一种双齿噁唑啉手性配体的制备方法与流程
本申请属于有机合成技术领域,具体涉及一种双齿噁唑啉手性配体的制备方法。
背景技术:
手性是自然界最重要的属性之一。自然界中构成生命体的基础物质核苷酸、氨基酸和单糖以及由它们构成的生物大分子核酸、蛋白质和糖类都具有独特的手性特征。许多物理、化学、生物功能的产生都起源于分子手性的精确识别和严格匹配,例如手性药物的手性对其生物应答关系等。在化学制药领域,药物化合物的手性直接关系到药物的药理作用、临床效果、毒副作用、药效发挥及药效时间等,例如不同的对映体表现出不同的活性(例如布洛芬)、一种对映体表现出活性而另一对映体则不具有活性(例如氯霉素)、两种对映体具有相反的活性(例如依托唑啉)、以及对映体表现出有毒或强副作用(例如反应停)。
大量的研究成果表明,每一个不对称反应的发现,都会促使一些新的手性药物走向市场,而手性药物的发展又促进着手性合成的发展。不对称催化合成是通过使用手性催化剂,把非手性原料转化为手性产物的方法。在不对称合成的诸多方法中,不对称催化合成是最理想方法,它具有手性增殖、高对映选择性、经济,易于实现工业化的优点,其中的手性实体仅为催化量。不对称催化合成所使用的手性催化剂,其由活性的金属中心和手性配体构成,而手性配体是影响反应立体选择性的关键因素。
发明人课题组的组员前期报道了一种锌催化的炔醇醚类底物与噁唑的[4+3]不对称催化环化反应,通过6π电环化途径,高立体选择性地获得2h-氮杂
然而,在该文献并未提及这种新颖而高效的双齿噁唑啉类手性配体(式ia)的制备方法。尽管一些典型的有机合成手段,例如经典人名反应已经被广泛应用于制备获得所需要的目标化合物,而发明人课题组基于目前所收到的反馈来看,有机合成领域的研究人员似乎在制备这类配体的过程中,仍然难以找到一种行之有效的合成策略,以保证配体的手性构型并以所需要的量来获得前述配体。基于此,发明人在本发明中,公开所述配体的全合成路线,并以期获得专利保护。
技术实现要素:
本发明的目的在于克服现有技术的不足,提供一种双齿噁唑啉手性配体的制备方法,该方法使用简单、经济廉价的起始原料和有机合成试剂,通过对合成路线的合理设计并优化反应条件,方便并以高产率地制备获得所述双齿噁唑啉手性配体。
作为本发明的第一个方面,本发明涉及如下具有式i-1~i-2所示的双齿噁唑啉手性配体:
作为本发明的第二个方面,本发明提供了一种式i所示的双齿噁唑啉手性配体的制备方法,当ar为苯基时,所述制备方法合成路线一如下:
其中,ar表示苯基;
或者当ar为2-萘基时,所述制备方法合成路线二如下:
根据本发明前述的制备方法,所述合成路线一和/或二中,所述步骤一反应条件包括:式a化合物:式a-1的arb(oh)2:碳酸钠:四(三苯基膦)钯的投料摩尔比为1:2.5:5:0.01;反应温度为120℃;反应时间为12h;反应溶剂为h2o/phme体积比为1:1的混合溶液;反应气氛为n2。
具体操作如下:在室温及n2保护下,向式a的3,5-二溴苯甲醛,式a-1的arb(oh)2,碳酸钠在h2o/phme中的溶液中,加入四(三苯基膦)钯,然后将反应混合物在120℃搅拌,并通过tlc检测反应进程,完成后,经后处理得到所需产物式b化合物。
根据本发明前述的制备方法,所述合成路线一和/或二中,所述步骤二反应条件包括:式b化合物:三甲基碘化锍:koh:h2o的投料摩尔比为1:1.2:2.0:0.25;反应温度为60℃;反应时间为7h;反应溶剂为mecn。
具体操作如下:在室温下将三甲基碘化锍添加到式b化合物,氢氧化钾和水的乙腈混合物中,在60℃下搅拌反应,并通过tlc监测反应进程,式b化合物反应完全后,经后处理得到所需产物式c化合物。
根据本发明前述的制备方法,所述合成路线一和/或二中,所述步骤三反应条件包括:式c化合物:浓硫酸的投料摩尔比为1:5;反应温度为回流;反应时间为10min;反应溶剂为thf/h2o按体积比为3:1的混合溶液。
具体操作如下:在0℃下,将浓硫酸缓慢加入到式c化合物的thf/h2o混合液中,然后将反应混合液加热至回流,经tlc监测原料消失,将碳酸氢钠溶液添加至反应体系中以中和酸,然后经后处理得到所需式d化合物(2.97g,10.23mmol),产率67%。
根据本发明前述的制备方法,所述合成路线一和/或二中,所述步骤四反应条件包括:1)式d化合物:叔丁基二甲基氯硅烷:咪唑投料摩尔比为1:1.1:3;反应溶剂为dcm;反应温度为室温;反应时间为12h进行反应;和随后2)按式d化合物:戴斯-马丁试剂(dess-martinperiodinanecas:87413-09-0)投料摩尔比为1:1.2加入戴斯-马丁试剂;反应溶剂为dcm;反应温度为室温;反应时间为2h进行反应。
具体操作如下:在室温下,向式d化合物和咪唑在dcm中的溶液中缓慢加入叔丁基二甲基氯硅烷,并将反应混合物在室温搅拌12小时,完成后,将反应混合物用饱和氯化铵溶液(100ml)洗涤,分离有机层,用硫酸镁干燥并浓缩,粗产物无需进一步纯化即可用于下一步;
在室温下,将戴斯-马丁试剂(dess-martinperiodinanecas:87413-09-0)缓慢加入到粗产物在dcm中的混合物中,然后在室温下搅拌反应,并通过tlc检测反应进程,反应完成后,经后处理得到所需产物式e化合物。
根据本发明前述的制备方法,所述合成路线一和/或二中,所述步骤五反应条件包括:式e化合物:钛酸异丙酯和(s)-叔丁基亚磺酰胺的投料摩尔比为1:4:1.5;反应温度为85℃;反应时间为12h;反应溶剂为无水thf;反应气氛为n2。
具体操作如下:将钛酸异丙酯在室温、氮气气氛下,添加到式e化合物和(s)-叔丁基亚磺酰胺在无水thf中的混合液中,随后将反应混合液在85℃下搅拌反应,并通过tlc监测反应进程,完成后,经后处理得到所需产物式f化合物。
根据本发明前述的制备方法,所述合成路线一和/或二中,所述步骤六反应条件包括:式f化合物:二异丁基氢化铝的投料摩尔比为1:3;反应温度为-78℃;反应时间为1h;反应溶剂为无水thf;反应气氛为n2。
具体操作如下:用注射泵在n2气氛、-78℃下将二异丁基氢化铝缓慢加入到式f化合物的无水thf溶液中,当tlc显示反应完成时,加入水以在-78℃下淬灭反应,并在室温下再搅拌15分钟,然后经后处理得到所需产物式g化合物。
根据本发明前述的制备方法,所述合成路线一中,步骤七反应条件包括:式g化合物:四丁基氟化铵三水合物的投料摩尔比为1:1.1;反应温度为室温;反应时间为5min;反应溶剂为thf。
具体操作如下:在室温下将四丁基氟化铵三水合物添加到式g化合物的thf混合液中,然后将反应搅拌5分钟,从tlc可以清楚地区分这两种非对映异构体,经后处理得到光学纯的产物式h化合物。
根据本发明前述的制备方法,所述合成路线一中,步骤八的反应条件包括:一定量的浓盐酸;反应温度为室温;反应时间为30min;反应溶剂为甲醇。
具体操作如下:在室温下将浓盐酸加入到式h化合物的meoh溶液中,在室温下搅拌反应,并通过tlc监测反应进展,完成后,加入碳酸氢钠溶液以中和酸,然后经后处理得到所需产物式j化合物。
根据本发明前述的制备方法,所述合成路线一中,步骤九的反应条件包括:式j化合物:二甲基丙二酰二氯:三乙胺的投料摩尔比为1:0.5:2.5;反应温度为0℃~室温;反应时间为24h;反应溶剂为无水dcm;反应气氛为n2。
具体操作如下:在n2气氛、0℃下将二甲基丙二酰二氯缓慢加入到式j化合物和三乙胺的干燥dcm混合液中,在室温下搅拌反应,并通过tlc检测反应的进程,当起始原料消失时,经后处理得到所需产物式k化合物。
根据本发明前述的制备方法,所述合成路线一中,步骤十的反应条件包括:式k化合物:对甲苯磺酰氯:4-二甲基氨基吡啶:三乙胺的投料摩尔比为1:2.2:0.1:4.4;反应温度为室温;反应时间为24h;反应溶剂为dcm。
具体操作如下:将三乙胺缓慢添加到式k化合物,对甲苯磺酰氯和4-二甲基氨基吡啶的dcm溶液中,然后在室温下搅拌反应,并通过tlc监测进展,式k化合物完全消耗后,经后处理得到所需式i所示的双齿噁唑啉手性配体。
根据本发明前述的制备方法,所述合成路线二中,步骤七的反应条件包括:一定量的浓盐酸;反应温度为室温;反应时间为1h;反应溶剂为甲醇。
具体操作如下:在室温下将浓盐酸加入到式g化合物的meoh溶液中,在室温下搅拌反应,并通过tlc监测进展,完成后,加入碳酸氢钠溶液以中和酸,然后经后处理得到所需产物式j化合物。
根据本发明前述的制备方法,所述合成路线二中,步骤八的反应条件包括:式j化合物:二甲基丙二酰二氯:三乙胺的投料摩尔比为1:0.5:2.5;反应温度为0℃~室温;反应时间为24h;反应溶剂为无水dcm;反应气氛为n2。
具体操作如下:在n2气氛、0℃下,将二甲基丙二酰二氯缓慢加入到式j化合物和三乙胺在无水dcm中的混合物中,在室温下搅拌反应,并通过tlc检测反应的进程,当起始原料消失时,经后处理得到所需产物式k化合物。
根据本发明前述的制备方法,所述合成路线二中,步骤九的反应条件包括:式k化合物:对甲苯磺酰氯:4-二甲基氨基吡啶:三乙胺的投料摩尔比为1:2.2:0.1:4.4;反应温度为室温;反应时间为24h;反应溶剂为dcm。
具体操作如下:将三乙胺缓慢添加至式k化合物、对甲苯磺酰氯和4-二甲氨基吡啶的dcm溶液中,然后在室温下搅拌反应,并通过tlc监测进展,式k化合物完全消耗后,经后处理得到所需产物式i所示的双齿噁唑啉手性配体。
本发明的方法取得了如下的有益效果:本发明首次提供了一种具有式i所示的双齿噁唑啉手性配体的制备方法全合成路线,该方法使用简单、经济廉价的起始原料和有机合成试剂,通过对合成路线的合理设计并优化反应条件,方便并以高产率地制备获得所述双齿噁唑啉手性配体。
附图说明
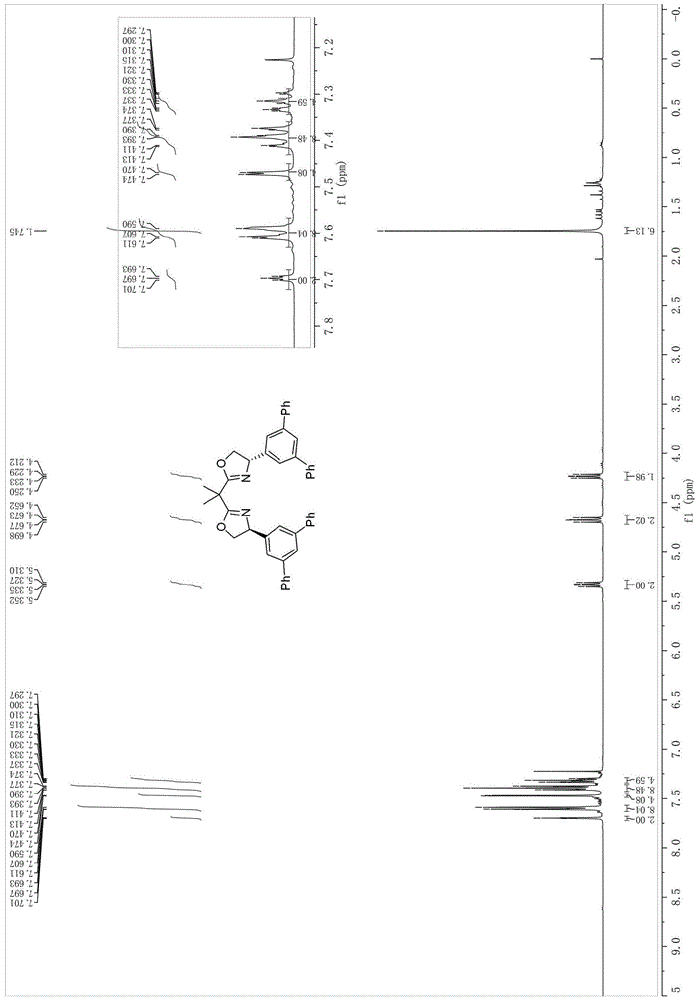
图1为实施例1制备的式i-1所示的双齿噁唑啉手性配体的核磁氢谱图。
图2为实施例1制备的式i-1所示的双齿噁唑啉手性配体的核磁碳谱图。
图3为实施例2制备的式i-2所示的双齿噁唑啉手性配体的核磁氢谱图。
图4为实施例2制备的式i-2所示的双齿噁唑啉手性配体的核磁碳谱图。
具体实施方式
以下结合具体实施例,对本发明作进一步的详述。在下文的具体实施例中,如无特殊说明,所用到的试剂均可以由本领域的技术人员通过常规的商业途径即可购买获得。
实施例1式i-1所示的双齿噁唑啉手性配体的合成(合成路线一)
步骤一式b化合物的合成(ar=ph)
在室温及n2保护下,向3,5-二溴苯甲醛29(5.28g,20mmol),苯硼酸(6.10g,50mmol),碳酸钠(10.60g,100mmol)在h2o/phme(50ml/50ml)中的溶液中加入四(三苯基膦)钯(0.23g,0.2mmol)。然后将反应混合物在120℃搅拌,并通过tlc检测反应进程。完成后,将混合物用etoac萃取,然后分离有机层,用硫酸镁干燥并浓缩。最后,混合物通过硅胶色谱法纯化(洗脱剂:正己烷/乙酸乙酯),得到所需产物式b化合物(ar=ph)。黄色固体(4.75g,18.4mmol),产率92%。
步骤二式c化合物的合成(ar=ph)
在室温下将三甲基碘化锍(4.51g,22.08mmol)添加到式b化合物(ar=ph)(4.75g,18.4mmol),氢氧化钾(2.06g,36.8mmol)和水(83μl,4.6mmol)的乙腈(40ml)混合物中,在60℃下搅拌反应,并通过tlc监测反应进程。式b化合物(ar=ph)反应完全后,将混合物浓缩,然后用水稀释并用etoac萃取。分离有机层,用硫酸镁干燥并浓缩。最后,将粗产物通过硅胶色谱法纯化(洗脱剂:正己烷/乙酸乙酯),得到所需产物式c化合物(ar=ph)。黄色固体(4.16g,15.27mmol),产率83%。
步骤三式d化合物的合成(ar=ph)
在0℃下,将浓硫酸(4.1ml,76.35mmol)缓慢加入到式c化合物(ar=ph)(4.16g,15.27mmol)的thf/h2o(60ml/20ml)混合液中,然后将反应混合液加热至回流。经tlc监测原料消失,将碳酸氢钠溶液添加至反应体系中以中和酸。然后将混合物浓缩并用etoac萃取。分离有机层,用硫酸镁干燥并浓缩。粗产物通过硅胶色谱法纯化(洗脱液:dcm/meoh),得到所需黄色油状的式d化合物(ar=ph)(2.97g,10.23mmol),产率67%。
步骤四式e化合物的合成(ar=ph)
在室温下,向式d化合物(ar=ph)(2.97g,10.23mmol)和咪唑(2.09g,30.69mmol)在dcm(30ml)中的溶液中缓慢加入叔丁基二甲基氯硅烷(1.70g,11.25mmol),并将反应混合物在室温搅拌12小时。完成后,将反应混合物用饱和氯化铵溶液(100ml)洗涤,分离有机层,用硫酸镁干燥并浓缩。残余物无需进一步纯化即可用于下一步。
在室温下,将戴斯-马丁试剂(dess-martinperiodinanecas:87413-09-0)(5.21g,12.28mmol)缓慢加入到粗产物(10.23mmol)在dcm(30ml)中的混合物中。然后在室温下搅拌反应,并通过tlc检测反应进程。反应完成后,将混合物过滤并将有机层浓缩。粗产物通过硅胶色谱法纯化(洗脱剂:正己烷/乙酸乙酯),得到所需产物式e化合物(ar=ph),无色油状(3.54g,8.80mmol),收率86%。
步骤五式f化合物的合成(ar=ph)
将钛酸异丙酯(10.00g,35.2mmol)在室温、氮气气氛下,添加到式e化合物(ar=ph)(3.54g,8.80mmol)和(s)-叔丁基亚磺酰胺(1.60g,13.2mmol)在无水thf(30ml)中的混合液中,随后将反应混合液在85℃下搅拌反应,并通过tlc监测反应进程。完成后,将溶液在室温下用水(1.9ml,105.6mmol)淬灭,然后过滤以除去固体沉淀物。然后将残余物浓缩并通过硅胶色谱法纯化(洗脱液:正己烷/乙酸乙酯),得到所需产物式f化合物(ar=ph),黄色固体(1.82g,3.61mmol),收率41%。
步骤六式g化合物的合成(ar=ph)
用注射泵在n2气氛、-78℃下将二异丁基氢化铝(1.5m的thf溶液,7.2ml,10.83mmol)缓慢加入到式f化合物(ar=ph)(1.82g,3.61mmol)的无水thf(15ml)溶液中。当tlc显示反应完成时,加入水(0.6ml,32.49mmol)以在-78℃下淬灭反应,并在室温下再搅拌15分钟。然后过滤残余物,并浓缩有机层。最后,将粗产物通过硅胶色谱法纯化(洗脱剂:正己烷/乙酸乙酯),得到所需产物式g化合物(ar=ph)。黄色固体(0.93g,1.84mmol),dr≈25∶1,产率51%。
步骤七式h化合物的合成(ar=ph)
在室温下将四丁基氟化铵三水合物(0.64g,2.02mmol)添加到式g化合物(ar=ph)(0.93g,1.84mmol)的thf(10ml)混合液中,然后将反应搅拌5分钟。从tlc可以清楚地区分这两种非对映异构体。将混合物浓缩,然后用水稀释,并用etoac萃取。分离有机层,用硫酸镁干燥并浓缩。粗产物通过硅胶色谱法纯化(洗脱剂:正己烷/乙酸乙酯),然后重结晶,得到光学纯的产物式h化合物(ar=ph)。白色固体(0.59g,1.51mmol),产率82%。
步骤八式j化合物的合成(ar=ph)
在室温下将浓盐酸(1.5ml)加入到式h化合物(ar=ph)(0.59g,1.51mmol)的meoh(10ml)溶液中。在室温下搅拌反应,并通过tlc监测反应进展。完成后,加入碳酸氢钠溶液以中和酸。用etoac萃取粗产物,然后分离有机层,经硫酸钠干燥并浓缩。最后,残余物通过硅胶色谱法纯化(洗脱液:dcm/meoh),得到所需产物式j化合物(ar=ph)。白色固体(0.30g,1.04mmol),收率69%。
步骤九式k化合物的合成(ar=ph)
在n2气氛、0℃下将二甲基丙二酰二氯(87.9mg,0.52mmol)缓慢加入到式j化合物(ar=ph)(0.30g,1.04mmol)和三乙胺(0.26g,2.6mmol)的干燥dcm(10ml)混合液中。在室温下搅拌反应,并通过tlc检测反应的进程。当起始原料消失时,加入饱和氯化铵溶液以淬灭反应。粗产物用dcm萃取,分离有机层,经硫酸镁干燥并浓缩。最后,混合物通过硅胶色谱法纯化(洗脱液:dcm/meoh),得到所需产物式k化合物(ar=ph)。白色固体(0.24g,0.35mmol),收率67%。
步骤十式i-1所示的双齿噁唑啉手性配体的合成
将三乙胺(0.16g,1.54mmol)缓慢添加到式k化合物(ar=ph)(0.24g,0.35mmol),对甲苯磺酰氯(0.15g,0.77mmol)和4-二甲基氨基吡啶(4.3mg,0.035mmol)的dcm(5ml)溶液中。然后在室温下搅拌反应,并通过tlc监测进展。式k化合物(ar=ph)完全消耗后,将反应液浓缩,并将残余物通过硅胶色谱法纯化(洗脱液:正己烷/乙酸乙酯),得到所需式i-1所示的双齿噁唑啉手性配体。白色固体(0.17g,0.27mmol),产率为76%。
实施例2式i-2所示的双齿噁唑啉手性配体的合成(合成路线二)
步骤一式b化合物的合成(ar=2-萘基)
在室温及氮气保护下,向3,5-二溴苯甲醛29(5.28g,20mmol),2-萘基硼酸(8.60g,50mmol),碳酸钠(10.60g,100mmol)的h2o/phme(50ml/50ml)混合液中,加入四(三苯基膦)钯(0.23g,0.2mmol)。然后将反应在120℃搅拌,并通过tlc检测反应进程。完成后,将混合物用etoac萃取,然后分离有机层,用硫酸镁干燥并浓缩。最后,将混合物通过硅胶色谱法纯化(洗脱液:己烷/乙酸乙酯),得到所需产物式b化合物(ar=2-萘基)。黄色固体(6.81g,19.0mmol),收率95%。
步骤二式c化合物的合成(ar=2-萘基)
在室温下将三甲基碘化锍(4.65g,22.8mmol)添加到式b化合物(ar=2-萘基)(6.81g,19.0mmol),氢氧化钾(2.13g,38.0mmol)和水(86μl,4.75mmol)的乙腈(40ml)混合物中。在60℃下搅拌反应,并通过tlc监测反应进程。在式b化合物(ar=2-萘基)被完全消耗之后,将混合物浓缩,然后用水稀释并且用etoac萃取。分离有机层,用硫酸镁干燥并浓缩。最后,将粗产物通过硅胶色谱法纯化(洗脱液:正己烷/乙酸乙酯),得到所需产物式c化合物(ar=2-萘基)。黄色固体(6.44g,17.29mmol),收率91%。
步骤三式d化合物的合成(ar=2-萘基)
在0℃下,将浓硫酸(4.6ml,86.45mmol)缓慢地加入到式c化合物(ar=2-萘基)(6.44g,17.29mmol)的thf/h2o(60ml/20ml)体系中,然后将反应加热至回流。经tlc监测判断出原料消失,将碳酸氢钠溶液添加至反应混合液中以中和酸。然后将混合物浓缩并用etoac萃取。分离有机层,用硫酸镁干燥并浓缩。粗产物通过硅胶色谱法纯化(洗脱液:dcm/meoh),得到所需产物式d化合物(ar=2-萘基)。黄色油(5.13g,13.14mmol),产率76%。
步骤四式e化合物的合成(ar=2-萘基)
在室温下,向式d化合物(ar=2-萘基)(5.13g,13.14mmol)和咪唑(2.68g,39.42mmol)在dcm(30ml)中的溶液中缓慢加入叔丁基二甲基氯硅烷(2.18g,14.45mmol),并将反应混合物在室温搅拌12小时。完成后,将反应混合物用饱和氯化铵溶液(100ml)洗涤,分离有机层,用硫酸镁干燥并浓缩。残余物无需进一步纯化即可用于下一步。
在室温下,将戴斯-马丁试剂(dess-martinperiodinanecas:87413-09-0)(6.69g,15.77mmol)缓慢加入到粗产物(13.14mmol)在dcm(30ml)中的混合物中。然后在室温下搅拌反应,并通过tlc检测进展。反应完成后,将混合物过滤并将有机层浓缩。粗产物通过硅胶色谱法纯化(洗脱剂:正己烷/乙酸乙酯),得到所需产物式e化合物(ar=2-萘基)。无色油(4.95g,9.86mmol),产率75%。
步骤五式f化合物的合成(ar=2-萘基)
将钛酸异丙酯(11.21g,39.44mmol)在室温和氮气气氛下添加到无水thf(30ml)中的式e化合物(ar=2-萘基)(4.95g,9.86mmol)和(s)-叔丁亚磺酰胺(1.79g,14.79mmol)的混合物中,在85℃下搅拌反应,并通过tlc监测反应进程。完成后,将溶液在室温下用水(2.1ml,118.32mmol)淬灭,然后过滤以除去固体沉淀物。然后将残余物浓缩并通过硅胶色谱法纯化(洗脱液:正己烷/乙酸乙酯),得到所需产物式f化合物(ar=2-萘基)。黄色固体(4.18g,6.90mmol),产率70%。
步骤六式g化合物的合成(ar=2-萘基)
用注射泵在n2气氛、-78℃下将二异丁基氢化铝(1.5m的thf,13.8ml,20.7mmol)缓慢加入到式f化合物(ar=2-萘基)(4.18g,6.90mmol)的无水thf(15ml)溶液中。当tlc显示反应完成时,在-78℃下加入水(1.1ml,62.1mmol)使反应淬灭,并在室温下再搅拌15分钟。然后过滤残余物,并浓缩有机层。最后,将粗产物通过硅胶色谱法纯化(洗脱液:正己烷/乙酸乙酯)(dr≈25:1)并重结晶,得到光学纯的产物式g化合物(ar=2-萘基)。黄色固体(2.94g,4.83mmol),产率70%。
步骤七式j化合物的合成(ar=2-萘基)
在室温下将浓盐酸(4.8ml)加入到式g化合物(ar=2-萘基)(2.94g,4.83mmol)的meoh(15ml)溶液中。在室温下搅拌反应,并通过tlc监测进展。完成后,加入碳酸氢钠溶液以中和酸。用etoac萃取粗产物,然后分离有机层,经硫酸钠干燥并浓缩。最后,残余物通过硅胶色谱法纯化(洗脱液:dcm/meoh),得到所需产物式j化合物(ar=2-萘基)。白色固体,收率90%(1.69g,4.35mmol)。
步骤八式k化合物的合成(ar=2-萘基)
在n2气氛、0℃下,将二甲基丙二酰二氯(0.37g,2.18mmol)缓慢加入到式j化合物(ar=2-萘基)(1.69g,4.35mmol)和三乙胺(1.10g,10.88mmol)在无水dcm(10ml)中的混合物中。在室温下搅拌反应,并通过tlc检测反应的进程。当起始原料消失时,加入饱和氯化铵溶液以淬灭反应。粗产物用dcm萃取,分离有机层,经硫酸镁干燥并浓缩。最后,混合物通过硅胶色谱法纯化(洗脱液:dcm/meoh),得到所需产物式k化合物(ar=2-萘基)。白色固体(1.81g,2.07mmol),收率95%。
步骤九式i-2所示的双齿噁唑啉手性配体的合成
将三乙胺(0.92g,9.11mmol)缓慢添加至式k化合物(ar=2-萘基)(1.81g,2.07mmol),对甲苯磺酰氯(0.87g,4.55mmol)和4-二甲氨基吡啶(25.3mg,0.207mmol)的dcm(5ml)溶液中。然后在室温下搅拌反应,并通过tlc监测进展。式k化合物(ar=2-萘基)完全消耗后,将反应液浓缩,并将残余物通过硅胶色谱法纯化(洗脱液:正己烷/乙酸乙酯),得到所需产物式i-2所示的双齿噁唑啉手性配体。白色固体(1.22g,1.45mmol),产率为70%。
以上所述实施例1和实施例2仅为发明人的经过大量的试验筛选之后确定的优选实施例,而并非本发明可行实施的穷举。对于本领域技术人员而言,在不背离本发明合成路线的前提下,对其所作出的任何显而易见的改动,都应当被认为包含在本发明的权利要求保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!