一种6-溴酞嗪的合成方法与流程
本发明涉及有机化工中间体合成技术领域,具体涉及一种6-溴酞嗪的合成方法。
背景技术:
6-溴酞嗪是一种酞嗪类的杂环化合物,是一类极其重要的药物中间体,应用前景广阔。该中间体及其衍生物被用作构建众多医药药物活性分子。如制备用于治疗增殖性疾病蛋白激酶抑制剂,用于治疗神经紊乱eph受体调节剂,用于治疗癌症的pi3激酶抑制剂等。
目前,合成6-溴酞嗪的合成路线主要有两种方法:
1.以4-溴邻苯二甲酸为原料制备
此方法是以4-溴邻苯二甲酸为原料,经三步:1)硼氢化钠/三氟化硼乙醚还原酯;2)swernoxidation氧化醇到醛;3)水合肼关环合成6-溴酞嗪。对于该路线,首先第一步反应产生大量的氢气,有非常大的安全隐患;其次第二步的swernoxidation需要在深冷(-70℃以下)进行,反应条件要求很苛刻,更为重要的是二醛化合物不稳定,该步收率在放大时降至10%以下。再次原料4-溴邻苯二甲酸的价格比较贵,不利于工业化生产6-溴酞嗪。
2.采用溴化铝/氯化铝高温关环制备
专利wo2009155121a2对该方法进行了报道,此方法路线中存在以下不足:采用高温+路易斯酸条件,对反应设备,配套设施及生产人员的要求很苛刻;选择性不好,得到的是6-溴酞嗪与5-溴酞嗪的混合物,比例为4:1,在专利wo2009155121a2中采用反相柱制备分离,该分离方法成本很高,不适合放大生产。
因此,现有技术中合成6-溴酞嗪的方法存在原料昂贵、反应条件苛刻、收率低的技术问题。
技术实现要素:
本发明的目的在于:提供一种6-溴酞嗪的合成方法,解决现有技术中反应收率低、原料贵、反应条件苛刻、不宜放大的技术缺陷。
本发明为实现上述目的所采用的技术方案如下:
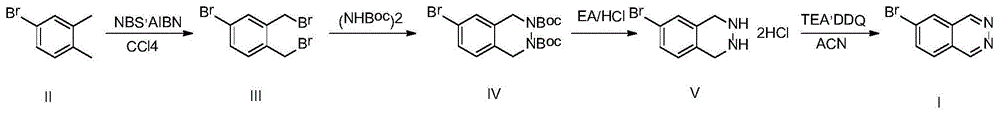
一种6-溴酞嗪的合成方法,合成路线如下:
所述合成方法包括以下四个步骤:
1)4-溴-1,2-二甲苯为原料,采用自由基反应合成化合物ⅲ;
2)步骤1)得到的化合物ⅲ与肼基二甲酸叔丁酯,在50%的氢氧化钠水溶液,相转移催化剂teab的作用下,生成化合物ⅳ;
3)步骤2)得到的化合物ⅳ在ea/hcl的作用下,生成化合物ⅴ;
4)步骤3)得到的化合物ⅴ在三乙胺,ddq(2,3-二氯-5,6-二氰基-1,4-苯醌)的作用下生成化合物ⅰ。
上述6-溴酞嗪的合成方法中,
步骤1)的具体过程为:将4-溴-1,2-二甲苯,nbs和aibn加入到四氯化碳中,升温至回流,在该条件下反应3小时,降至室温,过滤,滤液旋干后用石油醚打浆,过滤,减压除去溶剂得到化合物iii。
优选地,步骤1)中的4-溴-1,2-二甲苯与nbs的摩尔比是1:2.5-1:3.0;4-溴-1,2-二甲苯与aibn的摩尔比是1:0.1-1:0.2。
步骤2)的具体过程为:将化合物ⅲ与肼基二甲酸叔丁酯溶解到10倍体积的甲苯中,再向其中加入相转移催化剂teab及5倍体积的50%的氢氧化钠水溶液,随后升温至100℃,反应10小时,反应完毕后,降至室温,分层,有机相水洗,饱和食盐水洗涤干燥后旋干,将粗品加入到甲基叔丁基醚中,升温至回流,搅拌1小时后自然冷却至室温,烘干后得到化合物ⅳ。
优选地,步骤2)的化合物ⅲ与肼基二甲酸叔丁酯的摩尔比是1:1.05-1:1.1,化合物ⅲ与teab的摩尔比是1:0.05-1:0.1。
步骤3)的具体过程为:将化合物ⅳ溶解到5倍体积的乙酸乙酯,降温至0℃,控温0-5℃,向其中滴加5倍体积的乙酸乙酯/hcl溶液(4m),滴加完毕后,缓慢升至室温,反应2小时后,过滤即得化合物ⅴ,粗品直接用于下步即可。
所述步骤4)的具体过程为:将化合物ⅴ加入到10倍体积的乙腈中,降温至0℃,控温0-10℃,向其中滴加三乙胺,完毕后继续控温0-10℃向其中分批加入ddq,缓慢升至室温,反应18小时。将反应液倒入冰水中,过滤,将粗品用甲基叔丁基醚重结晶基后得到6-溴酞嗪。
优选地,步骤4)的化合物ⅴ与三乙胺的摩尔比是1:3.0~1:3.5,化合物ⅴ与ddq的摩尔比是1:2.5~1:3.0。
优选地,步骤4)中重结晶步骤,溶剂甲基叔丁基醚的使用量为2.5v到3.0v。
以上合成方法中,四步总收率为52%。中间体化合物ⅲ,化合物ⅳ和化合物ⅴ都只是经过简单的后处理即可达到所需纯度。终产物6-溴酞嗪采用甲基叔丁基醚重结晶后,即可得到纯品,纯度达到98%。
与现有技术相比,本发明的有益效果如下:
1.本发明以4-溴-1,2-二甲苯为原料通过自由基反应,关环,脱保护及氧化反应得到目标产物6-溴酞嗪。本发明解决了现有技术中原料贵,中间体不稳定,操作条件苛刻以及整体收率低的问题。该路线反应条件温和,操作简单,以较高的收率和高纯度制得终产物。
2.本发明提供6-溴酞嗪的路线,每步都不需要繁琐的柱层析,只是通过简单的后处理,显著地提高了收率,总收率达到52%,使得该合成路线适合工艺放大。
附图说明
附图1为本发明的工艺流程示意图。
具体实施方式
下面将对本发明的技术方案进行清楚、完整地描述,显然,所描述的实施例是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
以下通过具体实施例来说明本发明的技术方案。本发明中所用的原料和试剂均市售可得。
实施例1
将4-溴-1,2-二甲苯(200g,1.08mol,1.0eq),nbs(500g,2.81mol,2.6eq)和aibn(19.7g,0.12mol,0.11eq)加入到2l的四氯化碳中,升温至回流,在该条件下反应3小时,降至室温,过滤,滤液旋干后用400ml石油醚打浆,过滤,减压除去溶剂得到化合物ⅲ(297g,收率80.1%);
实施例2
将化合物ⅲ(280g,0.82mol,1.0eq)与肼基二甲酸叔丁酯(189.7g,0.82mol,1.0eq)溶解到2.8l甲苯中,再向其中加入相转移催化剂teab(5.8g,0.04mol,0.05eq)及1.4l的50%的氢氧化钠水溶液,随后升温至100℃,反应10小时,反应完毕后,降至室温,分层,有机相水洗,饱和食盐水洗涤干燥后旋干,向粗品中加入500ml甲基叔丁基醚,打浆0.5小时,过滤,烘干后得到化合物ⅳ(310g,收率91.8%)。
实施例3
将化合物ⅳ(300g,0.73mol,0.0eq)溶解到1.5l的乙酸乙酯中,降温至0℃,控温0-5℃,向其中滴加1.5l的乙酸乙酯/hcl溶液(4m),滴加完毕后,缓慢升至室温,反应2小时,过滤得到化合物ⅴ(198g,收率95.4%)。
实施例4
将化合物ⅴ(198g,0.69mol,1.0eq)加入到2l的乙腈中,降温至0℃,控温0-10℃,向其中滴加三乙胺(223g,2.21mol,3.2eq),滴加完毕后继续控温0-10℃,向其中分批加入ddq(438g,1.93mol,2.8eq),缓慢升至室温,反应18小时。将反应液倒入冰水中,过滤,将粗品加入到1l的甲基叔丁基醚中,升温至回流搅拌0.5小时,热过滤,滤液自然降至10-15℃,搅拌3小时后过滤,烘干后得到6-溴酞嗪(108g,74.6%)。
1hnmr(600mhz,cdcl3):9.58(s,1h),9.53(s,1h),8.19(s,1h),8.07(dd,j=8.5,1.4hz,1h),7.91(d,j=8.6,1h)。
应当说明的是,上述实施例均可根据需要自由组合。以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!