壳寡糖-N-芳樟醇共聚物及其制备方法与应用与流程
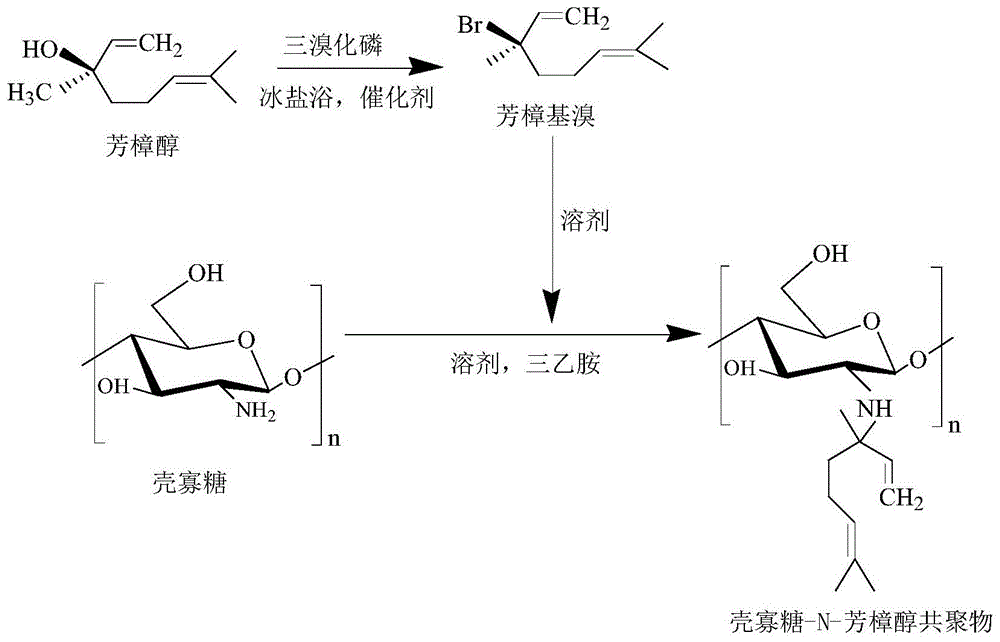
本发明涉及抗菌剂、抗炎剂技术领域,具体涉及一种壳寡糖-n-芳樟醇共聚物,其制备方法以及作为抗菌剂/抗炎剂的应用。
背景技术:
在自然界中存在大量由活生物产生并具有特殊生物学活性的天然产物。主要是从动物、植物组织、海洋生物和微生物体内的组成成分或其代谢产物以及人和动物体内提取出的许多内源性的化学成分。比如蛋白质、多肽、氨基酸、多糖、黄铜、萜类、酚类醌类、内酯、甾体化合物、鞣酸类、抗生素类等物质。由于诸多的天然产物具有一些特殊的功能特性,如抗氧化、抗菌、抗肿瘤、抗炎等活性,在医药、食品、化妆品和农业等领域被广泛应用,同时近年来,也收到广大研究人员的关注,并已成为研究热点。
壳寡糖是壳聚糖主链经物理、化学或酶降解断裂后得到的低分子量碱性氨基寡糖。它是从虾、蟹等甲壳类动物的外壳中提取出来的,具有生物可降解性、生物相容性、生物无毒性和化学反应活性,在食品工业中常被用作天然抗菌剂和抗氧化剂使用,除此之外,壳寡糖还显示出一定的抗结肠炎的活性,因此可以将其作为一种潜在的抗炎剂进行开发。然而,由于壳寡糖为天然大分子产物,当作为抗炎剂使用时,与传统的常用抗炎药物相比,仍有活性低的缺点,所以目前对其研究非常有限。因此,可以通过对壳寡糖的结构进行化学修饰,由于其分子链上具有化学反应活性的氨基和羟基,这些位点均是对壳寡糖进行化学改性的理想作用位点,可以进一步提高其功能活性,这也是目前国内外研究较多的有效方法。芳樟醇,学名是3,7-二甲基-1,6-辛二烯-3-醇,属于链状萜烯醇类属于链状萜烯醇类,有α-和β-两种异构体。还有左旋、右旋两种光异构体。在不同来源的精油中,多为异构体的混合物,并具有一定的抗菌、抗炎活性和抗肿瘤活性。
技术实现要素:
本发明要解决的技术问题是提供一种壳寡糖-n-芳樟醇共聚物,该共聚物具有良好是水溶性、热稳定性和抗炎活性,在食品、医药、化妆品和农业等领域具有很好的应用前景。
为了解决上述技术问题,本发明提供了如下所述的技术方案:
本发明第一方面提供了一种壳寡糖-n-芳樟醇共聚物,具有如下所示的结构式:
其中,4≤n≤20。
本发明第二方面提供了第一方面所述的壳寡糖-n-芳樟醇共聚物的制备方法,包括以下步骤:
在催化剂存在的条件下,将壳寡糖溶液滴加到芳樟基溴溶液中进行反应;反应结束后,分离产物,得到壳寡糖-n-芳樟醇共聚物。
壳寡糖分子链中有三个化学反应位点:c-2位的氨基和c-3、c-6的羟基,c-2位氨基氮上的电子云密度最大,亲核性最强,而c-3和c-6位羟基电子云密度较c-2位的小,且由于受到空间位阻效应的影响,所以烷基化反应首先发生在c-2位的氨基上。
进一步地,所述壳寡糖的分子量≤3000da,脱乙酰度为85%-95%;多分散性指数优选为0.88。
进一步地,溶解壳寡糖与芳樟基溴所采用的有机溶剂可为n,n-二甲基甲酰胺、二甲基亚砜、四氢呋喃、乙酸异丙酯中的一种或多种。优选地,所述有机溶剂的添加量与壳寡糖的体积质量比为20:1-60:1(ml:g)。
进一步地,所述壳寡糖与芳樟基溴的质量体积比为1:1-1:3(g:ml)。
进一步地,所述催化剂为三乙胺,其添加量与壳寡糖的体积质量比优选为0.6:1-1.8:1(ml:g)。
进一步地,所述反应的温度优选为45-60℃,反应的时间优选为6-8h。
进一步地,反应结束后,添加过量的有机溶剂终止反应,所述过量的有机溶剂优选为丙酮。
进一步地,所述分离产物具体为:反应结束后,离心收集沉淀,用有机溶剂对沉淀进行索氏抽提,真空干燥得到淡黄色粉末;将粉末于透析袋中透析一段时间,经浓缩后干燥,得纯化的壳寡糖-n-芳樟醇共聚物。其中,所述的有机溶剂可选择为丙酮、乙醇或石油醚,所述透析袋优选为截留分子量为77~100da的透析袋,透析时间优选为24h。
进一步地,合成产物壳寡糖-n-芳樟醇共聚物的取代度为0.7-0.9,得率为77%-85%。
进一步地,所述芳樟基溴的制备方法为:
在冰盐浴环境中,将三溴化磷溶液滴加到含有催化剂的芳樟醇溶液中,搅拌发生反应;反应结束后,依次用5%的碳酸氢钠溶液、去离子水及饱和食盐水洗涤,加入干燥剂干燥,过滤,旋转蒸发浓缩,获得淡黄色油状液体芳樟基溴。
进一步地,所述芳樟醇和三溴化磷的体积比优选为5:(2-3)。
进一步地,溶解芳樟醇与三溴化磷的有机溶剂可为无水乙醚、无水乙醇、丙二醇、二氯甲烷中的一种或多种。芳樟醇溶液中,有机溶剂与芳樟醇的体积比优选为(6-11):1;三溴化磷溶液中,有机溶剂与三溴化磷的体积比优选为(5-11):1。
进一步地,所述催化剂可为吡啶、三乙胺或二异丙基乙胺,催化剂的添加量与芳樟醇的体积比优选为(0.05-0.15):1。
进一步地,所述滴加的速度优选为1.0-1.5ml/min,所述搅拌的时间优选为45-90min。
进一步地,所述干燥剂可为无水硫酸钠、无水硫酸镁或无水碳酸钠。
进一步地,所述过滤采用的滤膜优选为0.22-0.45μm的滤膜。
进一步地,所述旋转蒸发浓缩的时间优选为30-45min,温度优选为30-50℃。
本发明第三方面还提供了第一方面所述的壳寡糖-n-芳樟醇共聚物作为抗菌剂或抗炎剂的应用。
与现有技术相比,本发明的有益效果在于:
1.本发明基于亚结构连接的思想,将具有抗炎活性的芳樟醇分子与壳寡糖的c2位活性氨基发生n烷基化反应,从而将芳樟醇引入到壳寡糖分子链中,得到壳寡糖-n-芳樟醇共聚物。抗菌实验结果显示,该壳寡糖-n-芳樟醇共聚物中的芳樟醇与壳寡糖分子能够产生协同作用,显著增强其抗菌活性;此外,该壳寡糖-n-芳樟醇共聚物还具有良好的抗炎活性。
2.本发明的壳寡糖-n-芳樟醇共聚物的制备过程和纯化过程简单,且具有低毒性、良好的水溶性、热稳定性和抗炎活性,在食品、医药、化妆品和农业等领域具有很好的应用前景。
附图说明
图1是壳寡糖-n-芳樟醇共聚物的合成路线图;
图2是壳寡糖、壳寡糖-n-芳樟醇共聚物的红外光谱图;
图3是壳寡糖、芳樟醇、壳寡糖-n-芳樟醇共聚物的1h-nmr图;
图4是壳寡糖、壳寡糖-n-芳樟醇共聚物的热重图;
图5是壳寡糖-n-芳樟醇聚合物对raw264.7细胞增殖的影响图;
图6是壳寡糖-n-芳樟醇聚合物对炎症因子的影响图。
具体实施方式
下面结合附图和具体实施例对本发明作进一步说明,以使本领域的技术人员可以更好地理解本发明并能予以实施,但所举实施例不作为对本发明的限定。
除非另有定义,本文所使用的所有的技术和科学术语与属于本发明的技术领域的技术人员通常理解的含义相同。本文中在本发明的说明书中所使用的术语只是为了描述具体的实施例的目的,不是旨在于限制本发明。本文所使用的术语“及/或”包括一个或多个相关的所列项目的任意的和所有的组合。
下述实施例中所使用的实验方法如无特殊说明,均为常规方法,所用的材料、试剂等,如无特殊说明,均可从商业途径得到。
实施例一:合成壳寡糖-n-芳樟醇共聚物
1、芳樟基溴的制备
先将5ml芳樟醇和2ml三溴化磷分别用30ml的无水乙醚溶解,在三口烧瓶中先加入芳樟醇溶液和0.45ml的吡啶,置于冰盐浴环境中快速搅拌,再将三溴化磷溶液以1.0ml/min的速度加入到上述反应体系中,滴加完毕后,继续快速45min,再分别依次用5%的碳酸氢钠溶液、去离子水及饱和食盐水洗涤3次,加入无水硫酸钠干燥,用。0.22μm的微孔滤膜过滤,在30℃下旋转蒸发浓缩30min获得淡黄色油状液体芳樟基溴。
2、壳寡糖-n-芳樟醇共聚物的制备
将1g壳寡糖与2ml芳樟基溴用n,n-二甲基甲酰胺分别溶解后,加入三乙胺作为催化剂,再用恒压漏斗将壳寡糖溶液逐滴缓慢加入芳樟基溴溶液中,在45℃下反应6h,反应结束后添加过量丙酮终止反应,离心收集沉淀。用乙醇进行索氏抽提12h,真空干燥得到褐色粉末,将该粉末在截留分子量为100da的透析袋中透析24h,经浓缩后干燥,最终获得纯化的壳寡糖-n-芳樟醇共聚物。
该壳寡糖-n-芳樟醇共聚物的得率为78%,使用红外光谱、核磁共振和热重分析对其进行表征。
图2a为壳寡糖的红外光谱图,其中,3422.36cm-1为o-h与n-h的伸缩振动吸收峰,2923.83cm-1为c-h伸缩振动的吸收峰,1628.76cm-1为nh2的弯曲振动吸收峰,1155.97cm-1与1071.52cm-1为c-o伸缩振动的吸收峰,893.24cm-1为环伸缩振动吸收峰。
图2b是本实施例中壳寡糖-n-芳樟醇共聚物的红外光图谱,其中,
1620cm-1处的双键吸收峰变显著,这是因为芳樟醇中的c=c吸收峰和壳寡糖的c=o吸收峰重合;1520cm-1处的氨基吸收峰减小;2920cm-1处的甲基吸收峰增加,3400cm-1处的特征吸收峰峰宽变小且强度减弱,这表明壳寡糖-n-芳樟醇共聚物合成成功。
图2a是本实施例中壳寡糖的1h-nmr图,δ=2.05-2.25ppm可归属为壳寡糖乙酰氨基残基上-ch3的质子峰;δ=3.19可归属为壳寡糖氨基葡萄糖残基和乙酰氨基残基上h2的质子峰;δ=3.21-3.92ppm可归属为氨基葡萄糖和乙酰氨基葡萄糖上相应的h-3,h-4,h-5,h-6处的质子峰。
图2c是本实施例中壳寡糖-n-芳樟醇共聚物的1h-nmr图,化学位移出现在2.04-2.22ppm处的峰对应的是乙酰氨基残基上的-ch3的质子峰;3.50-3.96ppm处的峰对应的是h2,h3,h4,h5,h6处的质子峰;4.60ppm处的峰对应的是h1处的质子峰;2.72ppm处的峰为溶剂峰;1.26-1.86ppm处的峰对应的是3,7-二甲基-1,6-辛二烯-3-醇骨架上h-4,h-5,h-8,h-9处的质子峰;5.16-5.45ppm处的峰对应的是3,7-二甲基-1,6-辛二烯-3-醇骨架上h-1,h-2,h-6处的质子峰。
因此,通过以上化学位移的归属并结合红外图谱解析,可以证明,壳寡糖-n-芳樟醇共聚物制备成功。
图3a为本实施例中壳寡糖的热稳定性图,第一阶段在30-100℃,热失重率约7%,主要是样品本身带有的水分蒸发所致;第二个热分解区间130-380℃之间,热失重率约52%,是一个比较复杂的分解过程,包括糖环的降解和样品中大分子链的分解等。在第二个热分解区间,出现了tmax,即最大降解速率时的温度,tmax=200℃。
图3b为本实施例中壳寡糖-n-芳樟醇共聚物的热稳定性图,热失重过程也由两个阶段构成,但热分解温度却有所不同。在100℃之前的第一阶段,约失重6%,为样品中吸附水和结晶水和其他小分子物质的挥发所致。在130-380℃之间的第二个热分解区间,失重约49%,在这个过程中,发生了壳寡糖糖环类的脱水、解聚和烷基化单元的聚合物的分解等。其中,tmax=220℃。热重实验的结果表明,壳寡糖-n-芳樟醇共聚物的热稳定性高于原料壳寡糖,由此推断在壳寡糖的糖链中引芳樟醇之后,壳寡糖分子排列发生变化,结晶性增强,结构更加规整、一致,得到壳寡糖-n-芳樟醇共聚物的结晶结构变得更加规整和有序,因而结晶性增强,热稳定性增强。
实施例二:合成壳寡糖-n-芳樟醇共聚物
1、芳樟基溴的制备
先将5ml芳樟醇和2.5ml三溴化磷分别用40ml的无水乙醚溶解,在三口烧瓶中先加入芳樟醇溶液和0.5ml的吡啶,置于冰盐浴环境中快速搅拌,再将三溴化磷溶液以1.2ml/min的速度加入到上述反应体系中,滴加完毕后,继续快速50min,再分别依次用5%的碳酸氢钠溶液、去离子水及饱和食盐水洗涤3次,加入无水硫酸钠干燥,用。0.22μm的微孔滤膜过滤,在35℃下旋转蒸发浓缩40min获得淡黄色油状液体芳樟基溴。
2、壳寡糖-n-芳樟醇共聚物
将1g壳寡糖与3ml芳樟基溴用二甲基亚砜分别溶解后,加入三乙胺作为催化剂,再用恒压漏斗将壳寡糖溶液逐滴缓慢加入芳樟基溴溶液中,在50℃下反应7h,反应结束后添加过量丙酮终止反应,离心收集沉淀。用乙醇进行索氏抽提12h,真空干燥得到淡黄色粉末,将该粉末在截留分子量为100da的透析袋中透析24h,经浓缩后干燥,最终获得纯化的壳寡糖-n-芳樟醇共聚物。
该壳寡糖衍生物的得率为80%,使用红外光谱、核磁共振和热重分析对其进行表征。
图1c是本实施例中壳寡糖-n-芳樟醇共聚物的红外光图谱,其中,1623cm-1处的双键吸收峰变显著是由于芳樟醇中的c=c吸收峰和壳寡糖的c=o吸收峰重合;1522cm-1处的氨基吸收峰减小;2922cm-1处的-ch3吸收峰增加,3402cm-1处的特征吸收峰峰宽变小且强度减弱均表明壳寡糖-n-芳樟醇共聚物合成成功。
因此,通过以上化学位移的归属并结合红外图谱解析,可以证明,壳寡糖-n-芳樟醇共聚物制备成功。
图3c为本实施例中壳寡糖-n-芳樟醇共聚物的热稳定性图,热失重过程也由两个阶段构成,但热分解温度却有所不同。在100℃之前的第一阶段,约失重5.8%,为样品中吸附水和结晶水和其他小分子物质的挥发所致。在130-380℃之间的第二个热分解区间,失重约47%,在这个过程中,发生了壳寡糖糖环类的脱水、解聚和烷基化单元的聚合物的分解等。其中,tmax=219℃。热重实验的结果表明,壳寡糖-n-芳樟醇共聚物的热稳定性高于原料壳寡糖,由此推断在壳寡糖的糖链中引入芳樟醇之后,壳寡糖分子排列发生变化,结晶性增强,结构更加规整、一致,得到壳寡糖-n-芳樟醇共聚物的结晶结构变得更加规整和有序,因而结晶性增强,热稳定性增强。
实施例三:合成壳寡糖-n-芳樟醇共聚物
1、芳樟基溴的制备
先将5ml芳樟醇和3ml三溴化磷分别用50ml的无水乙醇溶解,在三口烧瓶中先加入芳樟醇溶液和0.5ml的三乙胺,置于冰盐浴环境中快速搅拌,再将三溴化磷溶液以1.5ml/min的速度加入到上述反应体系中,滴加完毕后,继续快速80min,再分别依次用5%的碳酸氢钠溶液、去离子水及饱和食盐水洗涤3次,加入无水硫酸钠干燥,用0.45μm的微孔滤膜过滤,在45℃下旋转蒸发浓缩50min获得淡黄色油状液体芳樟基溴。
2、壳寡糖-n-芳樟醇共聚物的制备
将1g壳寡糖与5ml芳樟基溴用四氢呋喃分别溶解后,加入三乙胺作为催化剂,再用恒压漏斗将壳寡糖溶液逐滴缓慢加入芳樟基溴溶液中,在45℃下反应8h,反应结束后添加过量丙酮终止反应,离心收集沉淀。用石油醚进行索氏抽提12h,真空干燥得到褐色粉末,将该粉末在截留分子量为100da的透析袋中透析24h,经浓缩后干燥,最终获得纯化的壳寡糖-n-芳樟醇共聚物。
该壳寡糖衍生物的得率为85%,使用红外光谱、核磁共振和热重分析对其进行表征。
图1d是本实施例中壳寡糖-n-芳樟醇共聚物的红外光图谱,其中,1621cm-1处的双键吸收峰变显著是由于芳樟醇中的c=c吸收峰和壳寡糖的c=o吸收峰重合;1521cm-1处的氨基吸收峰减小;2925cm-1处的-ch3吸收峰增加,3402cm-1处的特征吸收峰峰宽变小且强度减弱均表明壳寡糖-n-芳樟醇共聚物合成成功。
因此,通过以上化学位移的归属并结合红外图谱解析,可以证明,壳寡糖-n-芳樟醇共聚物制备成功。
图4d为本实施例中壳寡糖-n-芳樟醇共聚物的热稳定性图,热失重过程也由两个阶段构成,但热分解温度却有所不同。在100℃之前的第一阶段,约失重5.5%,为样品中吸附水和结晶水和其他小分子物质的挥发所致。在130-380℃之间的第二个热分解区间,失重约47%,在这个过程中,发生了壳寡糖糖环类的脱水、解聚和烷基化单元的聚合物的分解等。其中,tmax=218℃。热重实验的结果表明,壳寡糖-n-芳樟醇共聚物的热稳定性高于原料壳寡糖,由此推断在壳寡糖的糖链中引芳樟醇之后,壳寡糖分子排列发生变化,结晶性增强,结构更加规整、一致,得到壳寡糖-n-芳樟醇共聚物的结晶结构变得更加规整和有序,因而结晶性增强,热稳定性增强。
实施例四:抗菌实验
(1)实验菌种:
两种革兰氏阳性菌:金黄色葡萄球菌(atcc25923),酿脓链球菌(atcc12344);两种革兰氏阴性菌:大肠杆菌(atcc25922),鼠伤寒沙门氏菌(cmcc50013)。
(2)操作步骤:
本实验采用lb培养基,将供试菌先移入相应的培养基,于37℃下培养24小时,用挑种环挑取少许菌体,放入装有无菌水的试管内,振动摇匀,职称菌悬液,采用比浊法计数,调整菌悬液浓度,使其含菌数约为107个/毫升,备用。
取直径为6毫米的滤纸片放入一定浓度的抑菌溶液中浸泡6小时,取出干燥。将各种供试菌悬液取0.2毫升分别注入直径为120毫米的无菌培养皿中培养,再倒入20毫升lb培养基,混匀后冷却,用无菌镊子夹取含浸出液的滤纸片贴在含菌平板上,每皿贴6片,每组重复3次,置于37℃培养24小时,测定滤纸片周围抑菌圈直径大小,观察抑菌效果。在上述试验条件下,使用同样浓度的苯甲酸钠、山梨酸钾进行对比实验,实验结果见表1。
表1抗菌实验结果
从表1的结果可知,壳寡糖-n-芳樟醇共聚物对金黄色葡萄球菌、酿脓链球菌、枯草芽孢杆菌和大肠杆菌均具有很好的抗菌活性,与苯甲酸钠、山梨酸钾相比,实施例的抗菌效果明显优于其他组,因此在食品防腐方面具有潜在的应用价值。
实施例五:对raw264.7细胞增殖的影响
将对数生长期生长良好的小鼠巨噬细胞(raw264.7),通过传代处理,调整细胞密度为5×105个/ml,接种于96孔板,每孔100μl。细胞均匀贴壁后,吸去培养液,给药组加入不同浓度梯度的含药完全培养基100μl,每个浓度平行6个复孔。于培养箱培养24h后,吸弃上清液,每孔加入100μl含mtt的培养基,37℃培养4h。小心吸弃上清,每孔加入150μldmso,震摇10min混匀,酶标仪490nm测定吸光度,计算细胞存活率。所得结果如图5所示。
由图5可以看出,不同浓度的壳寡糖-n-芳樟醇聚合物对raw264.7细胞的毒性仍然较小,细胞存活率较高。给药浓度即使到600μm时,仍有至少93%的存活率。
实施例六:抗炎实验
将对数生长期生长良好的小鼠巨噬细胞(raw264.7),通过传代处理,调整细胞密度5×105个/ml,接种于96孔板,每孔100μl,于37℃、5%co2培养箱中稳定培养24h,小心吸弃上清,分组给药,给药1h后,炎症模型组与给药组加入适量lps,使其终浓度为5μg/ml,空白组给予等量的pbs,继续培养24h,收集上清液,-20℃储存备用。分别取各给药组细胞上清液以及各浓度标准品稀释液50μl于96孔板中,向其中加入50μla液(磺胺10mg/ml+0.06%浓磷酸),再加入50μlb液(n-1-萘乙二胺盐酸盐,1mg/ml),振荡10min,酶标仪测定546nm处吸光度值,用elisacalc软件进行计算。同法使用lps诱导细胞炎症模型,收集上清液,按照各炎症因子elisa试剂盒说明书操作,测定炎性因子no的含量。
如图6所示,正常raw264.7细胞在未受lps刺激的情况下几乎不分泌炎性介质no,然而,细胞在受到lps刺激后,炎症模型组较对照组分泌大量的no,而阳性药布洛芬组no浓度较炎症模型组显著性降低(p<0.01),说明此炎症模型造模成功。通过聚合物干预之后,各实验组中no的分泌均受到不同程度的抑制作用,并且对no的抑制作用与阳性药布洛芬几乎相当。因此本发明的壳寡糖-n-芳樟醇聚合物在体外抗炎活性方面达到了增效减毒的作用。
以上所述实施例仅是为充分说明本发明而所举的较佳的实施例,本发明的保护范围不限于此。本技术领域的技术人员在本发明基础上所作的等同替代或变换,均在本发明的保护范围之内。本发明的保护范围以权利要求书为准。
- 还没有人留言评论。精彩留言会获得点赞!