一种盐酸左布比卡因的杂质及其制备和分析方法与流程
本发明涉及医药技术领域,尤其涉及一种盐酸左布比卡因的杂质及其制备和分析方法。
背景技术:
盐酸左布比卡因是盐酸布比卡因的单一异构体,属于新型长效酰胺类局部麻醉药,英文名为levobupivacainehydrochloride,其化学名为s-(-)-1-丁基-n-(2,6-二甲基苯基)-2-哌啶甲酰胺盐酸盐,它最早由美国purdue公司开发,适用于外周神经阻滞、硬脊膜外阻滞和蛛网膜下腔阻滞,低毒的盐酸左布比卡因被用于过去不能采用的局部麻醉手术中。盐酸左布比卡因分子式为c18h28n2o·hcl,分子量为324.94,化学结构式为:
药品杂质主要来源于合成和降解,盐酸左布比卡因未收录于任何药典中,但盐酸布比卡因的质量标准在英国药典、欧洲药典、美国药典中有收载,药典中公开了盐酸布比卡因6个相关杂质a-f,其结构式如下:
受不同制备工艺的影响,可能的工艺杂质的出现是不可避免的,对于新工艺杂质的研究,是一个动态发展和不断推进的过程,而对盐酸左布比卡因具新颖杂质结构的研究与分析,可以进一步完善盐酸左布比卡因的质量标准。
技术实现要素:
本发明在盐酸左布比卡因合成过程中发现了一个未知杂质g,属于工艺杂质,发现该杂质是n-(2′,6′-二甲苯基)-2-哌啶甲酰胺在高温,长时间与碳酸钾和1-溴丁烷共同作用下产生的,并且在英国药典(bp)、美国药典(usp)、欧洲药典(ep)等药典标准中均没有专门的论述和规定的限度。
鉴于对盐酸左布比卡因合成过程中发现的杂质g的结构还缺乏明确的表征,且亦缺乏其制备方法及分析检测方法,本发明对杂质g进行了研究,合成得到杂质g,鉴定了其结构,并建立了准确的杂质g分析检测方法。
为了实现上述目的,本发明采用了如下技术方案:
一种盐酸左布比卡因的杂质,命名为杂质g,其化学名为1-丁酰基-n-(2,6-二甲苯基)-2-哌啶甲酰胺,分子式为c19h28n2o3,具有如下的化学结构:
一种盐酸左布比卡因杂质g的制备方法,具体合成路线如下:
进一步的,制备方法包括以下反应步骤:
(1)将n-(2′,6′-二甲苯基)-2-哌啶甲酰胺溶于二氯甲烷中,其中二氯甲烷的体积是n-(2′,6′-二甲苯基)-2-哌啶甲酰胺的2~6倍;
(2)加入缚酸剂,-5~5℃条件下,滴加氯甲酸丁酯,滴毕回流反应,tlc监测反应进程,反应完成后得到反应液;
(3)将步骤(2)中的反应液经4mol/l稀盐酸调ph值至2.0~3.0,萃取分液,有机相水洗至中性,无水硫酸钠干燥,减压浓缩得该杂质粗品;
(4)将步骤(3)中的杂质粗品通过适当溶剂结晶精制步骤,得到该杂质g。
优选的,步骤(2)中的缚酸剂为碳酸钾、碳酸钠、三乙胺、吡啶中的一种。
优选的,步骤(2)中氯甲酸丁酯与n-(2′,6′-二甲苯基)-2-哌啶甲酰胺的摩尔比为1.0~1.5,优选1.2~1.5。
优选的,步骤(4)中的适当溶剂为石油醚-乙酸乙酯、异丙醚-乙酸乙酯、正己烷-乙酸乙酯、甲基叔丁基醚-乙酸乙酯中的一种。
进一步的,步骤(4)中得到的杂质g在盐酸左布比卡因有关物质检查项中作为杂质对照品的应用。
进一步的,步骤(2)中tlc监测用到的展开剂为石油醚和乙酸乙酯按照1:1的体积比混合后得到的混合液。
一种盐酸左布比卡因杂质g的分析方法,采用高效液相色谱检测分析,具体检测条件包括:
色谱柱:welchultimatexb-c18,规格为:3.9mm×150mm,5μm
检测波长:210nm
流速:1.0ml/min
进样体积:20μl
流动相:磷酸盐缓冲液-乙腈,磷酸盐缓冲液和乙腈的体积比为50︰50,磷酸盐缓冲液的制备方法为:取0.87%磷酸氢二钾溶液,用0.68%磷酸二氢钾溶液调节ph值至7.0。
与现有技术相比,本发明的有益效果是:
1、本发明发现了一种新的盐酸左布比卡因工艺杂质,该工艺杂质结构首次报道,是一个全新的化合物,本发明还设计了该杂质合成方法,且获得的杂质纯度大于95%,在盐酸左布比卡因的有关物质检查项中作为杂质对照品用于今后的质量研究与控制,可准确对该杂质进行定性与定量分析。
2、本发明提供的液相分析方法能快速检测出杂质g,选择杂质g的最大吸收波长作为检测波长,提高了该杂质的检出率,结果准确可靠,欧洲药典9.0盐酸布比卡因质量标准的有关物质检查项单个杂质限度为不大于0.10%,本发明对盐酸左布比卡因的工艺杂质进行了更深入的研究,通过对稳定性样品的检验,按照杂质限度0.10%进行控制,避免对人体产生不好的毒副作用。
3、本发明提供的分析方法中,盐酸左布比卡因工艺杂质g在规定的色谱条件下,能与极性相近的化合物完全分离开,分离度大于1.5,流动相在210nm检测波长下紫外吸收低,且210nm为杂质的最大吸收波长,保证了杂质的响应灵敏度,提高检出量。
附图说明
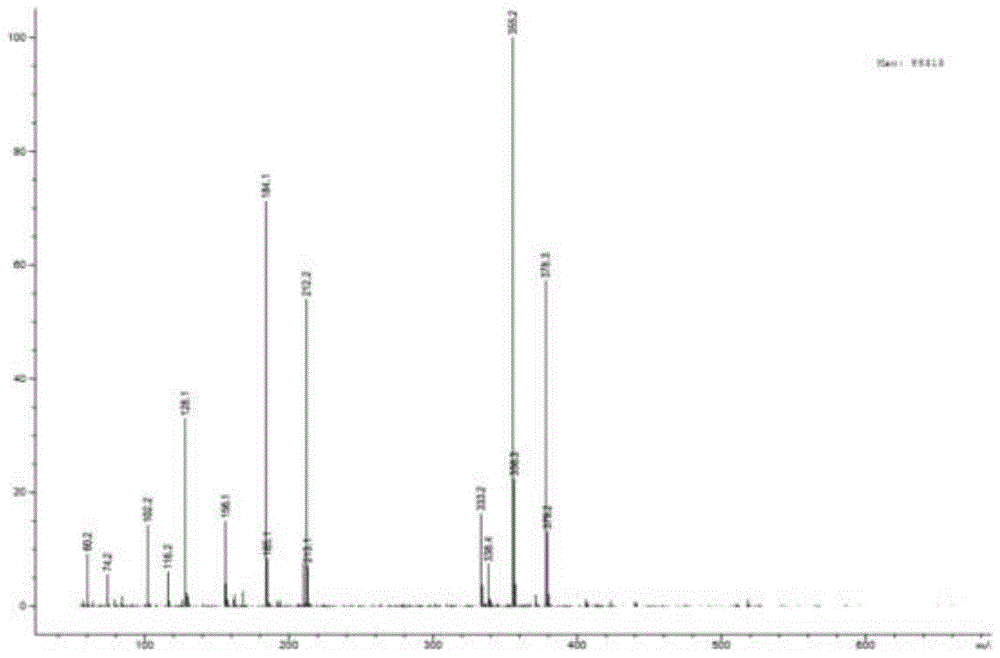
图1本发明实施例一中新工艺杂质g的质谱图;
图2本发明实施例一中新工艺杂质g的氢谱信号归属图;
图3本发明实施例三中新工艺杂质g的hplc图谱。
具体实施方式
下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。
实施例一
本发明的新杂质g的制备和结构确证:
(1)100ml反应瓶中依次加入二氯甲烷40ml,n-(2′,6′-二甲苯基)-2-哌啶甲酰胺9.06g,吡啶10g,-2~2℃低温下滴加氯甲酸丁酯6.92g,滴毕加热回流反应3h,tlc监控反应进程,反应完成后得到反应液,tlc监控时采用的展开剂为石油醚和乙酸乙酯按照1:1的体积比混合后得到的混合液;
(2)将步骤(1)中的反应液降至室温,滴加4mol/l稀盐酸调ph至2.0~3.0,萃取分液,用二氯甲烷水洗至中性,并用无水硫酸钠干燥,再经减压浓缩得到油状物;
(3)向步骤(2)中的油状物中加入18ml乙酸乙酯,搅拌均匀后,滴加甲基叔丁基醚60ml,析出大量白色固体,室温磁力搅拌5h,搅拌后进行抽滤,然后用鼓风机以45℃的温度烘干后得白色固体10.75g,其收率为82.9%,纯度为99.73%。
工艺杂质g的化学结构确证:
白色粉末,质谱如图1所示[m+na]+为m/z355,结合核磁特征可得分子式为c19h28n2o3;核磁共振氢谱信号及其归属如图2。综合可以确证该化合物的结构如下:
实施例二
杂质g的扩大制备
(1)1l反应瓶中依次加入二氯甲烷400ml,n-(2′,6′-二甲苯基)-2-哌啶甲酰胺92.8g,三乙胺120g,-5~0℃低温下滴加氯甲酸丁酯72g,滴毕加热回流反应4h,tlc监控反应进程,反应完成后得到反应液,tlc监控时采用的展开剂为石油醚和乙酸乙酯按照1:1的体积比混合后得到的混合液;
(2)将步骤(1)中的反应液降至室温,滴加4mol/l稀盐酸调ph至2.0~3.0,萃取分液,用二氯甲烷水洗至中性,并用无水硫酸钠干燥,再经减压浓缩得到油状物;
(3)向步骤(2)中的油状物中加入125ml乙酸乙酯,搅拌均匀后,滴加正己烷375ml,析出大量白色固体,室温磁力搅拌3h,搅拌后进行抽滤,然后用鼓风机以45℃的温度烘干后得112.5g白色固体,其收率为84.6%。
实施例三
本发明新杂质g的定性和定量分析
1、色谱条件:
色谱柱:welchultimatexb-c18,规格为:3.9mm×150mm,5μm
检测波长:210nm
流速:1.0ml/min
进样体积:20μl
流动相:磷酸盐缓冲液-乙腈,磷酸盐缓冲液和乙腈的体积比为50:50,磷酸盐缓冲液的制备方法为:取0.87%磷酸氢二钾溶液,用0.68%磷酸二氢钾溶液调节ph值至7.0。
2、溶液配制
(1)供试品溶液:取盐酸左布比卡因约100mg,精密称定,置100ml量瓶中,加流动相溶解并稀释至刻度,摇匀,作为供试品溶液。
(2)杂质g对照品溶液:精密称取杂质g10mg,加流动相溶解并稀释制成每lml中约含1μg的溶液,作为杂质g对照品溶液。
3、测定
精密量取供试品溶液、杂质g对照品溶液各20μl,分别注入液相色谱仪,记录色谱图,如图3。
4、杂质定性及定量
试验描述:将供试品溶液及杂质g对照品溶液进样检测并记录色谱图,见图3,供试品溶液中如有与杂质g出峰保留时间一致的杂质,可以定性为杂质g。按外标法以峰面积计算杂质g的含量,可以定量。
以上,仅为本发明较佳的具体实施方式,但本发明的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本发明揭露的技术范围内,根据本发明的技术方案及其发明构思加以等同替换或改变,都应涵盖在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!