一种聚酰胺酸及其制备方法和其聚酰亚胺、聚酰亚胺纤维与流程
1.本发明涉及聚酰亚胺领域,具体地说,是涉及一种聚酰胺酸及其制备方法和其聚酰亚胺、聚酰亚胺纤维。
背景技术:
2.聚酰亚胺(pi)是一类以酰亚胺环为特征结构的聚合物,作为一种综合性能突出的高分子材料,用途及其广泛,在航空航天、机械化工、原子能工业和国防军工等重要领域得到应用。pi的合成途径广,加工成型方法多,成为当今世界工程塑料产业中的开发热点。
3.在当前聚酰亚胺(pi)制备工艺中,两步合成法是最为广泛采用的合成方法。即首先在搅拌的适当溶剂中先后加入二胺和二酐单体,此时通过低温缩合聚合反应制备获取聚酰胺酸(paa)溶液。制备pi纤维时,paa溶液经过过滤和脱泡过程通过喷丝板形成聚合物溶液丝条,经过凝固浴形成初生纤维,经水洗和各阶段高温酰亚胺化过程,最后缠绕收卷。
4.为满足pi纤维高强高模的性能要求,加入第三单体,形成三元甚至多元共聚聚酰亚胺是一种常见的改性方法。根据聚合物的结构,可以将共聚聚酰亚胺分为无规共聚、交替共聚和嵌段共聚聚酰亚胺。由于嵌段共聚物分子链中有重复出现的规则链段,聚合物的性能具有较大的预见性,合成技术简单易控,能更好的控制结构序列、链段长度等重要参数,因此嵌段共聚是改善聚酰亚胺各项性能的重要手段。嵌段共聚物中可以含有两种或两种以上的链段,于是会出现两个或多个玻璃化转变温度(tg)。而无规共聚物中只含有一种链段,只有一个tg;嵌段共聚物和无规共聚物的分子量测试只有1个峰值,而共混物会有两个或多个峰值。结合dma和pl-gpc分析测试,当pi纤维存在两个或多个玻璃化转变温度(tg),而聚合溶液分子量只有1个峰值时,说明纺丝原液成功合成嵌段聚合物,而非无规共聚或共混。
5.目前高强高模型聚酰亚胺纤维普遍在聚合过程中通过无规共聚添加2-(4-氨基苯基)-5-氨基苯并咪唑(bia),引入氢键,增强分子链间作用力,调整二酐和二胺的种类和配比,提高纤维强度和模量,应用在航空航天等高端领域。但存在无规共聚导致分子链结构序列随机,缺乏预见性,纤维性能均一性和稳定性差,不利于提高力学强度和进一步加工的问题。
技术实现要素:
6.本发明主要解决现有技术问题之一是现有技术中存在高强高模型聚酰亚胺纤维普遍采用无规共聚导致分子链结构序列随机,缺乏预见性,纤维性能均一性和稳定性差,不利于提高力学强度和进一步加工的问题。
7.本发明涉及的高强高模聚酰亚胺纤维通过分子设计和嵌段共聚为主要手段,首先合成两种氨基封端的低聚物,避免酐基封端水解,充分混合后,再加入一定量的芳香族二酐单体,将两种氨基封端的低聚物连接起来形成嵌段共聚物,这样能更好的控制分子链结构序列,同时增加体系中氢键的密度,充分增强分子链间作用力,克服上述不足,进一步提高纤维的均一性和稳定性,力学性能好,有利于后续加工。
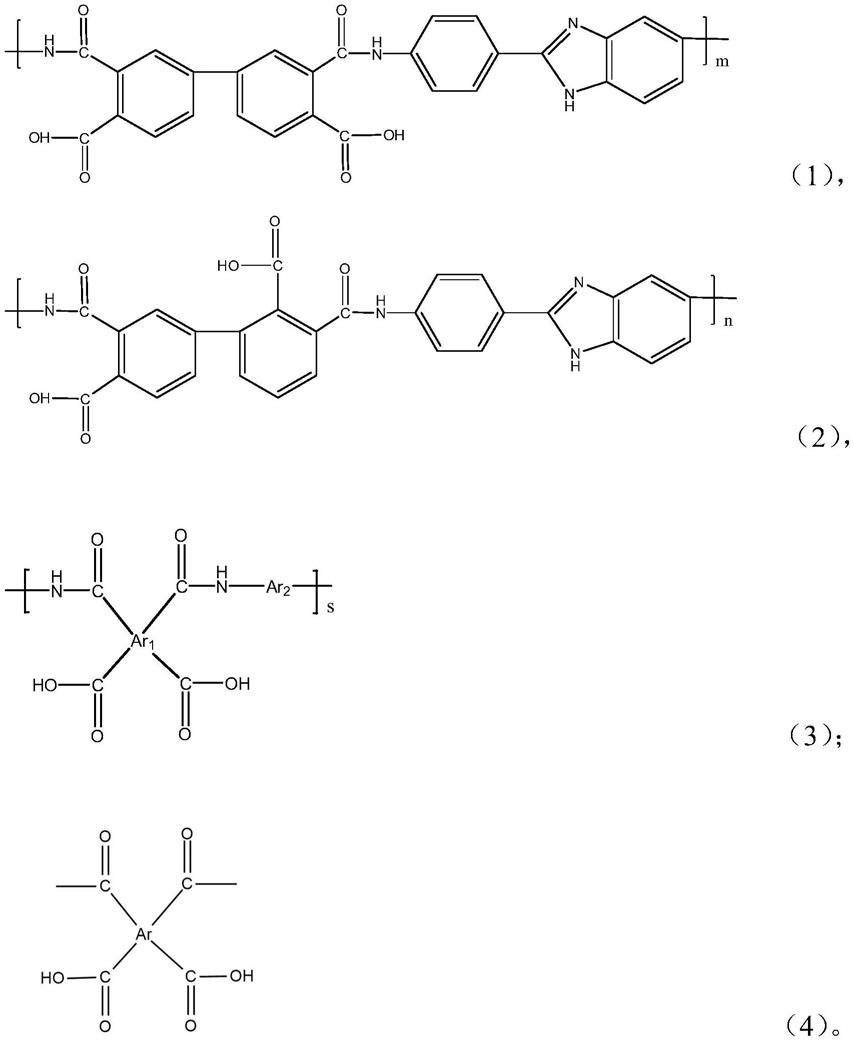
8.本发明目的之一为提供一种聚酰胺酸,包括链段a和链段b:
9.其中,链段a具有以下式1和式2所示结构中的至少一种,
[0010][0011][0012]
链段b具有以下式3所示结构,
[0013][0014]
其中,链段a与链段a之间、链段b与链段b之间、和/或:链段a和链段b之间的链接包含以下式4所示结构;链段a和链段b不相同;
[0015][0016]
以上m是的重复单元数;
[0017]
以上n是的重复单元数;其中m和n相同或不同;
[0018]
以上s是的重复单元数。
[0019]
本发明所述聚酰胺酸中,所述链段a占总链段的摩尔百分比为20~70%,优选为40~60%。
[0020]
ar1和ar各自独立为含有至少一个碳六元环的四价芳香族残基,ar和ar1可以相同或不同,优选以下结构式中所示的芳香族残基中的一种:
[0021][0022]
其中,r1为
[0023]
ar2为含有至少一个碳六元环的二价芳香族残基,优选以下结构式所示的芳香族残基中的一种:
[0024][0025]
其中,r2为h-、ch
3-、cl-、br-、f-、ch3o-。
[0026]
本发明目的之二为提供所述聚酰胺酸的制备方法,包括以下步骤:将2-(4-氨基苯基)-5-氨基苯并咪唑与联苯四甲酸二酐反应、将芳香族二胺与芳香族二酐反应,将以上两种反应所得产物混合并与芳香族二酐反应而得所述聚酰胺酸,其中联苯四甲酸二酐为3,3’4,4
’-
联苯四甲酸二酐和/或2,3,3’,4
’-
联苯二甲酸酐。
[0027]
优选地,所述制备方法包括:
[0028]
(1)在惰性的气体保护,将x摩尔的2-(4-氨基苯基)-5-氨基苯并咪唑溶解于有机溶剂中,加入y摩尔的联苯四甲酸二酐充分反应得到预聚体溶液1;
[0029]
(2)在惰性的气体保护,将z摩尔的芳香族二胺溶解于有机溶剂中,加入k摩尔的芳香族二酐充分反应得到预聚体溶液2;
[0030]
(3)将预聚体溶液1和预聚体溶液2混合充分搅拌得到预聚体溶液3;
[0031]
(4)将a摩尔的芳香族二酐加入到预聚体溶液3中充分反应得到所述聚酰胺酸。
[0032]
上述技术方案中,全部反应温度均为-10~40℃,优选为0-20℃;反应时间均为0.5~4h,优选为1~2h;搅拌时间为0.5~4h,优选为1~2h。
[0033]
上述技术方案中,步骤(2)中,所述芳香族二酐具有式4所示的结构:
[0034][0035]
其中,ar1为含有至少一个碳六元环的四价芳香族残基;
[0036]
步骤(2)中,所述芳香族二胺具有式6所示的结构:
[0037]
h2n-ar
2-nh2ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
(5),
[0038]
其中,ar2为含有至少一个碳六元环的二价芳香族残基。
[0039]
步骤(4)中,所述芳香族二酐具有式6所示的结构:
[0040][0041]
其中,ar为含有至少一个碳六元环的四价芳香族残基;
[0042]
ar1和ar各自独立为含有至少一个碳六元环的四价芳香族残基,ar和ar1可以相同或不同,优选以下结构式中所示的芳香族残基中的一种:
[0043][0044]
其中,r1为
[0045]
ar2为含有至少一个碳六元环的二价芳香族残基,优选以下结构式所示的芳香族残基中的一种:
[0046][0047]
其中,r2为h-、ch
3-、cl-、br-、f-、ch3o-。
[0048]
优选地,所述芳香族二酐为1,2,4,5-均苯四酸二酐,3,3’,4,4
’-
二苯甲酮四酸二酐,联苯四甲酸二酐(如3,3’4,4
’-
联苯四甲酸二酐、2,3,3’,4
’-
联苯二甲酸酐),3,3’,4,4
’-
二苯甲醚四酸二酐,4,4
’-
(六氟异丙基)双邻苯二甲酸二酐中的至少一种;
[0049]
优选地,所述芳香族二胺为4,4
’-
二氨基二苯醚、对苯二胺、3,4
’-
二氨基二苯醚、2-(4-氨基苯基)-5-氨基苯并咪唑中的至少一种。
[0050]
其中,步骤(2)中,所述芳香族二酐和芳香族二胺不同时为联苯四甲酸二酐和2-(4-氨基苯基)-5-氨基苯并咪唑。
[0051]
上述技术方案中,步骤(1)中,0.90≤y/x<1,优选为0.90≤y/x<0.98;
[0052]
步骤(2)中,0.90≤k/z<1,优选为0.90≤k/z<0.98;
[0053]
步骤(4)中,0.95≤a/(x-y+z-k)≤1.05,优选为0.98≤a/(x-y+z-k)≤1.02。
[0054]
进一步地,二酐与二胺的总摩尔比(y+k+a)/(x+z)为(0.95~1.05):1,优选为(0.98~1.02):1。
[0055]
上述技术方案中,本发明所述聚酰胺酸中链段a占总链段的摩尔百分比可以通过公式[2y/(x+y+z+k+a)]
×
100%计算得到。
[0056]
根据本发明一个优选的实施方式,所述聚酰胺酸溶液的制备方法,包括以下步骤:
[0057]
(1)在惰性的气体保护下,反应的温度为-10~40℃,优选为0~30℃,将x摩尔的2-(4-氨基苯基)-5-氨基苯并咪唑(bia)溶解于有机溶剂中,y摩尔的3,3’4,4
’-
联苯四甲酸二酐(bpda)加入到该二胺溶液中进行反应得到预聚体溶液1,其中0.90≤y/x<1;
[0058]
(2)与步骤(1)同样的聚合条件,将z摩尔的芳香族二胺溶解于有机溶剂中,k摩尔的芳香族二酐加入到该二胺溶液中,充分搅拌得到预聚体溶液2,其中0.90≤k/z<1;
[0059]
(3)将预聚体溶液1和预聚体溶液2充分混合得到预聚体溶液3;
[0060]
(4)将a摩尔的芳香族二酐加入到预聚体溶液3中充分搅拌反应得到酰胺酸,其中0.95≤a/(x-y+z-k)≤1.05;
[0061]
(5)聚合过程中二酐与二胺的总摩尔比(y+k+a)/(x+z)为(0.95~1.05):1,最后所得酰胺酸溶液固含量为5~30%,优选为10~20%。
[0062]
上述技术方案中,所述有机溶剂选自所述反应物料的良溶剂即可,优选为n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、n-甲基吡咯烷酮、二甲基亚砜和环丁砜等强极性非质子溶剂中的至少一种,优选为n,n-二甲基乙酰胺。
[0063]
本发明目的之三为提供一种聚酰亚胺,包括链段1和链段2:
[0064]
其中,链段1具有以下式8和式9所示结构中的至少一种,
[0065][0066]
链段2具有以下式10所示结构,
[0067][0068]
其中,链段1与链段1之间、链段2与链段2之间、和/或:链段1和链段2之间的链接包含以下式11所示结构;链段1和链段2不相同;
[0069][0070]
ar1和ar各自独立为含有至少一个碳六元环的四价芳香族残基,ar和ar1可以相同或不同,优选以下结构式中所示的芳香族残基中的一种:
[0071][0072]
其中,r1为
[0073]
ar2为含有至少一个碳六元环的二价芳香族残基,优选以下结构式所示的芳香族残基中的一种:
[0074][0075]
其中,r2为h-、ch
3-、cl-、br-、f-、ch3o-。
[0076]
所述的聚酰亚胺由以上所述聚酰胺酸或者所述制备方法得到的聚酰胺酸经亚胺化得到。
[0077]
本发明目的之四为提供一种聚酰亚胺纤维,包含所述的聚酰亚胺。
[0078]
进一步地,所述聚酰亚胺纤维由所述聚酰胺酸或者所述制备方法得到的聚酰胺酸
经亚胺化得到。
[0079]
上述技术方案中,所述聚酰亚胺纤维的单丝断裂强力大于3.0gpa,其cv≤5%;模量大于100gpa,其cv≤5%。
[0080]
本发明的聚酰亚胺纤维,通过分子设计和嵌段共聚为主要手段,首先合成两种氨基封端的低聚物,避免酐基封端水解,充分混合后,再加入一定量的芳香族二酐单体,将两种氨基封端的低聚物连接起来形成嵌段共聚物,这样能更好的控制分子链结构序列,同时增加体系中氢键的密度,充分增强分子链间作用力,得到均一性和稳定性好的pi纤维,力学性能好,有利于进一步加工。
[0081]
本发明目的之五为提供所述聚酰亚胺纤维的制备方法,包括以下步骤:将包含聚酰胺酸的纺丝原液经纺丝、亚胺化、热定型后得到聚酰亚胺纤维。
[0082]
根据本发明一个优选的实施方式,所述制备方法包括:
[0083]
(a)由联苯四甲酸二酐、2-(4-氨基苯基)-5-氨基苯并咪唑(bia)与其它芳香族二酐和二胺在有机溶剂中,通过本发明以上所述的分子设计和嵌段共聚的手段制备得到聚酰胺酸溶液,按二酐与二胺的总摩尔比(0.95~1.05):1进行配比,其中联苯四甲酸二酐/2-(4-氨基苯基)-5-氨基苯并咪唑嵌段占总链段的摩尔百分比为20~70%,优选为40~60%;
[0084]
(b)将聚酰胺酸溶液经脱泡、过滤后得到纺丝原液,采用湿法或干湿法工艺进行纺丝,经凝固、水洗、上油、干燥致密化、亚胺化、热定型后,得到均匀的聚酰亚胺纤维。
[0085]
上述技术方案中,对纺丝原液的纺丝、亚胺化、热定型工艺没有特别的限定,均为本领域通常的制备聚酰亚胺纤维的工艺。
[0086]
上述技术方案中,所述纺丝原液的固含量为5~30%,优选为12~18%。
[0087]
上述技术方案中,所述脱泡优选真空静置或脱泡塔流延的方式。
[0088]
上述技术方案中,所述的过滤方式优选为多道过滤,过滤精度优选为2~10μm,逐道增加过滤精度。
[0089]
上述技术方案中,所述凝固浴介质可采用本领域通常的凝固浴介质,优选为二甲基乙酰胺水溶液,凝固浴的浓度采用阶梯浓度凝固,浓度优选为3~35%,凝固浴的温度优选为0~40℃。
[0090]
上述技术方案中,所述水洗优选为多道水洗,水洗温度优选为40~65℃。
[0091]
上述技术方案中,所述上油优选采用压辊式上油。
[0092]
上述技术方案中,所述干燥致密化的温度优选为60~150℃。
[0093]
上述技术方案中,所述亚胺化处理优选采用多温区逐步升温的方式,亚胺化温度优选为100~550℃,更优选为150~480℃;所述亚胺化过程优选采用氮气保护,氧含量优选不大于1%。
[0094]
上述技术方案中,所述热定型的温度优选为200~300℃,更优选为200~280℃。
[0095]
本发明通过分子设计和嵌段共聚为主要手段,首先合成两种氨基封端的低聚物,避免酐基封端水解,充分混合后,再加入一定量的芳香族二酐单体,将两种氨基封端的低聚物连接起来形成嵌段共聚物,这样能更好的控制分子链结构序列,同时增加体系中氢键的密度,充分增强分子链间作用力,较好的解决了高强高模型聚酰亚胺纤维普遍采用无规共聚导致分子链结构序列随机,缺乏预见性,纤维性能均一性和稳定性差,不利于提高力学强度和进一步加工的问题。
[0096]
采用本发明的方案,得到的聚酰亚胺纤维,均一性和稳定性,力学性能好,有利于后续加工。聚酰亚胺纤维断裂强力大于3.0gpa,cv≤5%;模量大于100gpa,cv≤5%,取得了较好的技术效果。
[0097]
下面通过实施例对本发明做进一步的阐述:
附图说明
[0098]
图1为实施例1的gpc谱图。
[0099]
图2为实施例1的dma谱图。
[0100]
对实施例1的纺丝原液进行pl-gpc测试,对聚酰亚胺纤维进行dma测试,所得聚酰胺酸分子量为一个峰值(mw=187426),聚酰亚胺纤维玻璃化转变温度为两个峰值(318℃和464℃),说明此种聚合方法所得纺丝原液结构为均一的嵌段聚合物。
[0101]
图3为实施例1和比较例1的gpc谱图。
[0102]
图4为比较例1的dma谱图。
[0103]
对比较例1的纺丝原液进行pl-gpc测试,对聚酰亚胺纤维进行dma测试,所得聚酰胺酸分子量为一个峰值(111675),和实施例1比较,分子量较小,且分布较宽。聚酰亚胺纤维玻璃化转变温度为一个峰值(436℃),说明此种聚合方法所得纺丝原液结构并非嵌段聚合物。考虑到加料方式,纺丝原液应为无规共聚物。
具体实施方式
[0104]
下面结合具体实施例对本发明进行具体的描述,有必要在此指出的是以下实施例只用于对本发明的进一步说明,不能理解为对本发明保护范围的限制,本领域技术人员根据本发明内容对本发明做出的一些非本质的改进和调整仍属本发明的保护范围。
[0105]
本发明具体实施方式中所用原料为市售所得。
[0106]
本发明中所用的测试设备及测试条件为:
[0107]
纤维的力学性能:在全自动单纤维万能测试仪favimat+上进行单丝强力测试,分离长度在20mm以上的单丝,在初始应力是0.3cn,拉伸速度是10mm/min时进行测试。
[0108]
玻璃化转变温度:在氮气气氛下,以3℃/min的升温速率升温至600℃进行dma分析测试,得到纤维的玻璃化转变温度。
[0109]
分子量及分子量分布:使用安捷伦公司pl-gpc 200高温gpc,以dmf为流动相,配制paa为1mg/ml的dmf溶液样品,在35℃恒温条件下测试paa的分子量及其分布。
[0110]
嵌段共聚物中可以含有两种或两种以上的链段,于是会出现两个或多个玻璃化转变温度(tg)。而无规共聚物中只含有一种链段,只有一个tg;嵌段共聚物和无规共聚物的分子量测试只有1个峰值,而共混物会有两个或多个峰值。结合dma和pl-gpc分析测试,当pi纤维存在两个或多个玻璃化转变温度(tg),而聚合溶液分子量只有1个峰值时,说明纺丝原液成功合成嵌段聚合物,而非无规共聚或共混。
[0111]
【实施例1】(bpda/bia嵌段链段占总链段的摩尔百分比为70%)
[0112]
1、原液制备:(1)将5.48kg(24.44mol)2-(4-氨基苯基)-5-氨基苯并咪唑(bia)溶解于85kg n,n-二甲基乙酰胺(dmac)中,在25℃n2保护下搅拌,完全溶解后,加入6.47kg(22.00mol)3,3’,4,4
’-
联苯四甲酸二酐(bpda),加料完毕后继续搅拌1h,获得聚酰胺酸
(paa)预聚体溶液1;
[0113]
(2)同样的聚合条件,将0.75kg(6.97mol)对苯二胺(p-pda)溶解于n,n-二甲基乙酰胺中,1.37kg(6.27mol)均苯四酸二酐(pmda)加入到该二胺溶液中,充分搅拌1.5h得到预聚体溶液2;
[0114]
(3)将预聚体溶液1和预聚体溶液2充分混合1.5h得到预聚体溶液3;
[0115]
(4)将0.92kg(3.14mol)3,3’,4,4
’-
联苯四甲酸二酐(bpda)加入到预聚体溶液3中反应1.5h得到固含量为15%的纺丝原液;
[0116]
2、凝固成型:纺丝原液真空脱泡后,经过3μm过滤,然后通过喷丝头挤出进入第1道凝固浴,凝固浴温度0℃,浓度为35%,牵伸比为-50%,第2道凝固浴为30℃,浓度为20%,牵伸比为110%,第3道凝固浴为40℃,浓度为10%,牵伸比为105%,得到初生纤维。
[0117]
3、水洗:初生纤维经过10道水洗,水洗温度为45℃,水洗阶段牵伸1.1倍。
[0118]
4、上油及干燥致密化:将步骤3得到的纤维进行1道上油后进行干燥致密化,温度为85℃,随后再进行一次上油,上油后,进行第2道干燥致密化,干燥致密化的温度为130℃。
[0119]
5、亚胺化:将步骤4得到的原丝进行亚胺化处理,亚胺化过程共经过10个温区,采用逐步升温的方式,第一温区为100℃,第二温区为150℃,第三温区为200℃,第四温区为260℃,第五温区为280℃,第六温区为300℃,第七温区为310℃,第八温区为320℃,第九温区为330℃,第十温区为350℃,在亚胺化过程中采用氮气保护,氧含量小于100ppm。
[0120]
6、热牵伸、热定型及收丝:将步骤5得到的纤维进行热牵伸后在200℃热定型收丝,得到聚酰亚胺纤维,其中热牵伸温度为480℃,牵伸比为1.6。
[0121]
所得聚酰亚胺纤维单丝断裂强力为3.8gpa,cv=3.5%;模量为130gpa,cv=4.2%。
[0122]
【实施例2】(bpda/bia链段占总链段的摩尔百分比为20%)
[0123]
1、原液制备:(1)将1.99kg(8.89mol)2-(4-氨基苯基)-5-氨基苯并咪唑(bia)溶解于85kg n,n-二甲基乙酰胺(dmac)中,在25℃n2保护下搅拌,完全溶解后,加入2.35kg(8.00mol)3,3’,4,4
’-
联苯四甲酸二酐(bpda),加料完毕后继续搅拌1h,获得聚酰胺酸(paa)预聚体溶液1;
[0124]
(2)同样的聚合条件,将3.36kg(31.12mol)对苯二胺(p-pda)溶解于n,n-二甲基乙酰胺中,6.11kg(28.01mol)均苯四酸二酐(pmda)加入到该二胺溶液中,充分搅拌1h得到预聚体溶液2;
[0125]
(3)将预聚体溶液1和预聚体溶液2充分混合1h得到预聚体溶液3;
[0126]
(4)将1.18kg(4.00mol)3,3’,4,4
’-
联苯四甲酸二酐(bpda)加入到预聚体溶液3中反应1h得到固含量为15%的纺丝原液;
[0127]
其余步骤按照实施例1,所得聚酰亚胺纤维单丝断裂强力为3.2gpa,cv=3.2%;模量为105gpa,cv=4.0%。
[0128]
【实施例3】(bpda/bia嵌段链段占总链段的摩尔百分比为50%)
[0129]
1、原液制备:(1)将3.98kg(17.74mol)2-(4-氨基苯基)-5-氨基苯并咪唑(bia)溶解于85kg n,n-二甲基乙酰胺(dmac)中,在25℃n2保护下搅拌,完全溶解后,加入4.70kg(15.97mol)3,3’,4,4
’-
联苯四甲酸二酐(bpda),加料完毕后继续搅拌1h,获得聚酰胺酸(paa)预聚体溶液1;
[0130]
(2)同样的聚合条件,将2.84kg(14.19mol)4,4
’-
二氨基二苯醚(oda)溶解于n,n-二甲基乙酰胺中,2.78kg(12.78mol)均苯四酸二酐(pmda)加入到该二胺溶液中,充分搅拌1h得到预聚体溶液2;
[0131]
(3)将预聚体溶液1和预聚体溶液2充分混合1h得到预聚体溶液3;
[0132]
(4)将0.69kg(3.19mol)均苯四酸二酐(pmda)加入到预聚体溶液3中反应1h得到固含量为15%的纺丝原液;
[0133]
其余步骤按照实施例1,所得聚酰亚胺纤维单丝断裂强力为3.5gpa,cv=4.0%,模量为105gpa,cv=4.5%。
[0134]
【实施例4】(bpda/bia嵌段链段占总链段的摩尔百分比为50%)
[0135]
1、原液制备:(1)将3.89kg(17.34mol)2-(4-氨基苯基)-5-氨基苯并咪唑(bia)溶解于85kg n,n-二甲基乙酰胺(dmac)中,在25℃n2保护下搅拌,完全溶解后,加入4.59kg(15.61mol)3,3’,4,4
’-
联苯四甲酸二酐(bpda),加料完毕后继续搅拌1h,获得聚酰胺酸(paa)预聚体溶液1;
[0136]
(2)同样的聚合条件,将3.11kg(13.87mol)2-(4-氨基苯基)-5-氨基苯并咪唑(bia)溶解于n,n-二甲基乙酰胺中,2.72kg(12.48mol)均苯四酸二酐(pmda)加入到该二胺溶液中,充分搅拌1h得到预聚体溶液2;
[0137]
(3)将预聚体溶液1和预聚体溶液2充分混合1h得到预聚体溶液3;
[0138]
(4)将0.68kg(3.12mol)均苯四酸二酐(pmda)加入到预聚体溶液3反应1h中得到固含量为15%的纺丝原液;
[0139]
其余步骤按照实施例1,所得聚酰亚胺纤维单丝断裂强力为3.3gpa,cv=4.2%;模量为135gpa,cv=4.5%。
[0140]
【实施例5】(bpda/bia嵌段链段占总链段的摩尔百分比为50%)
[0141]
1、原液制备:(1)将4.05kg(18.07mol)2-(4-氨基苯基)-5-氨基苯并咪唑(bia)溶解于85kg n,n-二甲基乙酰胺(dmac)中,在25℃n2保护下搅拌,完全溶解后,加入5.21kg(17.71mol)3,3’,4,4
’-
联苯四甲酸二酐(bpda),加料完毕后继续搅拌1h,获得聚酰胺酸(paa)预聚体溶液1;
[0142]
(2)同样的聚合条件,将3.47kg(17.35mol)4,4
’-
二氨基二苯醚(oda)溶解于n,n-二甲基乙酰胺中,3.71kg(16.99mol)均苯四酸二酐(pmda)加入到该二胺溶液中,充分搅拌1h得到预聚体溶液2;
[0143]
(3)将预聚体溶液1和预聚体溶液2充分混合1h得到预聚体溶液3;
[0144]
(4)将0.16kg(0.72mol)均苯四酸二酐(pmda)加入到预聚体溶液3中反应1h得到固含量为15%的纺丝原液;
[0145]
其余步骤按照实施例1,所得聚酰亚胺纤维单丝断裂强力为3.65gpa,cv=4.5%,模量为125gpa,cv=4.3%。
[0146]
【实施例6】(bpda/bia嵌段链段占总链段的摩尔百分比为50%)
[0147]
1、原液制备:(1)将3.58kg(15.97mol)2-(4-氨基苯基)-5-氨基苯并咪唑(bia)溶解于85kg n,n-二甲基乙酰胺(dmac)中,在25℃n2保护下搅拌,完全溶解后,加入4.22kg(14.37mol)联苯四甲酸二酐(bpda),加料完毕后继续搅拌1h,获得聚酰胺酸(paa)预聚体溶液1;
[0148]
(2)同样的聚合条件,将2.56kg(12.78mol)4,4
’-
二氨基二苯醚(oda)溶解于n,n-二甲基乙酰胺中,3.71kg(11.50mol)3,3
′
,4,4
′-
二苯酮四酸二酐(btda)加入到该二胺溶液中,充分搅拌1h得到预聚体溶液2;
[0149]
(3)将预聚体溶液1和预聚体溶液2充分混合1h得到预聚体溶液3;
[0150]
(4)将0.926kg(2.87mol)3,3
′
,4,4
′-
二苯酮四酸二酐(btda)加入到预聚体溶液3中反应1h得到固含量为15%的纺丝原液;
[0151]
其余步骤按照实施例1,所得聚酰亚胺纤维单丝断裂强力为3.32gpa,cv=4.8%,模量为113gpa,cv=4.2%。
[0152]
【比较例1】(配比和实施例1相同,但先加入两个二胺,再加入两个二酐,无规共聚)
[0153]
1、原液制备:(1)将1.99kg(8.89mol)2-(4-氨基苯基)-5-氨基苯并咪唑(bia)和3.36kg(31.12mol)对苯二胺(p-pda)溶解于85kg n,n-二甲基乙酰胺(dmac)中,在25℃n2保护下搅拌,完全溶解后,加入3.53kg(12.00mol)3,3’,4,4
’-
联苯四甲酸二酐(bpda)和6.11kg(28.01mol)均苯四酸二酐(pmda),加料完毕后继续搅拌1h,得到固含量为15%的纺丝原液;
[0154]
按照实施例1步骤2~6制备聚酰亚胺纤维,所得聚酰亚胺纤维单丝断裂强力为3.1gpa,cv=12.5%;模量为115gpa,cv=11.8%。
[0155]
因为纺丝原液为无规共聚,聚合溶液粘度不稳定,纤维性能均一性和稳定性差。
[0156]
【比较例2】(顺序加料,二胺+二酐+二胺+二酐)
[0157]
1、原液制备:(1)将4.24kg(18.90mol)2-(4-氨基苯基)-5-氨基苯并咪唑(bia)溶解于85kg n,n-二甲基乙酰胺(dmac)中,在25℃n2保护下搅拌,完全溶解后,加入5.01kg(17.01mol)3,3’,4,4
’-
联苯四甲酸二酐(bpda),加料完毕后继续搅拌1h,获得聚酰胺酸(paa)预聚体溶液1;
[0158]
(2)然后将2.04kg(18.90mol)对苯二胺(p-pda)溶解于预聚体1中,再加入3.71kg(17.01mol)均苯四酸二酐(pmda),充分搅拌得到固含量为15%的纺丝原液;
[0159]
其余步骤按照实施例1步骤2~6制备聚酰亚胺纤维,所得聚酰亚胺纤维单丝断裂强力为2.5gpa,cv=15.6%;模量为95gpa,cv=13.8%。聚合溶液粘度具有偶然性,因为第二种二胺pda的加入会造成预聚体1发生分解,有时会导致聚酰胺酸溶液粘度较低,不符合湿法或者干湿法的纺丝溶液要求。或聚合物分子量偏低引起力学强度下降。
[0160]
【比较例3】(一种溶液酐过量,一种溶液胺过量)
[0161]
1、原液制备:(1)将3.97kg(17.68mol)2-(4-氨基苯基)-5-氨基苯并咪唑(bia)溶解于85kg n,n-二甲基乙酰胺(dmac)中,在25℃n2保护下搅拌,完全溶解后,加入5.46kg(18.56mol)3,3’,4,4
’-
联苯四甲酸二酐(bpda),加料完毕后继续搅拌1h,获得聚酰胺酸(paa)预聚体溶液1;
[0162]
(2)同样的聚合条件,将1.91kg(17.68mol)对苯二胺(p-pda)溶解于有机溶剂中,3.66kg(16.79mol)均苯四酸二酐(pmda)加入到该二胺溶液中,充分搅拌1h得到预聚体溶液2;
[0163]
(3)将预聚体溶液1和预聚体溶液2充分混合1h得到固含量为15%的纺丝原液;
[0164]
其余步骤按照实施例1步骤2~6制备聚酰亚胺纤维,所得聚酰亚胺纤维单丝断裂强力为2.8gpa,cv=16.2%;模量为102gpa,cv=15.4%。聚合溶液粘度具有偶然性,因为预
聚体溶液1酐过量容易发生酐水解为酸造成封端,混合后不进一步发生反应,容易造成溶液粘度较低或均匀性较差。
[0165]
【比较例4】(bpda/bia嵌段链段占总链段的摩尔百分比为10%)
[0166]
1、原液制备:(1)将1.06kg(4.70mol)2-(4-氨基苯基)-5-氨基苯并咪唑(bia)溶解于85kg n,n-二甲基乙酰胺(dmac)中,在25℃n2保护下搅拌,完全溶解后,加入1.24kg(4.23mol)3,3’,4,4
’-
联苯四甲酸二酐(bpda),加料完毕后继续搅拌1h,获得聚酰胺酸(paa)预聚体溶液1;
[0167]
(2)同样的聚合条件,将4.07kg(37.63mol)对苯二胺(p-pda)溶解于有机溶剂中,7.39kg(33.87mol)均苯四酸二酐(pmda)加入到该二胺溶液中,充分搅拌得到预聚体溶液2;
[0168]
(3)将预聚体溶液1和预聚体溶液2充分混合得到预聚体溶液3;
[0169]
(4)将1.24kg(4.23mol)3,3’,4,4
’-
联苯四甲酸二酐(bpda)加入到预聚体溶液3中得到固含量为15%的纺丝原液;
[0170]
其余步骤按照实施例1步骤2~6制备聚酰亚胺纤维,所得聚酰亚胺纤维单丝断裂强力为2.2gpa,cv=5.6%;模量为93gpa,cv=4.4%。
[0171]
【比较例5】(bpda/bia嵌段链段占总链段的摩尔百分比为80%)
[0172]
1、原液制备:(1)将6.01kg(26.77mol)2-(4-氨基苯基)-5-氨基苯并咪唑(bia)溶解于85kg n,n-二甲基乙酰胺(dmac)中,在25℃n2保护下搅拌,完全溶解后,加入7.09kg(24.1mol)3,3’,4,4
’-
联苯四甲酸二酐(bpda),加料完毕后继续搅拌1h,获得聚酰胺酸(paa)预聚体溶液1;
[0173]
(2)同样的聚合条件,将0.36kg(3.35mol)对苯二胺(p-pda)溶解于有机溶剂中,0.66kg(3.01mol)均苯四酸二酐(pmda)加入到该二胺溶液中,充分搅拌得到预聚体溶液2;
[0174]
(3)将预聚体溶液1和预聚体溶液2充分混合得到预聚体溶液3;
[0175]
(4)将0.89kg(3.01mol)3,3’,4,4
’-
联苯四甲酸二酐(bpda)加入到预聚体溶液3中得到固含量为15%的纺丝原液;
[0176]
其余步骤按照实施例1步骤2~6制备聚酰亚胺纤维,所得溶液bpda/bia嵌段含量较高,氢键密度较大,容易导致纺丝原液粘度过高,不利于纺丝。
[0177]
【比较例6】(最后补充的二酐a/(x-y+z-k)=0.9)
[0178]
1、原液制备:(1)将2.01kg(8.96mol)2-(4-氨基苯基)-5-氨基苯并咪唑(bia)溶解于85kg n,n-二甲基乙酰胺(dmac)中,在25℃n2保护下搅拌,完全溶解后,加入2.37kg(8.07mol)3,3’,4,4
’-
联苯四甲酸二酐(bpda),加料完毕后继续搅拌1h,获得聚酰胺酸(paa)预聚体溶液1;
[0179]
(2)同样的聚合条件,将3.39kg(31.37mol)对苯二胺(p-pda)溶解于有机溶剂中,6.16kg(28.23mol)均苯四酸二酐(pmda)加入到该二胺溶液中,充分搅拌得到预聚体溶液2;
[0180]
(3)将预聚体溶液1和预聚体溶液2充分混合得到预聚体溶液3;
[0181]
(4)将1.07kg(3.63mol)3,3’,4,4
’-
联苯四甲酸二酐(bpda)加入到预聚体溶液3中得到固含量为15%的纺丝原液;
[0182]
其余步骤按照实施例1步骤2~6制备聚酰亚胺纤维,此比较例中a/(x-y+z-k)<0.95,纺丝原液粘度不稳定,出现粘度下降,聚合不均匀,聚合物分子量偏低,引起力学强度下降,纤维不均匀。所得聚酰亚胺纤维单丝断裂强力为2.3gpa,cv=8.6%,模量为85gpa,cv
=14.4%。
[0183]
【比较例7】(最后补充的二酐a/(x-y+z-k)=1.1)
[0184]
1、原液制备:(1)将1.98kg(8.82mol)2-(4-氨基苯基)-5-氨基苯并咪唑(bia)溶解于85kg n,n-二甲基乙酰胺(dmac)中,在25℃n2保护下搅拌,完全溶解后,加入2.34kg(7.94mol)3,3’,4,4
’-
联苯四甲酸二酐(bpda),加料完毕后继续搅拌1h,获得聚酰胺酸(paa)预聚体溶液1;
[0185]
(2)同样的聚合条件,将3.34kg(30.88mol)对苯二胺(p-pda)溶解于有机溶剂中,6.06kg(27.79mol)均苯四酸二酐(pmda)加入到该二胺溶液中,充分搅拌得到预聚体溶液2;
[0186]
(3)将预聚体溶液1和预聚体溶液2充分混合得到预聚体溶液3;
[0187]
(4)将1.28kg(4.37mol)3,3’,4,4
’-
联苯四甲酸二酐(bpda)加入到预聚体溶液3中得到固含量为15%的纺丝原液;
[0188]
其余步骤按照实施例1步骤2~6制备聚酰亚胺纤维,此比较例中a/(x-y+z-k)>1.05,纺丝原液粘度不稳定,出现粘度下降,聚合不均匀,聚合物分子量偏低,引起力学强度下降,纤维不均匀。所得聚酰亚胺纤维单丝断裂强力为2.1gpa,cv=9.3%,模量为82gpa,cv=12.7%。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1