一种氟比洛芬酯的精制方法与流程
1.本发明属于医药合成技术领域,具体涉及一种氟比洛芬酯的精制方法。
背景技术:
2.氟比洛芬酯(英文名flurbiprofen axetil),其化学名为(
±
)2-(2-氟-4-联苯基)丙酸-1-乙酰氧基乙酯,分子式c
19h19
fo4,分子量330.36,密度1.169g/cm3。
3.结构式如下:
[0004][0005]
氟比洛芬酯是一种非甾体抗炎药,是氟比洛芬的前体药物,进入人体后可以靶向聚集在手术切口及炎症部位,水解生成氟比洛芬。氟比洛芬酯具有一定的靶向性、缓释性和脂溶性,较氟比洛芬具有更轻的胃肠道副作用、更强的药效和更长的药效持续时间。该药目前已取代氟比洛芬广泛应用于临床炎症性疼痛、癌痛及术后疼痛的治疗。
[0006]
氟比洛芬酯产品形状为油状液体,容易包合杂质,导致纯度不高,生产中也不能通过重结晶等相对简单的操作来对其进行精制(或纯化、提纯)。现有精制方法包括欧洲专利申请ep83108796.0公开的减压蒸馏法、中国专利申请cn201210574448.5公开的分子蒸馏法、中国专利申请cn201310638812.4、cn201510023365.0和cn201810609832.1公开的硅胶柱层析法、以及中国专利申请cn201110404346.4公开的硅胶吸附法等。
[0007]
减压蒸馏是液体产品纯化的常用方法,氟比洛芬酯最初也是采用该法进行精制,理论上收集173℃~175℃/0.8mmhg的馏分,由于长时间暴露于高温下,氟比洛芬酯容易发生分解反应,导致产品纯度和收率都不高。中国专利申请cn201210574448.5曾重复欧洲专利申请ep83108796.0公开的减压蒸馏方法,多次平行试验只得到了收率为73%~78%、纯度为96.5%~98.1%的产品。更让人遗憾地是,实际生产中真空度往往还达不到0.8mmhg的水平,这意味着氟比洛芬酯在大生产时可能要在更高的温度才能被收集,由此得到的产品很难直接用于药物制剂。
[0008]
为了改进氟比洛芬酯的纯化方法,中国专利申请cn201210574448.5公开了一种分子蒸馏法。分子蒸馏是一种在高真空条件下利用分子间平均自由程差异来实现分离的方法,其设备体积和设备复杂程度都远超常规蒸馏设备,相应的配套设备也多,因此在大规模生产应用中有不少困难,事实上中国也尚未见大规模运用。还有一些直接基于减压蒸馏的改进,如中国专利申请cn201310079429.x公开的一种氟比洛芬酯的制备方法,它将氟比洛芬酯粗品经减压蒸馏提纯后、再水洗和脱色,可以得到纯度大于99.7%的氟比洛芬精品。然而,该方法操作复杂、重现性不好,又因其以减压蒸馏为主要提纯手段,设备投资大、产品收率低、生产成本高等问题都限制了其在工业生产上的推广。
[0009]
当然,也有多篇文献报道硅胶柱层析法或硅胶吸附法可改进氟比洛芬酯纯度,但
本领域技术人员都知晓,若在最后精制的过程中使用硅胶既会增加用药安全性风险,又会因为硅胶废弃和溶剂回收污染环境。本发明人按照中国专利zl201110404346.4所提供方法制得的精制品在进行(25℃
±
2℃,60%
±
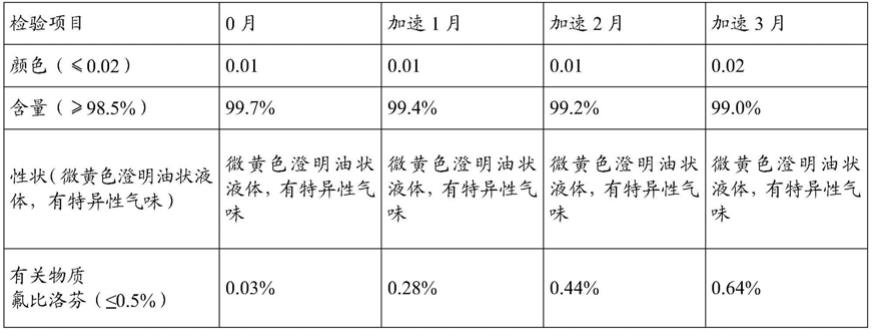
5%)加速稳定性试验时,2个月单杂氟比洛芬的含量已达1.77%,超出标准规定限度(≤0.5%),hplc结果见图1;而采用中国专利zl201310638812.4所提供方法制得的产品在3个月时单杂氟比洛芬的含量也已经为0.64%,超出标准规定限度(≤0.5%),hplc结果见图2。
[0010]
可见,现有的氟比洛芬酯精制(或纯化、提纯)方法都有其明显的弊端,将它们用于实验室研究尚可,但若用于工业大生产,均存在重现性差、设备庞大、操作复杂、环境污染和成本高企的问题。在环境保护的迫切需求和药品利润的缩窄趋势下,目前的氟比洛芬酯生产厂家都需要寻找到更高效、更环保、更经济的精制方法,以期获得纯度高、收率高、成本低的氟比洛芬酯纯品。
技术实现要素:
[0011]
以下是对本文详细描述的主题的概述。本概述并非是为了限制本技术的保护范围。
[0012]
本发明提供了一种氟比洛芬酯的精制方法,该方法操作简单、条件温和、生产成本低,经本方法精制后的氟比洛芬酯产品纯度高、收率高。
[0013]
本发明提供了一种氟比洛芬酯的精制方法,所述精制方法包括如下步骤:
[0014]
(1)将氟比洛芬酯粗品分散在非亲水性非质子有机溶剂中,完全溶解后,加入碱性水溶液形成混合液,将混合液在低于所述非亲水性非质子有机溶剂沸点的温度下搅拌,搅拌时间不少于10小时;
[0015]
(2)将步骤(1)搅拌后混合液,经静置,分液后,所得有机相先用水洗至ph=6.0~8.5后,再用饱和食盐水洗涤,所得有机相经干燥、过滤、浓缩后,得到氟比洛芬酯精制品。
[0016]
在本发明的上述实施方案中,步骤(1)中所述非亲水性非质子有机溶剂与氟比洛芬酯粗品的比例为1ml/g-10ml/g,可选地,1.5ml/g-10ml/g,1.5ml/g-5ml/g,或3ml/g-10ml/g。
[0017]
在本发明的上述实施方案中,步骤(1)中所述非亲水性非质子有机溶剂与所述碱性水溶液的体积比为1-8,优选地,为3-5。
[0018]
在本发明的上述实施方案中,步骤(1)中所述低于所述非亲水性非质子有机溶剂沸点的温度为15℃~49℃,优选地,为20℃~45℃。
[0019]
在本发明的上述实施方案中,步骤(1)中所述搅拌的转速为200rpm-600rpm,优选地,为300rpm-600rpm。
[0020]
在本发明的上述实施方案中,步骤(1)中所述搅拌的时间为10小时-40小时,优选地,为12小时-36小时,更优选地,为16小时-24小时。
[0021]
在本发明的上述实施方案中,步骤(2)中所述干燥的时间不少于1小时,优选地,不少于3小时。
[0022]
在本发明的上述实施方案中,步骤(2)中所述浓缩的温度不超过60℃,优选地,不超过50℃。
[0023]
在本发明的上述实施方案中,步骤(2)中所述静置的时间可以是5分钟至60分钟,
可选地,5分钟至40分钟,5分钟至30分钟,或者,5分钟、10分钟、15分钟、20分钟、25分钟、或30分钟。
[0024]
在本发明的上述实施方案中,所述碱性水溶液为氨水、氢氧化钠溶液、碳酸钠溶液、碳酸氢钠溶液、磷酸钠溶液、磷酸氢二钠溶液、亚硫酸钠溶液、草酸钠溶液、富马酸钠溶液、枸橼酸钠溶液、氢氧化钾溶液、碳酸钾溶液、碳酸氢钾溶液、磷酸钾溶液、磷酸氢二钾溶液、亚硫酸钾溶液、草酸钾溶液、富马酸钾溶液、枸橼酸钾溶液、氢氧化锂溶液、或碳酸锂溶液、或它们的组合。
[0025]
在一些实施方案中,所述碱性水溶液为饱和氨水。
[0026]
在一些实施方案中,所述碱性水溶液质量百分比为0.01%-0.4%的氢氧化钠溶液。
[0027]
在一些实施方案中,所述碱性水溶液质量百分比为10%至饱和的碳酸钠溶液。
[0028]
在一些实施方案中,所述碱性水溶液为饱和的碳酸氢钠溶液。
[0029]
在一些实施方案中,所述碱性水溶液质量百分比为0.01%-0.4%的氢氧化钾溶液。
[0030]
在一些实施方案中,所述碱性水溶液质量百分比为10%-20%的碳酸钾溶液。
[0031]
在一实施方案中,所述碱性水溶液质量百分比为10%-20%的碳酸氢钾溶液。
[0032]
在本发明的上述实施方案中,所述非亲水性非质子有机溶剂为乙醚、异丙醚、石油醚、正己烷、环己烷、正庚烷、异庚烷、辛烷、乙酸乙酯、甲酸乙酯、乙酸异丙酯、甲基乙基醚、甲基叔丁基醚、二氯甲烷、二氯乙烷、和三氯甲烷中的一种,或它们两种以上的混合溶剂。
[0033]
在一些实施方案中,所述非亲水性非质子有机溶剂为正庚烷、异庚烷、乙酸乙酯、甲基乙基醚、和甲基叔丁基醚中的一种,或它们两种以上的混合溶剂。
[0034]
在本发明的上述实施方案中,所述氟比洛芬酯精制品的纯度不小于99.9%。
[0035]
在本发明的上述实施方案中,所述氟比洛芬酯精制品的收率不小于95%。
[0036]
在本发明的上述实施方案中,所述氟比洛芬酯粗品采用氟比洛芬与溴乙基乙酸酯制备得到。也可参照中国专利zl201110404346.4、cn201310079429.x、cn201810609832.1等方法制备。
[0037]
本发明的有益效果是:
[0038]
本发明所提供氟比洛芬酯的精制方法工艺简单,极易操作,例如仅需室温搅拌,水洗浓缩就能得到成品。所得成品的纯度达到99.9%以上、收率达到95%以上,通过加速9月和长期2年考察质量检测均符合标准,产品稳定性好,生产过程中只需要一般的反应设备,重现性好;相比于减压蒸馏方式的收率提高了10%-25%,避免采用高温减压的蒸馏方式,能耗大幅度减少、成本大大降低,设备上避免使用昂贵的高精密高温减压蒸馏设备,能够广泛适用于工业化大生产,避免使用硅胶,降低了用药安全隐患以及解决了产品稳定性差、生产周期长的问题。
[0039]
本技术的其它特征和优点将在随后的说明书中阐述,并且,部分地从说明书中变得显而易见,或者通过实施本技术而了解。本技术的其他优点可通过在说明书以及附图中所描述的方案来实现和获得。
附图说明
[0040]
附图用来提供对本技术技术方案的理解,并且构成说明书的一部分,与本技术的实施例一起用于解释本技术的技术方案,并不构成对本技术技术方案的限制。
[0041]
图1为采用zl201110404346.4方法所得氟比洛芬酯成品的加速第2月hplc图谱(保留时间8.83min为氟比洛芬的峰,29.85和31.76min为氟比洛芬酯峰,其他为未知杂质峰);
[0042]
图2为采用zl201310638812.4方法所得氟比洛芬酯成品的加速第3月hplc图谱(保留时间8.84min为氟比洛芬的峰,29.97和31.88min为氟比洛芬酯峰,其他为未知杂质峰);
[0043]
图3为实施例1中氟比洛芬酯成品的hplc图谱(保留时间8.77min为氟比洛芬峰、15.5min为未知杂质峰,29.63和31.49min为氟比洛芬酯峰);
[0044]
图4为实施例2中氟比洛芬酯成品的hplc图谱(保留时间32.43min和34.56min为氟比洛芬酯峰,其他为未知杂质峰);
[0045]
图5为实施例3中氟比洛芬酯成品的hplc图谱(保留时间34.06min和36.34min为氟比洛芬酯峰,其他为未知杂质峰);
[0046]
图6为实施例4中氟比洛芬酯成品的hplc图谱(保留时间34.08min和36.35min为氟比洛芬酯峰,其他为未知杂质峰);
[0047]
图7为实施例5中氟比洛芬酯成品的hplc图谱(保留时间29.42min和31.27min为氟比洛芬酯峰)。
具体实施方式
[0048]
为使本技术的目的、技术方案和优点更加清楚明白,下文中将对本发明的实施例进行详细说明。需要说明的是,在不冲突的情况下,本技术中的实施例及实施例中的特征可以相互任意组合。
[0049]
在本发明的实施例中,本发明提供了一种氟比洛芬酯的精制方法,所述精制方法包括如下步骤:
[0050]
(1)将氟比洛芬酯粗品分散在非亲水性非质子有机溶剂中,完全溶解后,加入碱性水溶液形成混合液,将混合液在低于所述非亲水性非质子有机溶剂沸点的温度下搅拌,搅拌时间不少于10小时;
[0051]
(2)将步骤(1)搅拌后混合液,经静置,分液后,所得有机相先用水洗至ph=6.0~8.5后,再用饱和食盐水洗涤,所得有机相经干燥、过滤、浓缩后,得到氟比洛芬酯精制品;
[0052]
可选地,步骤(1)中所述非亲水性非质子有机溶剂与氟比洛芬酯粗品的比例为为1ml/g-10ml/g,可选地,1.5ml/g-10ml/g,1.5ml/g-5ml/g,或3ml/g-10ml/g;和/或
[0053]
步骤(1)中所述非亲水性非质子有机溶剂与所述碱性水溶液的体积比为1-8,优选地,为3-5;和/或
[0054]
步骤(1)中所述低于所述非亲水性非质子有机溶剂沸点的温度为15℃~49℃,优选地,为20℃~45℃;和/或
[0055]
步骤(1)中所述搅拌的转速为200rpm-600rpm,优选地,为300rpm-600rpm;和/或
[0056]
步骤(1)中所述搅拌的时间为10小时-40小时,优选地,为12小时-36小时,更优选地,为16小时-24小时;
[0057]
步骤(1)中所述碱性水溶液为饱和氨水、质量百分比为0.01%-0.4%的氢氧化钠
溶液、质量百分比为10%至饱和的碳酸钠溶液、饱和的碳酸氢钠溶液、质量百分比为10%-20%的碳酸钾溶液、或质量百分比为10%-20%的碳酸氢钾溶液;
[0058]
可选地,步骤(2)中所述静置的时间可以是5分钟至60分钟,可选地,5分钟至40分钟,5分钟至30分钟,或者,5分钟、10分钟、15分钟、20分钟、25分钟、或30分钟;
[0059]
步骤(2)中所述干燥的时间不少于1小时,优选地,不少于3小时;和/或
[0060]
步骤(2)中所述浓缩的温度不超过60℃,优选地,不超过50℃。
[0061]
在本发明中,采用hplc进行纯度检测的条件如下:
[0062]
色谱柱:十八烷基硅烷键合硅胶(色谱柱型号:xbridge@c18 4.6
×
250mm5um)
[0063]
柱温:35℃
[0064]
波长:254nm
[0065]
流速:1ml/min
[0066]
流动相:乙腈:水=500:500
[0067]
取样量:20μl
[0068]
检验方法:精密量取本品适量,用流动相溶解并稀释制成每l ml中约含0.5mg的溶液,作为供试品溶液;精密量取1ml,置100ml量瓶中,用流动相稀释至刻度,摇匀,作为对照溶液。另取氟比洛芬酯、用流动相溶解制成每l ml中含氟比洛芬酯0.5mg及各杂质5μg的溶液,作为系统适用性试验溶液。照含量测定项下色谱条件,取系统适应性溶液20μl注入液相色谱仪,另取对照溶液20μl,注入液相色谱仪,调节检测灵敏度,使主成分色谱峰的峰高约为满量程的20%。精密量取供试品溶液与对照溶液各20μl,分别注入液相色谱仪。
[0069]
本发明所述的氟比洛芬酯粗品由氟比洛芬与溴乙基乙酸酯制备得到,也可参照zl201110404346.4、cn201310079429.x、cn201810609832.1等方法制备。
[0070]
表一.专利样品(zl201310638812.4)(25℃
±
2℃,60%
±
5%)加速稳定性试验
[0071][0072]
表二.专利样品(zl201110404346.4)(25℃
±
2℃,60%
±
5%)加速稳定性试验
[0073]
[0074][0075]
表一是按照专利zl201310638812.4实例1制备;表二是按照专利zl201110404346.4的实例4制备。氟比洛芬酯粗品的制备:参照zl201110404346.4的实例4的工艺制备,具体如下,称取氟比洛芬600g加入反应釜中,加入丙酮6l搅拌溶解,称取无水碳酸钾350g和1-溴乙基乙酸酯550g,在50-60℃,回流反应2小时,过滤,用1l丙酮洗涤,浓缩至干即为粗品。
[0076]
实施例1
[0077]
取氟比洛芬酯粗品200g用乙酸乙酯600ml溶解,加入饱和的碳酸钠溶液1200ml,升温至35℃,在400rpm条件下搅拌28小时。
[0078]
静置5分钟分液,用水洗涤有机相,洗涤至测得水相ph至为7.1,用饱和的食盐水洗涤后,收集有机相,加入无水硫酸钠干燥3小时,过滤,在50℃条件下减压浓缩至溶剂残留合格(乙酸乙酯≤0.5%)即为成品。得到成品为193g,收率96.5%,hplc检测纯度99.99%,具体如图3所示。
[0079]
将所得到的氟比洛芬酯检测合格后,在(25℃
±
2℃,60%
±
5%)加速条件下的放3个月、6个月和9个月的样品;在(6℃
±
2℃)下进行长期稳定性考察放3月、6月、9月、12月、18月和24月的样品。检测了性质、有关、含量、颜色。
[0080]
所得检测结果如表1和表2所示:
[0081]
表1实施例1样品(25℃
±
2℃,60%
±
5%)加速稳定性试验
[0082][0083][0084]
表2实施例1样品(6℃
±
2℃)长期稳定性试验
[0085][0086]
从表1和表2可以看出,通过本发明合成出的成品,在经过(25℃
±
2℃,60%
±
5%)加速9月和(6℃
±
2℃)长期24月后产品质量几乎没有发生明显变化,说明产品的稳定性很好。
[0087]
实施例2
[0088]
将氟比洛芬酯粗品200g用正庚烷600ml溶解,加入20%的碳酸钾溶液1200ml,升温至30℃,在400转条件下搅拌24小时。
[0089]
静置10分钟分液,用水洗涤有机相,洗涤至测得水相ph至为7.3,用饱和的食盐水洗涤后,收集有机相,加入无水硫酸镁干燥4小时,过滤,在45℃条件下减压浓缩至溶剂残留合格(正庚烷≤0.5%)即为成品。得到成品为192g,收率96%,hplc纯度99.95%。
[0090]
将所得到的氟比洛芬酯检测合格后,在(25℃
±
2℃,60%
±
5%)加速条件下的放3个月、6个月和9个月的样品;在(6℃
±
2℃)下进行长期稳定性考察放3月、6月、9月、12月、18月和24月的样品。检测了性质、有关、含量、颜色。
[0091]
所得检测结果如表3和表4所示:
[0092]
表3实施例2样品(25℃
±
2℃,60%
±
5%)加速稳定性试验
[0093][0094][0095]
表4实施例2样品(6℃
±
2℃)长期稳定性试验
[0096][0097]
从表3和表4可以看出,通过本发明合成出的成品,在经过(25℃
±
2℃,60%
±
5%)加速9月和(6℃
±
2℃)长期24月后产品质量几乎没有发生明显变化,说明产品的稳定性很好。
[0098]
实施例3
[0099]
将氟比洛芬酯粗品200g用正庚烷600ml溶解,加入饱和的氨水溶液1200ml,升温至30℃,在600rpm条件下搅拌24小时。
[0100]
静置15分钟分液,用水洗涤有机相,洗涤至测得水相ph至为7.5,用饱和的食盐水洗涤后,收集有机相,加入无水硫酸镁干燥4小时,过滤,在45℃条件下减压浓缩至溶剂残留合格(正庚烷≤0.5%)即为成品。得到成品为195g,收率97.5%,hplc纯度99.97%。
[0101]
将所得到的氟比洛芬酯检测合格后,在(25℃
±
2℃,60%
±
5%)加速条件下的放3个月、6个月和9个月的样品;在(6℃
±
2℃)下进行长期稳定性考察放3月、6月、9月、12月、18月和24月的样品。检测了性质、有关、含量、颜色。
[0102]
所得检测结果如表5和表6所示:
[0103]
表5实施例3样品(25℃
±
2℃,60%
±
5%)加速稳定性试验
[0104][0105]
表6实施例3样品(6℃
±
2℃)长期稳定性试验
[0106][0107]
从表5和表6可以看出,通过本发明合成出的成品,在经过(25℃
±
2℃,60%
±
5%)加速9月和(6℃
±
2℃)长期24月后产品质量几乎没有发生明显变化,说明产品的稳定性很好。
[0108]
实施例4
[0109]
将氟比洛芬酯粗品200g用甲基叔丁基醚600ml溶解,加入饱和碳酸氢钠溶液1200ml,升温至45℃,在600rpm条件下搅拌36小时。
[0110]
静置20分钟分液,用水洗涤有机相,洗涤至测得水相ph至为7.0,用饱和的食盐水洗涤后,收集有机相,加入无水硫酸镁干燥4小时,过滤,在45℃条件下减压浓缩至溶剂残留合格(甲基叔丁基醚≤0.5%)即为成品。得到成品为196g,收率98%,hplc纯度99.95%。
[0111]
将所得到的氟比洛芬酯检测合格后,在(25℃
±
2℃,60%
±
5%)加速条件下的放3个月、6个月和9个月的样品;在(6℃
±
2℃)下进行长期稳定性考察放3月、6月、9月、12月、18月和24月的样品。检测了性质、有关、含量、颜色。
[0112]
所得检测结果如表7和表8所示:
[0113]
表7实施例4样品(25℃
±
2℃,60%
±
5%)加速稳定性试验
[0114][0115]
表8实施例4样品(6℃
±
2℃)长期稳定性试验
[0116][0117]
从表7和表8可以看出,通过本发明合成出的成品,在经过(25℃
±
2℃,60%
±
5%)加速9月和(6℃
±
2℃)长期24月后产品质量几乎没有发生明显变化,说明产品的稳定性很好。
[0118]
实施例5
[0119]
将氟比洛芬酯粗品200g用甲基叔丁基醚600ml溶解,加入10%碳酸钠溶液900ml,升温至40℃,在600rpm条件下搅拌28小时。
[0120]
静置25分钟分液,用水洗涤有机相,洗涤至测得水相ph至为7.2,用饱和的食盐水洗涤后,收集有机相,加入无水硫酸镁干燥3小时,过滤,在45℃条件下减压浓缩至溶剂残留合格(甲基叔丁基醚≤0.5%)即为成品。得到成品为192g,收率96%,hplc纯度99.97%。
[0121]
将所得到的氟比洛芬酯检测合格后,在(25℃
±
2℃,60%
±
5%)加速条件下的放3个月、6个月和9个月的样品;在(6℃
±
2℃)下进行长期稳定性考察放3月、6月、9月、12月、18月和24月的样品。检测了性质、有关、含量、颜色。
[0122]
所得检测结果如表9和表10所示:
[0123]
表9实施例5样品(25℃
±
2℃,60%
±
5%)加速稳定性试验
[0124][0125]
表10实施例5样品(6℃
±
2℃)长期稳定性试验
[0126]
[0127][0128]
从表9和表19可以看出,通过本发明合成出的成品,在经过(25℃
±
2℃,60%
±
5%)加速9月和(6℃
±
2℃)长期24月后产品质量几乎没有发生明显变化,说明产品的稳定性很好。
[0129]
本技术描述了多个实施例,但是该描述是示例性的,而不是限制性的,并且对于本领域的普通技术人员来说显而易见的是,在本技术所描述的实施例包含的范围内可以有更多的实施例和实现方案。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1