手性三氟甲基取代的马来酰亚胺衍生物及其制备方法与流程
本发明涉及有机合成领域,尤其涉及一种手性三氟甲基取代的马来酰亚胺衍生物及其制备方法。
背景技术:
多官能团化的手性马来酰亚胺作为重要的五元杂环化合物,在临床药物存在广泛,此外,某些天然产物中也发现具有含有此类骨架结构。目前的研究发现,这类化合物具有很好的生物活性,例如杀菌,消毒,灭虫、消炎、抗疟疾、抗结核等。另外,本领域知晓的,在药物分子中氟原子或含氟基团的引入能够提高药物效力并可以改变药物分子的代谢渗透性稳定性,并可以调节药物的pka及脂溶性;因此,近年来,含氟药物及相关中间体的开发研究十分活跃。
所以,寻找简单便捷的方法来合成含有氟原子的手性马来酰亚胺化合物不仅仅能够丰富含氟化合物的分子库,也可以为药物候选分子的合成提供新路线,这也是本领域亟需解决的问题。
此外,目前通过2-烯基琥珀酰亚胺合成手性马来酰亚胺类化合物的方法较少,目前仅有两例(chem.eur.j.,2010,16,12534;j.org.chem.,2012,77,6600),其合成路线如下:
从上述合成路线可以看出,两条合成路线中只有烯基端位没有任何取代基时,才能够发生1,3-氢迁移并生成手性马来酰亚胺化合物,这极大的限制了底物的普适性。当烯基端位连有芳香取代基时,反应仅仅得到琥珀酰亚胺。
另外,对于含三氟甲基的手性马来酰亚胺衍生物的合成方法目前还未有相关报道。因此,发展高效的和合成方法来构建手性马来酰亚胺化合物,特别是含有三氟甲基的手性马来酰亚胺衍生物是非常必要的。这不仅能够丰富该类化合物的分子库,为药物活性化合物的发现提供充足的化合物源,还能为具有该类骨架化合物的合成提供一定的参考。
技术实现要素:
本发明的目的之一,就在于提供一种手性三氟甲基取代的马来酰亚胺衍生物及其制备方法,以解决上述问题。
为了实现上述目的,本发明采用的技术方案是这样的:手性三氟甲基取代的马来酰亚胺衍生物,具有如下结构式(ⅲ)所示的结构:
上述结构式(ⅲ)中,r1基选自各种类型保护基,r2基选自芳基、杂芳基、稠芳基,r3基选自芳基、杂芳基、烷基。
作为优选的技术方案:所述r1基团选自叔丁氧羰基、甲基、苄基或苯基中的一种;所述r2选自苯基、邻甲基苯基、邻甲氧基苯基、邻氟苯基、邻氯苯基、邻溴苯基、间甲基苯基、间氯苯基、对甲基苯基、对甲氧基苯基、对氟苯基、对氯苯基、对溴苯基、2-萘基、2-噻吩基中的一种;所述r3选自苯基、邻甲基苯基、间甲基苯基、对甲基苯基、邻氟苯基、间氟苯基、间氯苯基、间溴苯基、对氟苯基、对氯苯基、对溴苯基、2-萘基、2-吡啶基、苄基中的一种。
本发明提供了一类全新的含有三氟甲基手性马来酰亚胺类化合物,该类化合物所具有的三氟甲基及马来酰亚胺骨架广泛存在于临床药物分子中,极大丰富了手性药物候选分子库。
本发明的目的之二,在于提供上述的手性三氟甲基取代的马来酰亚胺衍生物的制备方法,以解决前述的现有合成方法中适用范围窄、化合物中无法引入关键的含氟基团等问题;
采用的技术方案为,分别称取如下式(ⅰ)的2-芳烯基琥珀酰亚胺、手性有机催化剂,溶于有机溶剂中,搅拌下加入如下式(ⅱ)的β-三氟甲基烯酮,继续搅拌,反应完全后,分离纯化,即得;
反应通式如下:
上述反应式中的“手性有机催化剂”即环己二胺衍生的硫脲催化剂;后文实施例中的“手性有机催化剂”也是指环己二胺衍生的硫脲催化剂;需要说明的是,实施例中所述的“手性催化剂a”、“手性催化剂b”、“手性催化剂c”和“手性催化剂d”,是指与上式方框内的a、b、c、d对应的取代化合物。另外,本领域技术人员可以理解的,本申请所采用的手性催化剂,并不限于上面的四种,凡是环己二胺硫脲为母体,r取代基做类似的变换,也可以实现本发明。
所述2-芳烯基琥珀酰亚胺(ⅰ)具有如下结构:
其中,r1基选自各种类型保护基,r2基选自芳基、杂芳基、稠芳基;
所述β-三氟甲基烯酮(ⅱ)具有如下结构:
其中,r3基选自芳基、杂芳基、烷基。
本发明通过不对称非共轭michael加成反应/1,3-氢迁移,一步实现了含三氟甲基的手性马来酰亚胺衍生物的制备;
而且,本发明的方法,底物的烯基端位可以有多种类型的取代基,也能够发生1,3-氢迁移并生成手性马来酰亚胺化合物,底物的普适性大大提高。
作为优选的技术方案:所述有机溶剂选自甲苯、甲基叔丁基醚、乙醚、二氯甲烷、氯仿、四氢呋喃、乙酸乙酯及氯苯中的一种或者多种混合;
进一步优选二氯甲烷;因为在该溶剂下反应,反应的时间最短,收率最高且立体选择性最好。
作为优选的技术方案:所述手性催化剂用量最低为10mol%。可以节约手性催化剂的用量,节约成本。
作为优选的技术方案:所述式(i)的2-芳烯基琥珀酰胺与式(ⅱ)的β-三氟甲基烯酮的摩尔比为1:1~4:1;进一步优选为2:1;因为经过试验证明:摩尔比小于2:1时,收率降低,而摩尔比大于2:1时,收率没有提高,且浪费试剂。
作为优选的技术方案:所述(ⅱ)的β-三氟甲基烯酮的浓度为0.1~0.4mol/l,进一步优选0.33mol/l,因为在该浓度下反应,反应时间较短,收率最高,溶剂用量最少。
作为优选的技术方案:所述反应的温度为0-50℃,进一步优选反应温度为30℃,因为在该反应温度下,反应能耗较低,反应的收率及立体选择性最好。
作为优选的技术方案:所述反应的时间为1~3天。
与现有技术相比,本发明的优点在于:本发明首次实现了有机小分子催化剂催化的2-芳烯基琥珀酰亚胺与β-三氟甲基烯酮参与的不对称非共轭michael反应,通过不对称加成/1,3-氢迁移策略一步反应实现了含有三氟甲基手性马来酰亚胺类化合物的不对称构建;本方法具有操作简便,后处理简单,反应条件温和,收率及立体选择性高,具有原子经济性的优点。
附图说明
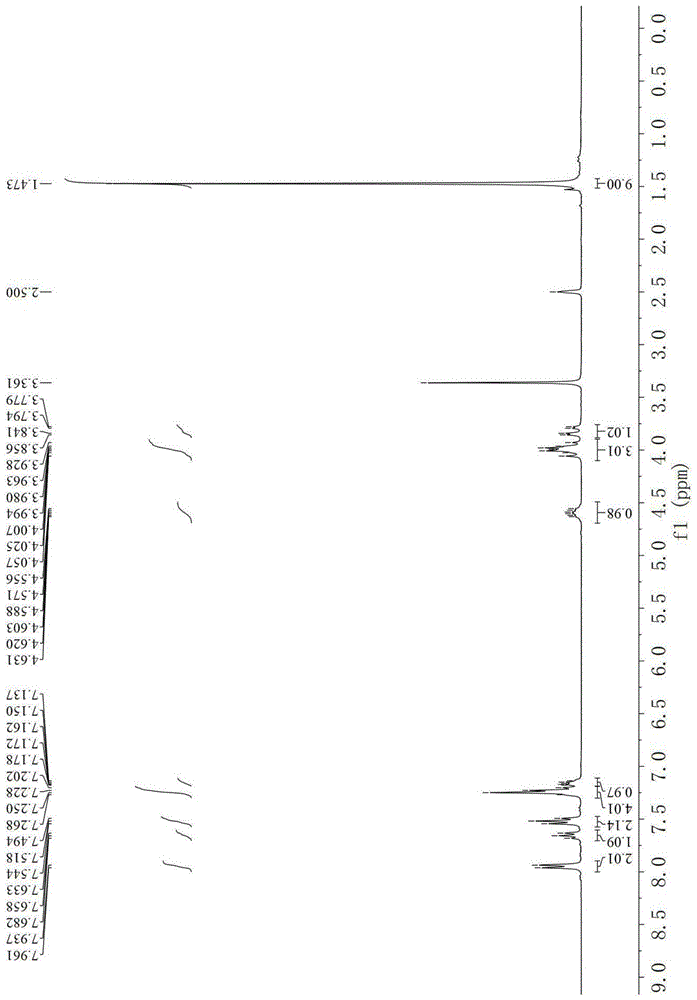
图1为实施例1制得的ⅲ-a的氢谱;
图2为实施例1制得的ⅲ-a的碳谱;
图3为实施例1制得的ⅲ-a的高分辨质谱图;
图4为实施例5制得的ⅲ-g的氢谱;
图5为实施例5制得的ⅲ-g的碳谱;
图6为实施例5制得的ⅲ-g的高分辨质谱图。
具体实施方式
下面将结合附图对本发明作进一步说明。
实施例1:合成化合物(ⅲ-a)
方法1:在反应试管中,将手性有机催化剂a(0.02mmol)及2-芳烯基琥珀酰亚胺i-a(0.2mmol)溶解在0.3ml二氯甲烷中;然后加入β-三氟甲基烯酮ⅱ-a(0.1mmol,反应物浓度为0.33mol/l),反应混合液在30℃下继续搅拌反应12小时(tlc监测)。经柱色谱分离纯化(石油醚:甲基叔丁基醚=20:1~6:1)得化合物ⅲ-a,收率为77%,98%ee;
方法2:在圆底烧瓶中,将手性有机催化剂a(0.8mmol)及2-苯烯基琥珀酰亚胺i-a(8.0mmol)溶解在12ml氯仿中;然后加入β-三氟甲基烯酮ⅱ-a(4.0mmol,反应物浓度为0.33mol/l),反应混合液在30℃下继续搅拌反应32小时(tlc监测);直接经柱色谱分离纯化(石油醚:甲基叔丁基醚=20:1~6:1)得化合物ⅲ-a,白色固体;收率为77%;95%ee.;[α]d20=-75.2(c1.00,ch2cl2);m.p.109.2-110.2℃;采用hplc(chiralpakod-h色谱柱)测定ee值(流动相:己烷/乙醇=85/15;流速:1.0ml/min;λ=254nm;tmajor=7.48min,tminor=9.46min);1hnmr(300mhz,dmso-d6)δ7.95(d,j=7.3hz,2h),7.66(t,j=7.3hz,1h),7.52(t,j=7.6hz,2h),7.27-7.20(m,4h),7.19-7.11(m,1h),4.63-4.56(m,1h),4.10-3.90(m,3h),3.82(dd,j=18.7,4.5hz,1h),1.47(s,9h);13cnmr(75mhz,dmso-d6)δ195.4,165.7,165.4,145.2,144.8,136.0,135.4,134.6,133.6,128.6,128.5,128.4,128.1,126.5,125.8(q,j=280.6hz),84.2,35.4,35.3(q,j=29.3hz),29.3,27.4;hrms(esi)calcd.forc26h24f3nnao5[m+na]+:510.1499;found:510.1487;如图1-3所示。
实施例2:合成化合物(ⅲ-b)
在反应试管中,手性有机催化剂b(0.02mmol)及2-邻甲基苯烯基琥珀酰亚胺i-b(0.4mmol)溶解在0.5ml乙酸乙酯中;然后加入β-三氟甲基烯酮ⅱ-a(0.1mmol,反应物浓度为0.20mol/l),反应混合液在30℃下继续搅拌反应20小时(tlc监测);经柱色谱分离纯化(石油醚:甲基叔丁基醚=20:1~6:1)得化合物ⅲ-b,黄色油状物;收率为79%;85%ee.;[α]d20=-21.5(c1.00,ch2cl2);m.p.46.9-48.2℃.采用hplc(chiralpakod-h色谱柱)测定ee值(流动相:己烷/乙醇=85/15;流速:1.0ml/min;λ=254nm;tmajor=4.51min,tminor=5.88min);1hnmr(300mhz,dmso-d6)δ7.96-7.88(m,2h),7.65(t,j=7.3hz,1h),7.51(t,j=7.6hz,2h),7.15(d,j=7.3hz,1h),7.10-6.90(m,3h),4.48(td,j=9.4,4.7hz,1h),4.00-3.86(m,3h),3.78(dd,j=18.7,4.7hz,1h),2.34(s,3h),1.48(s,9h);13cnmr(75mhz,dmso-d6)δ195.5,165.6,165.4,145.2,145.1,135.9,135.4,135.3,134.6,133.7,130.0,128.7,128.1,127.7,126.6,125.9,125.8(q,j=280.5hz),84.3,35.4,35.3(q,j=29.6hz).27.4,26.8,19.4;hrms(esi)calcd.forc27h26f3nnao5[m+na]+:524.1655;found:524.1652。
实施例3:合成化合物(ⅲ-c)
在反应试管中,手性有机催化剂d(0.01mmol)及2-邻氟苯烯基琥珀酰亚胺i-c(0.3mmol)溶解在0.3ml二氯甲烷中;然后加入β-三氟甲基烯酮ⅱ-a(0.1mmol,反应物浓度为0.33mol/l),反应混合液在40℃下继续搅拌反应12小时(tlc监测);经柱色谱分离纯化(石油醚:甲基叔丁基醚=20:1~6:1)得化合物ⅲ-c,黄色油状物;61%收率;72%ee.;[α]d20=-28.8(c1.00,ch2cl2);m.p.80.6-81.1℃.采用hplc(chiralpakod-h色谱柱)测定ee值(流动相:己烷/i-proh=90/10;流速:1.0ml/min;λ=254nm;tmajor=6.34min,tminor=10.57min);1hnmr(300mhz,dmso-d6)δ8.00-7.91(m,2h),7.72-7.60(m,1h),7.52(t,j=7.6hz,2h),7.27-7.21(m,2h),7.18-7.03(m,2h),4.55(td,j=9.7,4.6hz,1h),4.10-3.89(m,3h),3.81(dd,j=18.7,4.6hz,1h),1.47(s,9h);13cnmr(75mhz,dmso-d6)δ195.3,165.4,165.2,160.0(d,j=244.5hz),145.2,143.9,135.5,135.2,133.7,130.5(d,j=3.7hz),128.8(d,j=8.2hz),128.7,128.1,125.8(q,j=280.6hz),124.4(d,j=3.3hz),123.0(d,j=15.2hz),115.2(d,j=21.7hz),84.3,35.4,35.3(q,j=29.4hz),27.4,22.6(d,j=4.0hz);hrms(esi)calcd.forc26h23f4nnao5[m+na]+:528.1405;found:528.1410。
实施例4:合成化合物(ⅲ-d)
在反应试管中,手性有机催化剂d(0.02mmol)及2-间氯苯烯基琥珀酰亚胺i-d(0.2mmol)溶解在1.0ml四氢呋喃中;然后加入β-三氟甲基烯酮ⅱ-a(0.1mmol,反应物浓度为0.10mol/l),反应混合液在50℃下继续搅拌反应48小时(tlc监测);经柱色谱分离纯化(石油醚:甲基叔丁基醚=20:1~6:1)得化合物ⅲ-d,黄色固体;67%收率;79%ee.;[α]d20=-21.0(c1.00,ch2cl2);m.p.57.4-58.6℃;采用hplc(chiralpakod-h色谱柱)测定ee值(流动相:己烷/乙醇=85/15;流速:1.0ml/min;λ=254nm;tmajor=5.24min,tminor=7.43min);1hnmr(300mhz,dmso-d6)δ8.00-7.90(m,2h),7.66(t,j=7.3hz,1h),7.52(t,j=7.6hz,2h),7.36(s,1h),7.30-7.16(m,3h),4.61(td,j=9.1,4.3hz,1h),4.11-3.91(m,3h),3.81(dd,j=18.8,4.3hz,1h),1.47(s,9h).13cnmr(100mhz,dmso)δ195.5,165.7,165.4,145.2,138.7,135.4,135.1,133.8,133.1,130.2,128.8,128.6,128.1,127.4,126.6,125.9(q,j=279.3hz),84.3,38.9,35.4,35.3(q,j=28.5hz),28.9,27.5;hrms(esi)calcd.forc26h23clf3nnao5[m+na]+:544.1109;found:544.1124。
实施例5:合成化合物(ⅲ-e)
在反应试管中,手性有机催化剂c(0.02mmol)及2-萘烯基琥珀酰亚胺i-e(0.12mmol)溶解在0.3ml乙酸乙酯中;然后加入β-三氟甲基烯酮ⅱ-a(0.1mmol,反应物浓度为0.33mol/l),反应混合液在30℃下继续搅拌反应72小时(tlc监测);经柱色谱分离纯化(石油醚:甲基叔丁基醚=20:1~6:1)得化合物ⅲ-e,白色固体;收率为63%;95%ee.;[α]d20=-80.1(c1.00,ch2cl2);m.p.83.8-84.8℃;采用hplc(chiralpakod-h色谱柱)测定ee值(流动相:己烷/i-proh=90/10;流速:1.0ml/min;λ=254nm;tmajor=8.81min,tminor=12.38min);1hnmr(300mhz,dmso-d6)δ7.94-7.87(m,2h),7.84-7.79(m,2h),7.72-7.70(m,2h),7.63-7.58(m,1h),7.47-7.42(m,5h),4.72-4.56(m,1h),4.25-3.97(m,3h),3.83(dd,j=18.6,4.4hz,1h),1.47(s,9h);13cnmr(75mhz,dmso-d6)δ195.2,166.3,165.8,145.8,145.7,135.3,134.6,133.9,133.6,132.8,132.4,128.8,128.7,128.0,127.8,127.7,127.6,126.9,126.4,126.0,125.8(q,j=280.6hz),85.6,36.9(q,j=29.7hz),35.7,30.0,28.0;hrms(esi)calcd.forc30h26f3nnao5[m+na]+:560.1655;found:560.1662;如图4-6所示。
实施例6:合成化合物(ⅲ-f)
在反应试管中,手性有机催化剂a(0.015mmol)及2-萘烯基琥珀酰亚胺i-f(0.2mmol)溶解在0.8ml乙腈中;然后加入β-三氟甲基烯酮ⅱ-a(0.1mmol,反应物浓度为0.125mol/l),反应混合液在30℃下继续搅拌反应24小时(tlc监测);经柱色谱分离纯化(石油醚:甲基叔丁基醚=20:1~6:1)得化合物ⅲ-f,白色固体;收率为65%;96%ee.;[α]d20=-23.6(c1.00,ch2cl2);采用hplc(chiralpakod-h色谱柱)测定ee值(流动相:己烷/
i-proh=90/10;流速:1.0ml/min;λ=254nm;tmajor=7.64min,tminor=13.28
min);1hnmr(300mhz,dmso-d6)δ7.95-7.92(m,2h),7.66(t,j=7.3hz,
1h),7.52(t,j=7.6hz,2h),7.42(dd,j=4.9,2.9hz,1h),7.30-7.22(m,1h),7.02(dd,j=4.9,1.4hz,1h),4.55(td,j=9.6,4.6hz,1h),4.07-3.87(m,3h),3.81(dd,j=18.7,4.6hz,1h),1.48(s,9h);13cnmr(100mhz,dmso)δ195.4,165.8,165.5,145.3,144.47,135.4,135.3,134.1,133.8,128.7,128.3,128.1,126.2,125.9(d,j=280.6hz),122.6,84.3,35.5,35.1(q,j=29.2hz),27.5,24.3;hrms(esi)calcd.forc24h22f3nnao5s[m+na]+:516.1063;found:516.1075.
实施例7:合成化合物(ⅲ-g)
在反应试管中,手性有机催化剂b(0.02mmol)及2-苯烯基琥珀酰亚胺i-a
(0.2mmol)溶解在0.3ml二氯甲烷中;然后加入β-三氟甲基烯酮ⅱ-b(0.1mmol,反应物浓度为0.33mol/l),反应混合液在30℃下继续搅拌反应10小时(tlc监测);经柱色谱分离纯化(石油醚:甲基叔丁基醚=20:1~6:1)得化合物ⅲ-g,白色固体;收率为98%;86%ee.;[α]d20=-56.6(c1.00,ch2cl2);m.p.45.6-46.6℃.采用hplc(chiralpakod-h色谱柱)测定ee值(流动相:己烷/i-proh=90/10;流速:1.0ml/min;λ=254nm;tmajor=7.94min,tminor=8.81min);1hnmr(300mhz,dmso-d6)δ8.72(d,j=4.7hz,1h),8.01(td,j=7.7,1.7hz,1h),7.93(d,j=7.7hz,1h),7.74-7.63(m,1h),7.30-7.08(m,5h),4.62-4.54(m,1h),4.11(dd,j=19.0,9.5hz,1h),4.04-3.84(m,3h),1.47(s,9h).13cnmr(100mhz,dmso-d6)δ196.6,165.7,165.3,151.7,149.2,145.3,137.6,136.0,134.4,128.6,128.5,128.3,127.34,126.6,125.9(q,j=281.1hz),121.6,84.3,35.4(q,j=28.8hz),34.8,29.3,27.4;hrms(esi)calcd.forc25h23f3n2nao5[m+na]+:511.1451;found:511.1453。
实施例8:合成化合物(ⅲ-h)
在反应试管中,手性有机催化剂a(0.02mmol)及2-苯烯基琥珀酰亚胺i-a(0.2mmol)溶解在0.3ml氯苯中。然后加入β-三氟甲基烯酮ⅱ-c(0.1mmol,反应物浓度为0.33mol/l),反应混合液在30℃下继续搅拌反应48小时(tlc监测);经柱色谱分离纯化(石油醚:甲基叔丁基醚=20:1~6:1)得化合物ⅲ-g,白色固体;收率为43%;98%ee.;[α]d20=+13.2(c1.00,ch2cl2);采用hplc(chiralpakod-h色谱柱)测定ee值(流动相:己烷/i-proh=95/5;流速:1.0ml/min;λ=254nm;tminor=6.52min,tmajor=8.60min);1hnmr(300mhz,dmso-d6)δ7.34-7.18(m,8h),7.14-7.11(m,2h),4.37(td,j=9.5,4.8hz,1h),3.98-3.76(m,3h),3.68(d,j=16.4hz,1h),3.45(dd,j=18.4,9.1hz,1h),3.33-3.24(m,1h),1.48(s,9h);13cnmr(100mhz,dmso-d6)δ203.9,165.6,165.1,145.2,145.0,136.1,134.2,134.0,129.7,128.6,128.5,128.3,126.8,126.7,125.7(q,j=280.4hz),84.3,48.1,38.3,35.0(q,j=28.1hz),29.2,27.4;hrms(esi)calcd.forc27h26f3nnao5[m+na]+:524.1655;found:524.1655。
以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!