一种奎宁胺的制备方法与流程
1.本发明属于药物化学领域,具体涉及一种奎宁胺的制备方法。
背景技术:
2.顺式2-取代-3-奎宁胺化合物是一类重要的医药中间体,目前该类化合物的制备方法鲜有报道,尤其是2位为单取代基团的顺式3-奎宁胺的制备,尚缺少高效的合成方法。
3.已经被批准上市的药物马罗匹坦(英文名:maropitant),其化学结构式中即含有此类结构片段。因此类结构中含有两个手性中心,因此有1个对映异构体及2个非对映异构体,共3个光学异构体杂质,这些杂质会影响药品的质量等。
4.现有的拆分等方法工艺过程繁琐、成本高,不利于工业化生产。
技术实现要素:
5.本发明提供一种制备方法。本发明所述的制备方法,包括使用过渡金属催化剂,与磺酰胺化合物反应,一步反应构建两个手性中心,具有较高的选择性,简洁、高效,质量可控。
6.根据本发明的一个方面,本发明提供一种化合物iii的制备方法,其包括,任选惰性气体保护下,在有机溶剂中,在使用助化试剂,供氢试剂,任选碱试剂和过渡金属催化剂条件下,化合物i和化合物ii经过反应,反应完毕,经过后处理,制备得到化合物iii,
[0007][0008]
其中,r1为c1-c6(1个碳-6个碳)的烷基,苯基,或任意取代的苯基;r2为ch2r3或ch(r3)2,r3选自h,任意取代的或未取代的以下基团:烷基,环烷基,杂环烷基,芳基或杂芳基;*表示手性碳原子。
[0009]
在一些实施方式中,r3选自任意取代的或未取代的以下基团:c1-c6(1个碳-6个碳)的烷基,c3-c10(3个碳-10个碳)的环烷基,c2-c10(2个碳-10个碳)的杂环烷基,c6-c10(6个碳-10个碳)的芳基,或c4-c10(4个碳-10个碳)的杂芳基。
[0010]
在一些实施方式中,r3选自任意取代的或未取代的以下基团:甲基,乙基,丙基,异丙基,叔丁基,苯基,对甲氧基苯基,吡啶基,,吡咯基,2-呋喃基,2-噻吩基,3-甲基苯基,2,5-二氟苯基,3,4-(亚甲二氧基)苯基,3,5-二甲氧基苯基,3,4-二甲基苯基,1-萘基,2-萘基,4-联苯基,3-羟基-4-甲氧基苯基,4-氟苯基,或4-甲氧基苯基。
[0011]
在一些实施方式中,r1为苯基或任意取代的苯基,如对甲基苯基。在一些实施方式中,r1为甲基。
[0012]
所述助化试剂为钛酸四异丙酯,钛酸四乙脂,三异丙基硼酸酯,对甲苯磺酸或其水合物,樟脑磺酸,或乙酸,或其组合。在一些实施方式中,所述助化试剂包括或为钛酸四异丙酯,有利于反应进行和目标产物的获得。在一些实施方式中,所述助化试剂包括或为对甲苯磺酸或其水合物,有利于反应进行和目标产物的获得。
[0013]
所述助化试剂与化合物ii的投料摩尔比为3:1-1:1。在一些实施方式中,所述助化试剂与化合物ii的投料摩尔比为1.2:1-2.0:1,更有利于反应进行。
[0014]
所述化合物i和化合物ii的投料摩尔比可以为2.5:1-1:1。在一些实施方式中,所述化合物i与化合物ii的投料摩尔比为1.1:1-1.4:1,更有利于反应进行。
[0015]
所述供氢试剂可以是任意适宜的能够提供氢元素的试剂。在一些实施方式中,所述供氢试剂包括甲酸、甲酸铵、甲酸钠或甲酸钾中的任意一种。在一些实施例中,所述的供氢试剂为甲酸。
[0016]
所述供氢试剂与化合物ii的投料摩尔比为20:1-1:1。在一些实施方式中,所述供氢试剂与化合物ii的投料摩尔比为15:1-1:1。在一些实施方式中,所述供氢试剂与化合物ii的投料摩尔比为10:1-1:1。在一些实施方式中,所述供氢试剂与化合物ii的投料摩尔比为6:1-1:1,更有利于操作控制和目标产物的获得。
[0017]
所述制备方法中,可以加入碱试剂,也可以不加入碱试剂。所述碱试剂可为任意适宜的有机碱。在一些实施方式中,所述碱试剂包括三乙胺,二乙胺,n,n-二异丙基乙胺,或1,4-二氮杂二环[2.2.2]辛烷中的任意一种。在一些实施方式中,所述碱试剂为三乙胺,二乙胺,n,n-二异丙基乙胺,1,4-二氮杂二环[2.2.2]辛烷或其组合。在一些实施方式中,所述碱试剂为三乙胺,或二乙胺,或其组合,更有利于反应进行和目标产物的获得。
[0018]
所述碱试剂与化合物ii的投料摩尔比可以为12:1-1:1。在一些实施方式中,所述碱试剂与化合物ii的投料摩尔比为10:1-1:1。在一些实施方式中,所述碱试剂与化合物ii的投料摩尔比为5:1-1:1。在一些实施方式中,所述碱试剂与化合物ii的投料摩尔比为3:1-1:1,更有利于反应进行和控制。
[0019]
本发明提供的制备方法中,供氢试剂与碱试剂可以独立的分别投料,也可以按投料量混合后再加入到反应体系中。
[0020]
所述过渡金属催化剂如式cat所示:
[0021][0022]
其中,r4为c1-c6(1个碳-6个碳)的烷基,任意取代的c1-c6(1个碳-6个碳)的烷基,苯基或任意取代的苯基;r5,r6独立地为苯基或任意取代的苯基,或r5,r6一起为亚丁基;x不存在,或x为氯(cl),氢(h),三氟甲磺酸基(otf)或四氟硼酸基(bf4);m选自钌,铑或铱;m为钌时,ar为苯基或任意取代的苯基;m为铑或铱时,ar为环戊二烯基或任意取代的环戊二烯基;*表示手性碳原子。
[0023]
一些实施方式中,m为铱,更有利于目标产物的获得。
[0024]
一些实施例中,r4为2,4,6-三异丙基苯基,2,4,6-三甲基苯基,对甲基苯基,对三氟甲基苯基,3,5-二三氟甲基苯基,五氟苯基,甲基或三氟甲基。
[0025]
在一些实施例中,r5,r6均为苯基。
[0026]
在一些实施方式中,x为氯,或氢。
[0027]
在一些实施例中,所述的ar为苯基,4-甲基异丙基苯基,环戊二烯基或任意取代的环戊二烯基。一些实施方式中,ar为环戊二烯基,或任意取代的环戊二烯基,更有利于目标产物的获得。
[0028]
在一些实施方式中,r4为2,4,6-三异丙基苯基,2,4,6-三甲基苯基,对甲基苯基,对三氟甲基苯基,3,5-二三氟甲基苯基,五氟苯基,甲基或三氟甲基;r5,r6均为苯基;x为氯或氢。
[0029]
所述取代或任意取代可为被烷基,卤代烷基,或卤素任意取代。所述卤素可为氟、氯、溴或碘。
[0030]
所述杂环的杂原子可以为氮、氧或硫。
[0031]
在一些实施方式中,当过渡金属催化剂为(r,r)构型时,可获得(s,s)构型的化合物iii;当过渡金属催化剂为(s,s)构型时,可获得(r,r)构型的化合物iii。
[0032]
在一些实施方式中,一种化合物iii的制备方法,其包括,任选惰性气体保护下,在有机溶剂中,在使用助化试剂,供氢试剂,任选碱试剂和过渡金属催化剂条件下,化合物i和如下式所示的化合物ii经过反应,反应完毕,经过后处理,制备得到如下式所示的化合物iii,反应式如下式所示:
[0033]
或者
[0034][0035]
其中,r3如前述所定义;*表示手性碳原子,化合物iii中的两个手性碳可以为(r,r)构型,或(s,s)构型;过渡金属催化剂如下式cat所示:
[0036][0037]
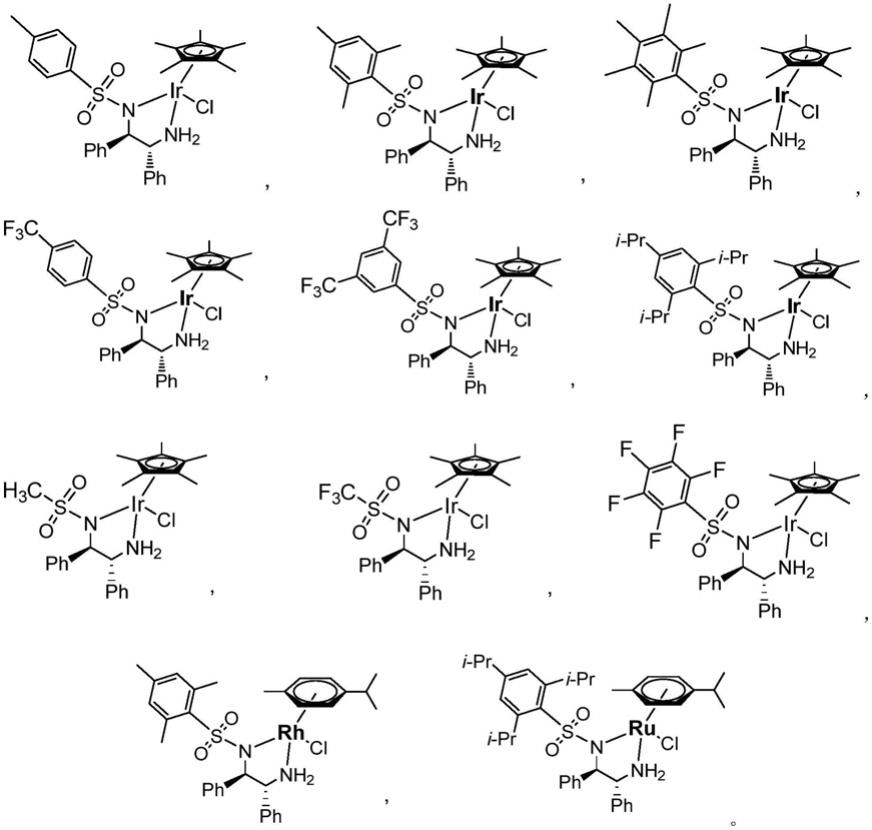
在一些实施方式中,所述过渡金属催化剂可以是下列结构的过渡金属催化剂中的至少之一:
[0038][0039]
所述过渡金属催化剂与化合物ii的投料摩尔比可为0.001:1-0.1:1。在一些实施方式中,所述过渡金属催化剂与化合物ii的投料摩尔比可为0.001:1-0.01:1。在一些实施方式中,所述过渡金属催化剂与化合物ii的投料摩尔比可为0.01:1-0.1:1。在一些实施方式中,所述过渡金属催化剂与化合物ii的投料摩尔比可为0.005:1-0.05:1。在一些实施方式中,所述过渡金属催化剂与化合物ii的投料摩尔比可为0.05:1-0.1:1。
[0040]
所述有机溶剂可为四氢呋喃,二氯甲烷,甲苯,n,n-二甲基甲酰胺或其组合。在一些实施方式中,所述有机溶剂为四氢呋喃或甲苯,更有利于反应进行和处理。
[0041]
每一克化合物ii,所述有机溶剂的用量为1ml-30ml。在一些实施方式中,每一克化合物ii,所述有机溶剂的用量为3ml-15ml,有利于控制和目标产物的获得。
[0042]
所述化合物iii的制备方法中,反应的反应温度为0℃-100℃。在一些实施方式中,所述反应的反应温度为0℃-50℃。在一些实施方式中,所述反应的反应温度为0-30℃。在一些实施方式中,所述反应的反应温度为15℃-35℃。在一些实施方式中,所述反应的反应温度为20℃-30℃。在一些实施方式中,所述反应的反应温度为25℃-35℃。在一些实施方式中,所述反应的反应温度为5℃,10℃,15℃,20℃,25℃,30℃,35℃或40℃。
[0043]
所述化合物iii的制备方法中,可以加入碱试剂,也可以不加入碱试剂,优选加入碱试剂。
[0044]
所述化合物iii的制备方法中,各反应物料或试剂,可以同时加入反应容器中,也可以分别先后加入反应容器中;如可以先在反应容器中加入有机溶剂,化合物i,化合物ii和助化试剂,反应一段时间后,再加入供氢试剂,任选碱试剂和过渡金属催化剂,反应至反应完毕;也可以将有机溶剂,化合物i,化合物ii,助化试剂供氢试剂,任选碱试剂和过渡金属催化剂都加入反应容器中,反应至反应完毕。
[0045]
根据本发明的一些实施例,在一些实施方式中,所述的一种化合物iii的制备方法包括,任选惰性气体保护下,有机溶剂中,化合物i,化合物ii和助化试剂混合,反应第一段时间;然后加入供氢试剂,碱试剂和过渡金属催化剂,或加入供氢试剂和碱试剂混合后的混合物和过渡金属催化剂,或加入供氢试剂和过渡金属催化剂,反应第二段时间;经过后处理,制备得到化合物iii。
[0046]
所述第一段时间可为0h-20h(0h表示化合物i,化合物ii,助化试剂,供氢试剂,任选碱试剂和过渡金属催化剂一起加入反应器中)。一些实施方式中,所述第一段时间可为0h-18h。一些实施方式中,所述第一段时间可为0h-14h。一些实施方式中,所述第一段时间可为1h-18h。一些实施方式中,所述第一段时间可为10h-20h。一些实施方式中,所述第一段时间可为10h-14h。一些实施方式中,所述第一段时间可为1h-8h。一些实施方式中,所述第一段时间可为1h-6h。一些实施方式中,所述第一段时间可为4h-8h。一些实施方式中,所述第一段时间可为8h,10h,12h,14h,15h,16h,或18h。
[0047]
所述第二段时间可为8h-30h。一些实施方式中,所述第二段时间可为12h-30h。一些实施方式中,所述第二段时间可为12h-24h。一些实施方式中,所述第二段时间可为12h-20h。一些实施方式中,所述第二段时间可为15h-24h。一些实施方式中,所述第二段时间可为15h-20h。一些实施方式中,所述第二段时间可为15h,18h,20h,22h,或24h。
[0048]
在制备化合物iii的过程中,任选使用惰性气体如氮气,氦气,氩气等保护。在一些实施方式中,在氮气保护下进行反应。
[0049]
在一些实施方式中,所述一种化合物iii的制备方法包括,任选惰性气体保护下,有机溶剂中,化合物i,化合物ii和助化试剂混合后,在0℃-50℃反应1小时-20小时;然后再加入供氢试剂,任选碱试剂和过渡金属催化剂,再在0℃-50℃反应8小时-30小时;经过后处理,制备得到化合物iii。
[0050]
在一些实施方式中,所述一种化合物iii的制备方法包括,任选惰性气体保护下,有机溶剂中,化合物i,化合物ii和助化试剂混合后,在0℃-50℃反应8小时-16小时;然后加入供氢试剂,碱试剂和过渡金属催化剂,或加入供氢试剂和碱试剂混合后的混合物和过渡
金属催化剂,或加入供氢试剂和过渡金属催化剂,在0℃-50℃继续反应12小时-24小时;经过后处理,制备得到化合物iii。
[0051]
根据本发明的一些实施例,在一些实施方式中,所述的一种化合物iii的制备方法包括,任选惰性气体保护下,有机溶剂中,化合物i,化合物ii,助化试剂,供氢试剂,任选碱试剂和过渡金属催化剂混合后,在0℃-50℃反应8小时-36小时;经过后处理,制备得到化合物iii。在一些实施方式中,一种化合物iii的制备方法包括,任选惰性气体保护下,有机溶剂中,化合物i,化合物ii,助化试剂,供氢试剂,任选碱试剂和过渡金属催化剂混合后,在0℃-50℃反应12小时-24小时;经过后处理,制备得到化合物iii。
[0052]
所述后处理包括:经过萃取和/或洗涤等,得粗品;粗品用合适的溶剂结晶,得到化合物iii。在一些实施方式中,所述后处理包括:任选将反应液除去溶剂,用酸调节ph至2-4,用有机溶剂洗涤,分出水相并降温至10℃-30℃,调节ph至10或以上;然后任选过滤,滤饼用有机溶剂洗涤,滤液加入有机溶剂萃取;所得有机相合并浓缩至干,得粗品;粗品用合适的溶剂结晶,得到化合物iii。在一些实施方式中,所述后处理包括:将反应液减压蒸馏除,用盐酸调节ph至2-4,用有机溶剂洗涤,分出水相并降温至10℃-20℃,调节ph至10以上;然后经过硅藻土过滤,滤饼用有机溶剂洗涤,滤液中加入有机溶剂萃取;合并有机相,所得有机相浓缩至干,得粗品;粗品用溶剂溶解,然后降温至-5℃-5℃,析出固体,过滤,干燥,得到化合物iii。
[0053]
所述萃取或洗涤所用有机溶剂可为甲苯、二氯甲烷、乙酸乙酯、乙酸异丙酯或其组合。所述的结晶所用溶剂可以为异丙醇,乙醇,甲醇,甲苯中的至少一种,或为异丙醇、乙醇、甲醇、甲苯中至少一种与正己烷和/或环己烷的组合。
[0054]
在一些实施方式中,所述后处理包括:将反应液减压蒸馏,用盐酸调节ph至2-4,用甲苯洗涤,分出水相并降温至10℃-20℃,调节ph至10以上;然后经过硅藻土过滤,滤饼用甲苯洗涤,滤液中加入甲苯萃取;合并有机相,所得有机相浓缩至干,得粗品;粗品用甲苯和正己烷加热溶解,然后降温至-5℃-5℃,析出固体,过滤,干燥,得到化合物iii。
[0055]
前述的方法,制备得到的化合物iii,其ee值达到95%或以上。在一些实施方式中,前述的方法,制备得到的化合物iii,其ee值达到98%或以上。在一些实施方式中,前述的方法,制备得到的化合物iii,其ee值达到99%或以上。
[0056]
前述的化合物iii经过反应,可以制备得到化合物iv,由化合物iv,可以制备获得其它需要的中间体化合物等,其中,r1,r2如前述所定义;*表示手性碳原子。
[0057][0058]
在一些实施方式中,前述的制备方法中,r3为任意取代的或未取代的以下基团:苯基,2-呋喃基,吡啶基,吡咯基,噻吩基,2-噻吩基,1-萘基,或2-萘基;r1为任意取代的或未取代的以下基团:甲基,乙基,正丙基,异丙基,正丁基,叔丁基,苯基,或苄基。
[0059]
在一些实施方式中,前述的制备方法中,r3为任意取代的或未取代的以下基团:苯基,对甲基苯基,对甲氧基苯基,2-呋喃基,或2-噻吩基;r1为甲基,苯基,或对甲基苯基。
[0060]
在一些实施方式中,前述的制备方法中,r1为甲基,苯基,对甲基苯基或对甲氧基苯基;r3为苯基,对甲基苯基,对甲氧基苯基,2-呋喃基,或2-噻吩基。
[0061]
本发明的提供的制备方法,能够一步反应高收率地引入两个手性中心,具有较高的选择性,使工艺简化和环境友好,同时产物的纯度高,收率高,有利于成本控制和工业化生产。
具体实施方式
[0062]
本发明所使用的试剂均可以从市场上购得或者可以通过本发明所描述的方法制备而得。
[0063]
本发明可以采用tlc(薄层色谱法)或hplc(高效液相色谱法)监测原料的反应程度,如采用hplc监测,当原料的峰面积小于4.0%或2.0%或1.0%或0.5%时,认为反应完毕。
[0064]
本发明中,thf表示四氢呋喃,naoh表示氢氧化钠,ph表示苯基,i-pr表示异丙基,me表示甲基;h表示小时,min表示分钟,ml或ml表示毫升,g表示克,mol表示摩尔,mpa表示兆帕;ee表示对映体过量(enantiomeric excess)。
[0065]
本发明中,如“化合物a”和“式a所示的化合物”的表述,表示的是同一个化合物。
[0066]
本发明中,涉及到干燥时,指干燥至恒重。“任选”指可以包括也可以不包括。
[0067]
在本说明书的描述中,参考术语“一个实施例”、“一些实施方式”、“示例”、“具体示例”、或“一些示例”等的描述意指结合该实施例或示例描述的具体特征、结构、材料或者特点包含于本发明的至少一个实施例或示例中。在本说明书中,对上述术语的示意性表述不必须针对的是相同的实施例或示例。而且,描述的具体特征、结构、材料或者特点可以在任一个或多个实施例或示例中以合适的方式结合。此外,在不相互矛盾的情况下,本领域的技术人员可以将本说明书中描述的不同实施例或示例以及不同实施例或示例的特征进行结合和组合。
[0068]
为了使本领域的技术人员更好地理解本发明的技术方案,下面进一步披露一些非限制实施例对本发明作进一步的详细说明。
[0069]
对比例1:制备化合物iii-0
[0070][0071]
向反应瓶中依次加入化合物ii-01(10g)、苄胺(6.0g)和30ml四氢呋喃,氮气保护下,加入钛酸四异丙酯(17.2g),25℃-30℃反应12-18h,向体系中加入1.1g cat01,加入三乙胺(12.7g)和甲酸(14.7g),25℃-30℃继续反应15h-20h,至原料化合物ii-01《2.0%,停止反应;40℃-50℃减压除去溶剂,加入2mol/l的盐酸水溶液调节ph至2-4,搅拌约30-60min,加入60ml甲苯洗涤1次,保留水相;水相降温至10℃-20℃,用50%naoh水溶液(质量分数)调节ph至12-13,经硅藻土过滤,滤饼用25ml甲苯洗涤1次,所得滤液分别用50ml甲苯萃取2次(2*50ml),合并有机相,经40-50℃碱压浓缩至干得油状粗品;粗品经异丙醇/正己
烷(40ml,7:1,体积比)重结晶得白色固体化合物iii-0:13.5g,收率95%,hplc纯度:98%,ee:81%。
[0072]
使用对甲氧基苯胺或二苯基甲胺替代苄胺,其它条件相同,发现反应未进行,无产物生成。
[0073]
对比例2:制备化合物iii-0
[0074][0075]
向反应瓶中依次加入化合物ii-01(10g)、苄胺(6.0g)和30ml四氢呋喃,氮气保护下,加入钛酸四异丙酯(17.2g),25℃-30℃反应12-18h,向体系中加入0.78g cat02,加入三乙胺(12.5g)和甲酸(15.1g),25℃-30℃继续反应15h-20h,至原料化合物ii-01《2.0%,停止反应;40℃-50℃减压除去溶剂,加入2mol/l的盐酸水溶液调节ph至2-4,搅拌约30-60min,加入60ml甲苯洗涤1次,保留水相;水相降温至10℃-20℃,用50%naoh水溶液(质量分数)调节ph至12-13,经硅藻土过滤,滤饼用25ml甲苯洗涤1次,所得滤液分别用50ml甲苯萃取2次(2*50ml),合并有机相,经40-50℃减压浓缩至干得油状粗品;粗品经异丙醇/正己烷(40ml,7:1,体积比)重结晶得白色固体化合物iii-0:12.7g,收率89%,hplc纯度:98%,ee:46%。
[0076]
对比例3:制备化合物iii-0
[0077][0078]
向反应瓶中依次加入化合物ii-01(10g)、苄胺(6.0g)和30ml四氢呋喃,氮气保护下,加入钛酸四异丙酯(17.2g),25℃-30℃反应12-18h,向体系中加入0.68g cat03,加入三乙胺(12.3g)和甲酸(14.9g),25℃-30℃继续反应15h-20h,至原料化合物ii-01《2.0%,停止反应;40℃-50℃减压除去溶剂,加入2mol/l的盐酸水溶液调节ph至2-4,搅拌约30-60min,加入60ml甲苯洗涤1次,保留水相;水相降温至10℃-20℃,用50%naoh水溶液(质量分数)调节ph至12-13,经硅藻土过滤,滤饼用25ml甲苯洗涤1次,所得滤液分别用50ml甲苯萃取2次(2*50ml),合并有机相,经40-50℃减压浓缩至干得油状粗品;粗品经异丙醇/正己烷(32ml,7:1,体积比)重结晶得白色固体化合物iii-0:9.5g,收率67%,hplc纯度:97%,ee:53%。
[0079]
实施例1:制备化合物iii-01
[0080][0081]
向反应瓶中依次加入化合物ii-01(10g)、对甲基苯磺酰胺(8.0g)和30ml甲苯,氮气保护下,加入钛酸四异丙酯(17.2g),25℃-30℃反应约12h,向体系中加入1.0g cat01,加入三乙胺(12.2g)和甲酸(13.9g),25℃-30℃继续反应15h-20h,至原料化合物ii-01《2.0%,停止反应;40℃-50℃减压除去溶剂,加入2mol/l的盐酸水溶液调节ph至2-4,搅拌约30min-60min,加入60ml甲苯洗涤1次,保留水相;水相降温至10℃-20℃,用50%naoh水溶液(质量分数)调节ph至12-13,经硅藻土过滤,滤饼用25ml甲苯洗涤1次,所得滤液分别用60ml甲苯萃取2次(2*60ml),合并有机相,经40℃-50℃减压浓缩至干得油状粗品;粗品经异丙醇/正己烷(40ml,7:1,体积比)重结晶得白色固体化合物iii-01:16.3g,收率95%,hplc纯度:99.3%,ee:98%。
[0082]
实施例2:制备化合物iii-02
[0083][0084]
向反应瓶中依次加入化合物ii-02(8g)、对甲基苯磺酰胺(6.2g)和30ml甲苯,氮气保护下,加入钛酸四异丙酯(10.2g),25℃-30℃反应约12h,向体系中加入0.55g cat01,加入三乙胺(7.8g)和甲酸(8.8g),25℃-30℃继续反应15h-20h,至原料化合物ii-02《2.0%,停止反应;40℃-50℃减压除去溶剂,加入2mol/l的盐酸水溶液调节ph至2-4,搅拌约30-60min,加入40ml甲苯洗涤1次,保留水相;水相降温至10℃-20℃,用50%naoh水溶液(质量分数)调节ph至12-13,经硅藻土过滤,滤饼用20ml甲苯洗涤1次,所得滤液分别用40ml甲苯萃取2次(2*40ml),合并有机相,经40-50℃减压浓缩至干得油状粗品;粗品经异丙醇/正己烷重结晶(40ml,7:1,体积比)得白色固体化合物iii-02:11.9g,收率97%,hplc纯度:99.2%,ee:99%。
[0085]
实施例3:制备化合物iii-03
[0086][0087]
向反应瓶中依次加入化合物ii-03(10g)、对甲基苯磺酰胺(7.3g)和30ml甲苯,氮
气保护下,加入钛酸四异丙酯(15.7g),25℃-30℃反应12-18h,向体系中加入0.96g cat01,加入三乙胺(12.5g)和甲酸(14.5g),25℃-30℃继续反应15h-20h,至原料化合物ii-03《2.0%,停止反应;40℃-50℃减压除去溶剂,加入2mol/l的盐酸水溶液调节ph至2-4,搅拌约30-60min,加入50ml甲苯洗涤1次,保留水相;水相降温至10℃-20℃,用50%naoh水溶液(质量分数)调节ph至12-13,经硅藻土过滤,滤饼用25ml甲苯洗涤1次,所得滤液分别用60ml甲苯萃取2次(2*60ml),合并有机相,经40-50℃减压浓缩至干得油状粗品;粗品经异丙醇/正己烷(32ml,7:1,体积比)重结晶得白色固体化合物iii-03:15.3g,收率92%,hplc纯度:99%,ee:96%。
[0088]
实施例4:制备化合物iii-04
[0089][0090]
向反应瓶中依次加入化合物ii-04(10g)、对甲基苯磺酰胺(8.3g)和30ml甲苯,氮气保护下,加入钛酸四异丙酯(18g),25℃-30℃反应约12h,向体系中加入1.1g cat01,加入三乙胺(13.8g)和甲酸(15.8g),25℃-30℃继续反应15h-20h,至原料化合物ii-04《2.0%,停止反应;40℃-50℃减压除去溶剂,加入2mol/l的盐酸水溶液调节ph至2-4,搅拌约30-60min,加入50ml甲苯洗涤1次,保留水相;水相降温至10℃-20℃,用50%naoh水溶液(质量分数)调节ph至12-13,经硅藻土过滤,滤饼用25ml甲苯洗涤1次,所得滤液分别用50ml甲苯萃取2次(2*50ml),合并有机相,经40-50℃减压浓缩至干得油状粗品;粗品经异丙醇/正己烷(32ml,7:1,体积比)重结晶得白色固体化合物iii-04:16.7g,收率95%,hplc纯度:99.1%,ee:95%。
[0091]
实施例5:制备化合物iii-05
[0092][0093]
向反应瓶中依次加入化合物ii-05(10g)、对甲基苯磺酰胺(7.0g)和30ml甲苯,氮气保护下,加入钛酸四异丙酯(15.1g),25℃-30℃反应约12h,向体系中加入1.0g cat01,加入三乙胺(11.5g)和甲酸(13.2g),25℃-30℃继续反应15h-20h,至原料化合物ii-05《2.0%,停止反应;40℃-50℃减压除去溶剂,加入2mol/l的盐酸水溶液调节ph至2-4,搅拌约30-60min,加入50ml甲苯洗涤1次,保留水相;水相降温至10℃-20℃,用50%naoh水溶液(质量分数)调节ph至12-13,经硅藻土过滤,滤饼用25ml甲苯洗涤1次,所得滤液分别用50ml甲苯萃取2次(2*50ml),合并有机相,经40-50℃减压浓缩至干得油状粗品;粗品经异丙醇/正
己烷重(40ml,7:1,体积比)结晶得白色固体化合物iii-05:15.7g,收率96%,hplc纯度:99.3%,ee:99%。
[0094]
本发明的方法已经通过较佳实施例进行了描述,相关人员明显能在本发明内容、精神和范围内对本文所述的方法和应用进行改动或适当变更与组合,来实现和应用本发明技术。本领域技术人员可以借鉴本文内容,适当改进工艺参数实现。特别需要指出的是,所有类似的替换和改动对本领域技术人员来说是显而易见的,它们都被视为包括在本发明内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1