的分子量(mn)为9.5kda,分散度(mw/mn)为1.77。在聚合反应3h时,pmma的分子量(mn)为12.8kda,分散度(mw/mn)为1.75。图6为实施例三中加样摩尔比链终止剂:链转移剂=10:1条件下,mma的raft聚合的聚合动力学,以及聚合产物分散度、分子量的变化过程。实施例三中数据表明,在加样摩尔比链终止剂:链转移剂=10:1条件下,mma的raft聚合在聚合时间为2h时即失去了聚合活性,反应转化率停留在50%左右。聚合物的分子量分布mw/mn最大值由加样摩尔比链终止剂:链转移剂=5:1时的1.48进一步增加至1.77,分散度调控良好。实施例四加样摩尔比单体:链终止剂:链转移剂=200:15:1合成聚甲基丙烯酸甲酯平行进行多组实验,每组实验反应不同时间,具体方法如下:按摩尔比,[mma]0:[mbr]0:[cpdb]0:[accn]0=200:15:1:0.2,依次加入mbr(0.2490g)、cpdb(0.0209g)、accn(0.0046g)、mma(2.0ml)及dmf(2.0ml)于10ml的安瓿瓶中,加入搅拌子,经过3次标准的冷冻-抽气-解冻充气循环后,在无氧氛围下封管。将封管后的安瓿瓶置于磁力搅拌器中在110℃下按预定的时间进行反应,转速600rpm。反应结束后,取出封管,打开封管,用2~5ml的四氢呋喃溶解,倒入250ml的甲醇中,过夜放置后抽滤、烘干即可得到聚甲基丙烯酸甲酯。图7为实施例四中加样摩尔比链终止剂:链转移剂=15:1条件下,mma的raft聚合的gpc流出曲线图;gpc测试结果表明,在聚合反应0.5h时,pmma的分子量(mn)为8.2kda,分散度(mw/mn)为1.42。在聚合反应1h时,pmma的分子量(mn)为9.5kda,分散度(mw/mn)为1.77。在聚合反应3h时,pmma的分子量(mn)为10.5kda,分散度(mw/mn)为1.93。图8为实施例四中加样摩尔比链终止剂:链转移剂=15:1条件下,mma的raft聚合的聚合动力学,以及聚合产物分散度、分子量的变化过程。实施例四中数据表明,在加样摩尔比链终止剂:链转移剂=15:1条件下,mma的raft聚合在聚合时间约为1h时即失去了聚合活性,反应转化率停留在46%左右。聚合物的分子量分布mw/mn最大值由加样摩尔比链终止剂:链转移剂=10:1时的1.77进一步增加至1.93,分散度调控良好。实施例五加样摩尔比单体:链终止剂:链转移剂=200:30:1合成聚甲基丙烯酸甲酯平行进行多组实验,每组实验反应不同时间,具体方法如下:按摩尔比,[mma]0:[mbr]0:[cpdb]0:[accn]0=200:30:1:0.2,依次加入mbr(0.4980g)、cpdb(0.0209g)、accn(0.0046g)、mma(2.0ml)及dmf(2.0ml)于10ml的安瓿瓶中,加入搅拌子,经过3次标准的冷冻-抽气-解冻充气循环后,在无氧氛围下封管。将封管后的安瓿瓶置于磁力搅拌器中在110℃下按预定的时间进行反应,转速600rpm。反应结束后,取出封管,打开封管,用2~5ml的四氢呋喃溶解,倒入250ml的甲醇中,过夜放置后抽滤、烘干即可得到聚甲基丙烯酸甲酯。图9为实施例五中加样摩尔比链终止剂:链转移剂=30:1条件下,mma的raft聚合的gpc流出曲线图;gpc测试结果表明,在聚合反应0.5h时,pmma的分子量(mn)为6.3kda,分散度(mw/mn)为1.51。在聚合反应1h时,pmma的分子量(mn)为7.5kda,分散度(mw/mn)为1.69。在聚合反应3h时,pmma的分子量(mn)为8.0kda,分散度(mw/mn)为1.85。图10为实施例五中加样摩尔比链终止剂:链转移剂=30:1条件下,mma的raft聚合的聚合动力学,以及聚合产物分散度、分子量的变化过程。实施例五中数据表明,在加样摩尔比链终止剂:链转移剂=30:1条件下,mma的raft聚合在大量的链终止剂存在的情况下受到很大的抑制,在短时间的反应后,反应转化率停留在30%以下。同时,由于反应收到极大抑制,聚合物的分子量分布mw/mn最大值为1.85。可以推想至再次增加链终止剂的加样比,反应进一步被抑制。实施例六加样摩尔比单体:链终止剂:链转移剂=400:5:1或800:5:1合成聚甲基丙烯酸甲酯按摩尔比,[mma]0:[mbr]0:[cpdb]0:[accn]0=400:5:1:0.2或800:5:1:0.2,依次加入mbr(0.0415g)、cpdb(0.0105g)、accn(0.0023g)、mma(2.0或4.0ml)及dmf(4.0或10.0ml)于25ml的反应瓶中,加入搅拌子,经过3次标准的冷冻-抽气-解冻充气循环后,在无氧氛围下封管。将封管后的安瓿瓶置于磁力搅拌器中在110℃下反应6h,转速600rpm。反应结束后,取出封管,打开封管,用2~5ml的四氢呋喃溶解,倒入250ml的甲醇中,过夜放置后抽滤、烘干即可得到聚甲基丙烯酸甲酯。表1为实施例六中加样摩尔比单体:链终止剂:链转移剂=400:5:1或800:5:1条件下,mma的raft聚合产物分散度、分子量等聚合结果。表1不同单体摩尔比下对聚甲基丙烯酸甲酯分散度的调控[mma]0:[mbr]0:[cpdb]0:[accn]0转化率/%mn/g/molmw/mn400:5:1:0.264.1190001.53800:5:1:0.257.2295001.47以上结果表明,单体与链终止剂的摩尔比对分散度影响不大,只要控制链终止剂与链转移剂的摩尔比为5:1,即可控制最终产物的分散度为1.50左右。因此,需要制备一定分散度,更高分子量的聚合物时,仅需提高单体与链转移剂的比例即可。随着单体与链转移剂的摩尔比的升高,转化率有一定的下降趋势。实施例七加样摩尔比单体:链终止剂:链转移剂=200:(3.5~10):1合成聚苯乙烯按摩尔比,[st]0:[mbr]0:[cpdb]0:[accn]0=200:(3.5~10):1:0.2,依次加入mbr、cpdb(0.0194g)、accn(0.0043g)、st(2ml)及dmf(1.0ml)10ml的反应瓶中,mbr的用量根据其他物质的用量和摩尔比进行确定。向反应瓶中加入搅拌子,经过3次标准的冷冻-抽气-解冻充气循环后,在无氧氛围下封管。将封管后的安瓿瓶置于磁力搅拌器中110℃下按预定的时间进行反应,转速600rpm。反应结束后,取出封管,打开封管,用2~5ml的四氢呋喃溶解,倒入250ml的甲醇中,过夜放置后抽滤、烘干即可得到聚苯乙烯。表2为实施例七中加样摩尔比单体:链终止剂:链转移剂=200:(3.5~10):1条件下,st的raft聚合产物分散度、分子量等聚合结果。表2不同链终止剂摩尔比对聚苯乙烯分散度的调控[st]0:[mbr]0:[cpdb]0:[accn]0反应时间/hmn/g/molmw/mn200:3.5:1:0.25876001.57200:5:1:0.27156001.70200:10:1:0.27132001.34以上结果表明,本发明同样适用于苯乙烯类单体的raft聚合。由于苯乙烯的活性低于甲基丙烯酸甲酯,所需要的聚合时间较长,且最终分子量较低。随着链终止剂用量的升高,分散度升高,调控效果较好;但链终止剂过高的用量会导致聚合完全失去活性,分子量较低,同时分散度不高,因此需要选择适宜的链终止剂和链转移剂的摩尔比。实施例八通过加入fmbr调控聚甲基丙烯酸甲酯分散度本实施例中,引发剂采用accn和abvn,链终止剂选择fmbr,按以下方法进行pmma的制备:按摩尔比,[mma]0:[fmbr]0:[cpdb]0:[accn]0:[abvn]0=(200~400):(10~20):1:0.2:0.2,依次加入fmbr、cpdb、accn、abvn、mma及dmf(2.0~4.0ml)于10ml的安瓿瓶中,加入搅拌子,经过3次标准的冷冻-抽气-解冻充气循环后,在无氧氛围下封管。最初将封管后的安瓿瓶置于磁力搅拌器中在40℃下反应一定时间,转速600rpm;然后转移至另一磁力搅拌器中在110℃下反应一定时间,转速600rpm。反应结束后,取出封管,打开封管,用2~5ml的四氢呋喃溶解,倒入250ml的甲醇中,过夜放置后抽滤、烘干即可得到聚甲基丙烯酸甲酯。图11为实施例八中加样摩尔比[mma]0:[fmbr]0:[cpdb]0:[accn]0:[abvn]0=400:10:1:0.2:0.2,程序变温40℃(46h)-110℃(5h)条件过程中,在第46h(曲线a)、40℃反应46h以及110℃反应1h(曲线b)、40℃反应46h以及110℃反应5h(曲线c)条件下,mma的raft聚合的gpc流出曲线图;gpc测试结果表明,曲线a对应的pmma的分子量(mn)为19.5kda,分散度(mw/mn)为1.24。曲线b对应的pmma的分子量(mn)为22.5kda,分散度(mw/mn)为1.42。曲线c对应的pmma的分子量(mn)为23.3kda,分散度(mw/mn)为1.43。图12为实施例八中加样摩尔比[mma]0:[fmbr]0:[cpdb]0:[accn]0:[abvn]0=400:10:1:0.2:0.2,程序变温40℃(46h)-110℃(5h)过程中,自反应第46h起,呋喃保护的2-溴代马来酰亚胺(fmbr)脱保护动力学、mma的raft聚合的聚合动力学,以及聚合产物分散度、分子量的变化过程。表3为不同加样摩尔比、不同程序控温条件所得产物的聚合情况测试结果。表3序号1-4为实施例八中在不同加样摩尔比以及在不同程序变温条件下产物的转化率、分散度、分子量等聚合结果。如序号1为在加样摩尔比[mma]0:[fmbr]0:[cpdb]0:[accn]0:[abvn]0=200:10:1:0.2:0.2、程序变温40℃(4h)-110℃(3h)条件下,mma在第一段温度下聚合后以及在第二段温度下聚合后产物转化率、分散度、分子量等聚合结果。其他序号代表含义同此处,在此不再赘述。表3通过fmbr的程序变温释放对聚甲基丙烯酸甲酯分散度的调控以上数据表明,通过对链终止剂2-溴代马来酰亚胺保护,使得低温下体系中不存在2-溴代马来酰亚胺,mma能够正常活性聚合;而升到高温时,fmbr中的2-溴代马来酰亚胺被释放,在此时间点以后,pmma开始增加分散度实现同一投料比、不同分子量、相近分散度的产品的获得,同时增加了单体的转化率。以上仅是本发明的优选实施方式,并不用于限制本发明,应当指出,对于本
技术领域:
的普通技术人员来说,在不脱离本发明技术原理的前提下,还可以做出若干改进和变型,这些改进和变型也应视为本发明的保护范围。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1