一种利那洛肽的合成方法与流程
本发明涉及药物合成技术领域,具体涉及一种采用液相一步环合制备利那洛肽的方法。
背景技术:
利那洛肽(商品名为linzess)是一种由ironwood公司开发的新型的gc-c(肠上皮细胞尿苷酸环化酶c)受体激动剂。它激活肠上皮细胞顶端的gc-c受体,导致细胞内和细胞外环磷酸鸟苷的增加。其作用是增加了氯和碳酸氢盐进入肠腔的分泌,进而导致液体分泌增加和大便加速,用于治疗慢传输型便秘和便秘型肠易激综合征(ibs-c)的成人患者。
利那洛肽由14个氨基酸组成,含有3对二硫键,其序列结构如下:
利那洛肽中三个二硫键的成环反应是该产品合成的难点。专利文献w02019113872a1公开了采用一步树脂环合法合成利那洛肽。这个方法是基于使用n-x-丁二酰亚胺中的一种形式,比如:n-氯丁二酰亚胺、n-溴丁二酰亚胺、n-碘丁二酰亚胺、n-羟基硫代丁二酰亚胺。虽然该方法具有低浓度、树脂环合的优点,但同时去除所有半胱氨酸保护基和一步形成三个二硫桥,会导致反应中出现多个杂质,进而导致纯化变得繁琐。
专利文献cn106831950a公开了采用分步氧化法,分两步合成利那洛肽。第一步先两个二硫键,第二步形成剩余的第三个二硫键。
专利文献cn103626849a公开了采用分步氧化法合成利那洛肽的方法,在单一溶液中借助于三种不同的半胱氨酸保护基如fmoc-cys(trt)-oh,fmoc-cys(acm)-oh,fmoc-cys(hmq)-oh进行了反应。然而,在分步氧化法中,半胱氨酸需要三对不同的保护基,这一复杂的操作步骤将导致产率低、成本高。
即使在同一溶液中对半胱氨酸采用两种或三种不同的保护基进行逐步氧化,获得的产率和纯度也较低,且成本较高。
随机氧化法是一步形成三个二硫桥的方法,其优点是该体系对所有六种半胱氨酸只需要一个类似的保护基团,氧化体系也易于操作,但这里的一个问题是可能产生错配异构体。
专利文献cn106167514a采用随机氧化法来合成利那洛肽,在盐酸胍氧化体系的帮助下,用胱氨酸/半胱氨酸一步环合形成了三个二硫桥。但该试剂体系即盐酸胍-胱氨酸/半胱氨酸氧化体系对三个二硫桥的形成并不有效,它会导致一些杂质的产生。
专利文献cn105884864a公开了采用随机氧化法合成利那洛肽,在较浓氧化体系的帮助下形成3个二硫桥。即每1.0g利那洛肽,用50-70ml碳酸铵/dmso/水溶液与对苯二酚或tcep一起,反应维持时间为1-2小时。然而,在该体系条件下,会产生更多的杂质,进而使净化变得繁琐。
专利文献cn106008674a公开了随机氧化法合成利那洛肽,采用了以二甲基亚砜/氨水体系为氧化剂,并用hmp-am树脂制备了线型利那洛肽。但hmp-am树脂不适合构建14个氨基酸的多肽,用这个方法得到的产物最大纯度仅为98.4%。除此之外,在氧化系统中使用乙腈是不可取的,为进一步纯化带来困难。
专利文献cn102875655b公开了随机氧化法合成利那洛肽。在gsh(谷胱甘肽)/gssh(氧化谷胱甘肽)氧化体系的辅助下获得利那洛肽的粗品。但该氧化体系不能准确定位三对二硫键的连接位置,导致大量杂质的产生。
专利文献us20170240599a1介绍了采用随机氧化法,在空气和过氧化氢的作用下合成利那洛肽。然而,过氧化氢的强氧化性会破坏二硫键,并形成二硫桥的错配。
因此,有必要开发有效的方法来解决上述问题,并找到适合商业化规模的最佳方法。
技术实现要素:
本发明的目的在于提供一种经济有效的方法来合成低外消旋度的,无二硫桥错配的利那洛肽,以满足商业化规模生产的要求。
为实现上述目的,本发明采用如下技术方案:
一种利那洛肽的合成方法,包括以下步骤:
(1)按照seqidno:1所示氨基酸序列从c端到n端顺序,依次将n端和侧链偶联有保护基的氨基酸连接到树脂固相载体上,固相合成利那洛肽树脂,其中半胱氨酸的c末端含有opfp酯;
(2)利用裂解试剂脱除利那洛肽树脂上所有保护基和树脂固相载体,得到线性利那洛肽;
(3)将线性利那洛肽溶于质量百分比浓度为2~10%的二甲亚砜水溶液中,制得线性利那洛肽浓度为1~5mg/ml的混合液,再加入相当于线性利那洛肽10~15倍当量的硫酸铵、氢氧化铵或碳酸铵,ph值控制在8.0±0.3,环合反应形成第1、6位cys的二硫键、第2、10位cys的二硫键以及第5、13位cys的二硫键,得到利那洛肽粗品,纯化后制得利那洛肽。
本发明采用一步氧化方法制备利那洛肽,防止肽链重复折叠,大大提高生产效率,并在正确的二硫键对上进行环合反应,减少错配副产物的产生,提高利那洛肽的纯度。
步骤(1),利用固相合成法制备树脂肽。
本发明对于所有六个半胱氨酸均使用相同的保护基,对于利那洛肽的合成,其cys侧链保护基的选择关系着线性利那洛肽的纯度,作为优选,cys侧链上偶联的保护基为氧杂蒽基(xan)、三苯甲基(trt)或叔丁基(tbu),更为优选,cys侧链上偶联的保护基为氧杂蒽基(xan),由于保护基的空间位阻,避免线性多肽链发生聚集,保证肽的稳定性。
对于其余需要保护侧链的氨基酸,本发明采用tbu作为tyr、thr侧链保护基、trt作为asn侧链保护基、otbu作为glu侧链保护基。
半胱氨酸是一种易外消旋的氨基酸,本发明在半胱氨酸的位置上使用opfp酯,可以避免这种情况。固相合成过程中,opfp酯偶联无需使用偶联试剂,且偶联效率更高,有效地防止了固相多肽合成中的β-sheet。
作为优选,fmoc-cys(xan)-opfp用于固相合成利那洛肽树脂。
具体地:将fmoc-tyr(tbu)-oh偶联到树脂固相载体上,再脱fmoc保护基得到h-tyr(tbu)-树脂固相载体;然后按照seqidno:1所示氨基酸序列顺序,依次将fmoc-cys(xan)-opfp、fmoc-thr(tbu)-gly-oh、fmoc-cys(xan)-opfp、fmoc-pro-ala-oh、fmoc-asn(trt)-oh、fmoc-cys(xan)-opfp、fmoc-cys(xan)-opfp、fmoc-tyr(tbu)-oh、fmoc-glu(otbu)-oh、fmoc-cys(xan)-opfp、boc-cys(xan)-opfp进行延伸偶联,得到boc-cys(xan)-cys(xan)-glu(otbu)-tyr(tbu)-cys(xan)-cys(xan)-asn(trt)-pro-ala-cys(xan)-thr(tbu)-gly-cys(xan)-tyr(tbu)-树脂固相载体。
为了降低合成成本和去除多肽杂质,两种二肽即fmoc-thr(tbu)-gly-oh、fmoc-pro-ala-oh被应用在合成中。
所述树脂固相载体可采用王树脂、2-氯三苯甲基氯树脂、phb树脂、hmpa树脂、hmpb树脂、rinkacid树脂、tentageltga树脂、tentagelsphb树脂。作为优选,所述树脂固相载体为wang树脂或ctc树脂,取代度为1.0~1.2mmol/g,这两种树脂的锚定效果好,操作简单且材料易得。
在第一个氨基酸即fmoc-tyr(tbu)-oh锚定后,使用10-30%哌啶在dmf、dmso、n,n-二乙基乙酰胺、nmp或任何其他合适的非质子溶剂中裂解fmoc基团。
在切断fmoc保护基后,游离胺基与序列中的下一个氨基酸偶联,即在dmf、dmso、n,n-二乙基乙酰胺、nmp或任何其他合适的非质子溶剂中的fmoc-cys(xan)-opfp。
在反应过程中,对于c-末端含酸基的氨基酸,如fmoc-thr(tbu)-gly-oh、fmoc-pro-ala-oh、fmoc-asn(trt)-oh、fmoc-tyr(tbu)-oh、fmoc-glu(otbu)-oh,采用肽偶联试剂进行偶联,所述肽偶联试剂为dic、hbtu、tbtu、pybop、hatu、hctu的任意一种,或者任意一种与diea或n-甲基吗啉联用。对于c末端含有opfp酯的氨基酸,不使用偶联试剂。
fmoc氨基酸和偶联试剂总是过量使用。作为优选,各fmoc氨基酸片段和肽偶联试剂的添加量均为树脂取代物的2-4倍当量。
固相合成过程中通过kaisertest和chloraniltest监测偶合和去偶合反应。
步骤(2),所有氨基酸的连接完成后,使用裂解试剂将肽链从树脂上切割下来。
所述裂解试剂为三氟乙酸(tfa)、乙二硫醇(edt)、三异丙基硅烷(tis)和水以不同比例混合。作为优选,裂解试剂为三氟乙酸、乙二硫醇、三异丙基硅烷和水以体积比为92.5:2.5:2.5:2.5混合所得。利那洛肽树脂与裂解试剂的混合比为200g:1l。
裂解条件为0~40℃下反应2~5小时。作为优选,10~20℃下反应3~4小时。
裂解反应结束后,收集滤液,加入乙醚、二异丙基醚或甲基叔丁基醚,5-10℃下放置以沉淀产物,将沉淀产物过滤,用醚洗涤3-5次并干燥。
步骤(3),随机氧化法在同一溶液体系中依次完成利那洛肽中三对二硫键的形成。具体地,将线性利那洛肽溶于二甲亚砜水溶液中,再加入硫酸铵、氢氧化铵或碳酸铵,用氨水调节反应体系ph值在8.0±0.3,室温下保持反应10~12小时。
本发明研究表明,选择合适的氧化剂、适当稀释的氧化溶液和合适的ph值可以避免二硫桥的错配。氧化体系采用硫酸铵/2-10%dmso水溶液(ph=7-10)、碳酸铵/2-10%dmso水溶液(ph=7-10)或氢氧化铵/2-10%dmso水溶液(ph=7-10),作为优选,氧化体系采用硫酸铵/2%dmso水溶液。
优选地,所述混合液中线性利那洛肽浓度为2mg/ml,硫酸铵、氢氧化铵或碳酸铵的用量为线性利那洛肽的15倍当量。
反应结束后,采用反向高效液相色谱法分离纯化氧化产物。
所述纯化分两步进行,第一步:将利那洛肽粗品通过c18反相柱,用含体积比0.1%的乙酸水溶液洗涤,再以含体积比0.5%乙酸和0.05%三氟乙酸的水溶液为流动相a,乙腈为流动相b进行洗脱,收集目标峰馏分;第二步:将收集的收集目标峰馏分装入c18反相柱,用含质量百分比0.15%的醋酸铵水溶液洗涤,再以含体积比0.1%的乙酸水溶液为流动相a,乙腈为流动相b进行洗脱,收集目标产物。
本发明具备的有益效果:
(1)本发明采用c末端含有opfp酯的半胱氨酸用于固相多肽合成,可以避免dic、hbtu、tbtu、pybop、hatu、hctu等偶联试剂的使用。使用opfp酯的偶联效率也更高,有效地防止了固相多肽合成中的β-sheet。
(2)半胱氨酸是一种易外消旋的氨基酸,在半胱氨酸的位置上使用opfp酯,比如fmoc-cys(xan)-opfp可以避免这种情况,大大减少消旋体杂质生成。另外,由于作为半胱氨酸保护基(氧杂蒽基)的空间位阻,可以避免聚集。
(3)在ph值8.0±0.3的硫酸铵/2%dmso水溶液氧化体系下,线性利那洛肽的浓度为1~2mg/ml,采用随机氧化法,即在1-6、2-10、5-13三个位置一步形成三个二硫桥,不存在二硫桥错配现象。氧化反应后的粗水溶液直接进行纯化,得到纯度为99.5%且杂质<0.1%的利那洛肽。
(4)本发明提供的合成方法工艺操作简单,一步环合即可形成正确的二硫键对,二硫键的错配减少,提高了纯度和收率,大大提高了生产效率,降低制造成本。
附图说明
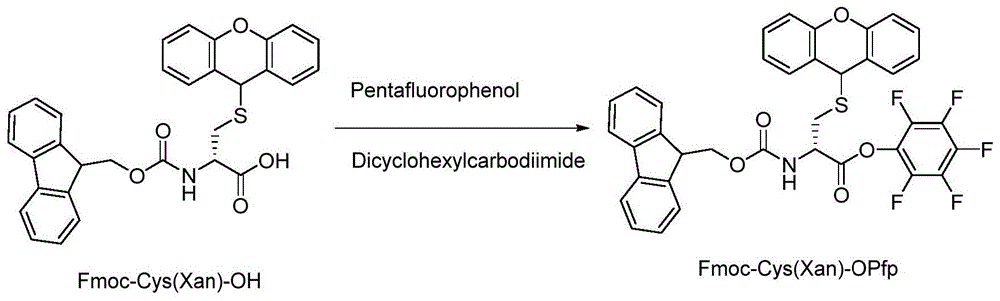
图1为实施例1中fmoc-cys(xan)-opfp的合成反应示意图。
图2为实施例1中fmoc-thr(tbu)-gly-oh的合成反应示意图。
图3为实施例1中利那洛肽王树脂的制备示意图。
图4为实施例1中线性利那洛肽的合成反应示意图。
图5为实施例1中随机氧化法合成带有三个二硫桥的利那洛肽的反应示意图。
具体实施方式
下面结合具体实施例对本发明做进一步说明,但本发明并不局限于此。
实施例中采用的原料、试剂均为市场化商品。
缩写说明:
hbtu:o-benzotriazole-n,n,n',n'-tetramethyl-uronium-hexafluorophosphate,中文名称:o-苯并三氮唑-四甲基脲六氟磷酸酯;
hobt:hydroxybenzotriazole,中文名称:1-羟基苯并三唑;
tbtu:o-(benzotriazol-1-yl)-n,n,n',n'-tetramethyluroniumtetrafluoroborate,中文名称:o-苯并三氮唑-n,n,n',n'-四甲基脲四氟硼酸酯;
dcc:n,n'-dicyclohexylcarbodiimide,中文名称:n,n-二环己基碳二亚胺;
dic:diisopropylcarbodiimide,中文名称:n,n'-二异丙基碳二亚胺;
pybop:benzotriazole-1-yl-oxytripyrrolidinophosphoniumhexafluorophosphate,中文名称:六氟磷酸苯并三唑-1-氧基三(吡咯烷并)磷;
dipea:n,n-diisopropylethylamine,中文名称:n,n-二异丙基乙胺;
dmf:n,n-dimethylformamide,n,n-二甲基甲酰胺;
dcm:dichloromethane,中文名称:二氯甲烷;
thf:tetrahydrofuran,中文名称:四氢呋喃;
νμρ:n-methylpyrrolidine,中文名称:n-甲基吡咯烷酮;
dmac:dimethylacetamide,中文名称:二甲基乙酰胺;
tfa:trifluoroaceticacid,中文名称:三氟乙酸;
edt:ethanedithiol,中文名称:乙二硫醇;
tis:triisopropylsilane,中文名称:三异丙基硅烷;
dmso:dimethylsulfoxide,中文名称:二甲亚砜;
mtbe:methyltert-butylether,中文名称:甲基叔丁基醚;
meoh:methanol,中文名称:甲醇;
ipa:isopropylalcohol,中文名称:异丙醇;
ctc:chlorotritylchloride,中文名称:2-氯三苯甲基氯;
trt:trityl,中文名称:三苯甲基;
xan:xanthenyl,中文名称:氧杂蒽基;
fmoc:9-fluorenylmethoxycarbonyl,中文名称:9-芴基甲氧羰基;
boc:tert-butoxycarbonyl,中文名称:叔丁氧羰基;
hctu:o-(6-chloro-1-hydrocibenzotriazol-1-yl)-1,1,3,3-tetramethyluroniumhexafluorophosphat,中文名称:o-(6-氯-1-氢苯并三唑-1-基)-1,1,3,3-四甲基铀六氟磷酸盐。
实施例1
一、fmoc-cys(xan)-opfp的合成
称取fmoc-cys(xan)-oh(100g,190mmol),加入含有700ml四氢呋喃的三口烧瓶中。将混合物在25±2℃下搅拌5分钟。然后把溶于100.0mlthf的42.18g(229.2mmol)五氟苯酚加入到里面,并搅拌5-10分钟。在另外一个圆底烧瓶中,取47.3g二环己基碳二亚胺(229.2mmol)溶于200ml四氢呋喃中。将此溶液于25±2℃下慢慢滴加(30-45min)到上述fmoc-cys(xan)-oh四氢呋喃溶液中。加入完毕后,在同一温度下搅拌3-4h,用薄层色谱法(tlc)监测原料的完全反应。反应进程如图1所示。
反应液经过滤去除尿素,溶剂四氢呋喃经旋转蒸发仪完全除去,得到胶状固体产物。将异丙醇(2000毫升)加入上述固体产物中,搅拌90分钟,然后过滤。经过滤和真空干燥,得到白色固体120.0g,收率91.1%。
二、fmoc-thr(tbu)-gly-oh的合成
a)fmoc-thr(tbu)-osu的合成
称取fmoc-thr(tbu)-oh(50.0g,125.8mmol)并加入含有400ml四氢呋喃的三口烧瓶中。将混合物在25±2℃下搅拌5分钟,然后加入17.37g(150.9mmol)n-羟基丁二酰亚胺,并搅拌5-10分钟。
另取一个圆底烧瓶,把31.14g二环己基碳二亚胺(150.9mmol)溶于100ml四氢呋喃。把该溶液于10±2℃下慢慢滴入到上述fmoc-thr(tbu)-oh的thf溶液中,时间为30-45分钟。滴加完成后,将体系升温至室温,再反应3小时,用薄层色谱法(tlc)对原料进行监测,直至完全反应。
反应液经过滤去除尿素,用旋转蒸发仪将四氢呋喃完全去除,得到胶状固体产物。向上述固体产物中加入400毫升异丙醚,搅拌30分钟,过滤。经过滤、真空干燥后,得到白色固体54.0g,收率86.7%。
b)fmoc-thr(tbu)-gly-oh
称取l-甘氨酸(9.1g,121.3mmol)加入含有300ml四氢呋喃和水(1:1)的三口瓶中。将混合物在25±2℃下搅拌5分钟,然后加入12.86g(121.3mmol)na2co3,并搅拌5-10分钟。
另取一个圆底烧瓶,将50.0gfmoc-thr(tbu)-osu(101.1mmol)溶于200ml四氢呋喃。然后把此溶液在25±2℃下慢慢滴加至上述溶液中,持续30-45分钟。滴加完成后,将体系升温至室温,再反应3h,用薄层色谱法(tlc)对原料进行监测,直至反应完全。
反应结束后,用旋转蒸发仪将四氢呋喃完全去除,并在水层中加入400ml乙酸乙酯,用10%柠檬酸溶液将ph值调节到3.5±0.5。分离水层,用乙酸乙酯进一步提取,减压下去除溶剂,得到白色固体。然后通过使用乙酸乙酯和丙酮打浆纯化,得到fmoc-thr(tbu)-gly-oh的白色干燥固体(40.0g,产率87%)。
上述反应进程如图2所示。
三、采用逐步固相合成法制备树脂肽。
1、取代度为0.8mmol/g的fmoc-tyr(tbu)-wangresin的合成
称取50.0g取代度为1.1mmol/g的wang树脂,加入固相多肽合成仪中。随后,用dmf洗涤树脂两次,并在dmf中膨胀30分钟。然后将50.54gfmoc-tyr(tbu)-oh和13.9gdic溶解在dmf中,并在冰水浴下搅拌5-10分钟,然后添加到上述装有树脂的合成器中。5min后,加入0.1g4-二甲氨基吡啶dmap,搅拌2小时,分别用dmf和dcm洗涤树脂3次,用醋酸酐/吡啶/dmf(15ml:15ml:500ml)封闭30min,再滤干。用甲醇把树脂收缩,甲醇再被干燥后得到取代度为0.8mmol/g的fmoc-tyr(tbu)-wangresin。
2、线性利那洛肽王树脂的制备
称取65.0g取代度为0.8mmol/g的fmoc-tyr(tbu)-wang树脂,加入反应器中。随后,使用dmf对fmoc-tyr(tbu)-wang树脂进行两次洗涤,并在dmf中膨胀30分钟。使用20%哌啶dmf溶液去除fmoc保护,然后使用dmf对树脂进行5次洗涤。采用茚三酮法对树脂进行测试,通过树脂颜色的出现来表明对fmoc的去除效果。
将62.83gfmoc-cys(xan)-opfp(120.0mmol)溶于dmf中,装入固相反应器,在室温下反应3-4小时,用茚三酮试验确定反应终点。
上述fmoc去保护步骤和相应的氨基酸偶联步骤基于利那洛肽的主链序列重复进行。fmoc-thr(tbu)-gly-oh、fmoc-cys(xan)-opfp、fmoc-pro-ala-oh、fmoc-asn(trt)-oh、fmoc-cys(xan)-opfp、fmoc-cys(xan)-opfp、fmoc-tyr(tbu)-oh、fmoc-glu(otbu)-oh、fmoc-cys(xan)-opfp、boc-cys(xan)-opfp相继按顺序偶合。
在反应过程中,对于c-末端含酸基的氨基酸,如fmoc-thr(tbu)-gly-oh、fmoc-pro-ala-oh、fmoc-asn(trt)-oh、fmoc-tyr(tbu)-oh、fmoc-glu(otbu)-oh,均采用hobt/dic体系,反应溶剂为dmf。然而对于在c-末端位置含有opfp酯基的氨基酸,如fmoc-cys(xan)-opfp,不需要偶合试剂和hobt系统。反应进程如图3所示。
所有氨基酸偶联后,用甲醇对树脂进行收缩,然后将树脂干燥后,得到200g的利那洛肽王树脂粗品。
四、线性利那洛肽的合成
以tfa/ethanedithiol/tis/water的体积比为92.5:2.5:2.5:2.5的比例配制0.9l裂解试剂,并装入三口瓶中。将裂解试剂冷却至15±2℃,加入180.0g利那洛肽王树脂,在15±2℃下反应3.5h。反应结束后,过滤树脂,收集滤液。树脂用少量tfa洗涤。将滤液混合,加入甲苯(3000ml)和异丙醚(5000ml),搅拌60分钟,过滤,然后用异丙醚(2×1200ml)进行两次打浆洗涤。经过滤和真空干燥后,得到纯度为70%的白色固体120.0g。上述反应进程如图4所示。
五、利那洛肽粗品的合成
用随机氧化法合成带有三个二硫桥的利那洛肽
线性利那洛肽(100g)溶于2%二甲基亚砜水溶液中(50l)。在化合物溶解后,加入硫酸铵(15当量,硫酸铵与线性利那洛肽的摩尔比为15:1),并用氨水将反应物的ph值调整为8.0±0.3,在室温下保持反应10-12小时,该反应使用uplc过程分析仪来监测。产物纯度分析结果表明,在上述氧化条件下制备的产物纯度达到75%。反应结束后,用反相高效液相色谱法分离纯化氧化产物。
上述反应进程如图5所示。
六、利那洛肽的纯化
第一阶段
将从实施例5获得的被氧化的粗肽溶液通过c18反相柱,用0.1%乙酸溶液洗涤60分钟。然后用以0.5%乙酸和0.05%tfa水溶液作为流动相a,以乙腈作为流动相b的体系来进行洗脱。
第二阶段
将从第一阶段得到的目标峰馏分装入反相c18柱,用0.15%醋酸铵溶液洗涤60分钟。然后以0.1%乙酸溶液为流动相a,以乙腈为流动相b的体系来洗脱。
纯度大于99%且最大单杂小于0.1%的组分被汇集并冻干以获得纯的利那洛肽。
产量:35.0g;高效液相色谱纯度:>99.5%。
对比例1
本对比例在实施例1的第三部分固相合成法制备树脂结合肽的步骤2中,氨基酸偶联时使用fmoc-cys(xan)-oh替代fmoc-cys(xan)-opfp,具体地,将62.83gfmoc-cys(xan)-oh(120.0mmol)、hobt(16.2g,120.0mmol)和dic(18.8ml,120.0mmol)溶解于dmf中,加载到固相反应合成器中,室温下反应3-4小时,通过茚三酮试验确定反应终点。以上fmoc去保护步骤和相应的氨基酸偶联步骤根据利那洛肽的肽骨架序列重复进行。fmocthr(tbu)-gly-oh、fmoc-cys(xan)-oh、fmoc-pro-ala-oh、fmoc-asn(trt)-oh、fmoc-cys(xan)-oh、fmoc-cys(xan)-oh、fmoc-tyr(tbu)-oh、fmoc-glu(otbu)-oh、fmoc-cys(xan)-oh、boc-cys(xan)-oh相继按顺序偶合。
其他操作步骤同实施例1。
利那洛肽王树脂裂解后,洗涤、过滤、干燥后得到纯度为60%的白色固体117.0g。
产物分析后发现,一个消旋体杂质的结构与利那洛肽相似,即d-cys2-利那洛肽,跟主峰挨得很近,其相对保留时间约为1.03,含量为2.3%。类似地,另一个消旋体杂质的结构也与利那洛肽相似,即d-cys5-利那洛肽,跟主峰也挨得很近,其相对保留时间约为1.05,含量为2.0%。d-cys2-利那洛肽和d-cys5-利那洛肽的相对保留时间都约为1.0,这会使得分离和纯化粗肽比较困难。
而实施例1中制备的线性利那洛肽,d-cys2-利那洛肽和d-cys5-利那洛肽的量从各自的2.3%和2.0%减少到可以忽略的量。可见,在合成过程中采用fmoc-cys(xan)-opfp,线性利那洛肽中的消旋体杂质大大减少。
对比例2
本对比例在实施例1的第五部分利那洛肽粗品的合成过程中,用水替换二甲亚砜水溶液,具体为:线性利那洛肽(100g)溶于水(50l)。在化合物溶解后,加入硫酸铵(15eq),并用氨水将反应物的ph值调整为8.0±0.3,室温下保持反应24-48小时,反应采用uplc过程分析仪监控。
结果显示,即使保持反应24-48小时,仍有50%的起始原料未反应,且含有多种杂质。
对比例3
本对比例在实施例1的第五部分利那洛肽粗品的合成过程中,用h2o2作为氧化剂,具体为,将线性利那洛肽(100g)溶解于水(50l)中,并用氨水将反应物的ph值调节为8.0±0.3。30%h2o2溶液(100.0ml30%h2o2溶于1000ml水中)在25±2℃条件下逐渐滴入到上述溶液中,滴加时间为30-45分钟,并在室温下保持反应10-12小时,反应通过uplc过程分析仪来监控。
在氧化反应中使用h2o2溶液,反应在3-4小时内完成,但产生降解杂质和多聚体杂质。因此,本发明最终把反应条件定为(2%二甲基亚砜水溶液和硫酸铵)的组合。
对比例4
本对比例在实施例1的第五部分利那洛肽粗品的合成过程中,改变反应体系ph值,具体为:线性利那洛肽(100g)溶于2%二甲基亚砜水溶液中(50l)。在化合物溶解后,加入硫酸铵(15eq),并用氨水将反应物的ph值调整为4.0±0.5,室温下保持反应24-48小时,反应采用uplc过程分析仪监控。类似地,并列设置了ph值为5.0±0.5、6.0±0.5和7.0±0.5的反应条件。
结果显示,尽管反应时间为24~48小时,没有任何产物在ph值为3.5~7.5的条件下生成。
对比例5
本对比例在实施例1的第五部分利那洛肽粗品的合成过程中,改变反应体系ph值,具体为:线性利那洛肽(100g)溶于2%二甲基亚砜水溶液中(50l)。在化合物溶解后,加入硫酸铵(15eq),将反应物的ph值调整为9.5±0.5,并在室温下保持反应10-12小时,反应用uplc过程分析仪来监控。类似地,并列设置了ph值为10.5±0.5的反应条件。
结果显示,当反应在高于9.0的ph下进行时,由于错误的折叠,观察到多聚体的形成。当反应在ph值为9.5±0.5时进行,产物纯度只有30%,并伴有多聚体杂质。当反应物ph值增加到10.5±0.5时,产物纯度只有15%,并出现了更多的硫桥错配和降解杂质。
通过质谱分析,确定了多聚体杂质、降解杂质和硫桥错配杂质的形成。
与对比例5相比,在实施例1的氧化条件下制得的利那洛肽粗品的纯度从30.0%增加到75%。
对比例6
本对比例在实施例1的第五部分利那洛肽粗品的合成过程中,改变氧化剂水溶液的用量,具体地,线性利那洛肽(100g)溶于2%二甲基亚砜水溶液(20l)(5mg/ml)中。在化合物溶解后,加入硫酸铵(15eq),并用氨水将反应物的ph值调整为8.0±0.3,室温下反应10-12小时,反应用uplc过程分析仪来监控。类似地,并列设置了不同的浓度条件,如1.0mg/ml、20mg/ml和100mg/ml。
结果显示,当以100mg/ml浓度(每100g线性肽溶于1.0l的2%二甲基亚砜水溶液)和20mg/ml浓度(每100g线性肽溶于5.0l的2%二甲基亚砜水溶液)进行氧化反应时,没有生成产物,但产生降解杂质。
当以5mg/ml浓度(每100g线性肽溶于20.0l的2%二甲基亚砜水溶液)进行氧化反应时,只有20%的收率,并有降解杂质。
当采用实施例1的浓度条件2mg/ml时,获得75%的高纯度,与1mg/ml浓度有相同的良好结果。为了便于工艺的实施,本发明最终确定了2mg/ml的浓度条件。
序列表
<110>台州吉诺生物科技有限公司
<120>一种利那洛肽的合成方法
<160>1
<170>siposequencelisting1.0
<210>1
<211>14
<212>prt
<213>人工序列(artificialsequence)
<400>1
cyscysglutyrcyscysasnproalacysthrglycystyr
1510
- 还没有人留言评论。精彩留言会获得点赞!