一种唑吡坦的制备方法与流程
1.本发明属于药物合成技术领域,具体涉及一种唑吡坦的制备方法。
背景技术:
2.酒石酸唑吡坦(zolpidem tartrate),化学名为2-(4-甲基苯基)-n,n,6-三甲基咪唑并[1,2-a]吡啶-3-乙酰胺酒石酸盐,是一种非苯二氮杂
□
类催眠药,商品名由法国synthelabo公司原研,1988年在法国首次上市。临床用于治疗严重的睡眠障碍疾病,如偶发性失眠症和暂时性失眠症;此外,本品对原发性失眠、抑郁症、精神病引起的失眠也都有显著的疗效;具有药效快、成瘾性低等特点。其化学结构式如下:
[0003][0004]
唑吡坦的合成工艺主要有以下几种:
[0005]
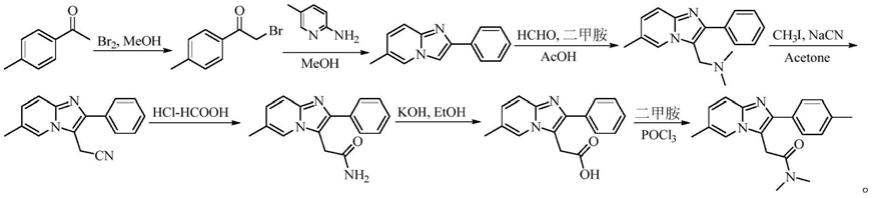
1.美国专利us20070027180、us4382938以对甲基苯乙酮为起始原料,先溴代生成2-溴-4'-甲基苯乙酮,再经缩合、mannich反应、氰基取代以及水解得2-[6-甲基-2-(对甲基苯基)咪唑并[1,2-a]吡啶-3-基]乙酸,最后再与大大过量的二甲胺反应得目标产品。但该工艺反应步骤长、操作繁琐;并且使用了剧毒的碘甲烷、氰化钠、甲醛,以及对环境不利的三氯氧磷和大大过量的二甲胺等试剂,不利于工业化生产。反应路线如下:
[0006][0007]
2.文献org.lett.,2012,14(17):4580-4583以对甲基-β-硝基苯乙烯为起始原料,经michael加成、酰化、关环以及水解反应后,在五氯化磷作用下与二甲胺缩合得目标产品。但该法起始原料价昂贵,生产成本较高;所用吡啶刺激性大,对健康及环境都不利。反应路线如下:
[0008][0009]
3.文献angew.chem.int.ed.engl.,2010,49(15):2743-2746中以对甲基苯甲醛为起始原料,先和2-氨基-5-甲基吡啶缩合生成相应的shiff碱,然后在三氟甲磺酸铜[cu(otf)2]和氯化亚铜的催化下与n,n-二甲基丙炔酰胺环合生成目标产品。但该法所用的n,n-二甲基丙炔酰胺和cu(otf)2价格极贵,大大增加了生产成本。反应路线如下:
[0010][0011]
4.文献j.med.chem.,1997,40(19):3109-3118中以n,n-二甲基-4-氧代-4-(对甲基苯基)丁酰胺为起始原料,与溴素溴代后,与2-氨基-5-甲基吡啶环合得目标产品。虽然该方法绿色化程度较高、原子经济性好,但仍存在以下不足:1)起始原料价昂贵,总收率较低,仅为17%;2)溴代化合物难以纯化,要进行2~3次精制才能得到95%以上的纯度,收率低(50%~60%);3)粗品反应中的杂质较多,产品的纯化难度大;除可利用产品溶于酸水的特点去掉一些杂质外,其他杂质只能靠多次精制除去,收率很低(30%)。反应路线如下:
[0012][0013]
综上,目前唑吡坦的制备方法在工艺安全、操作繁琐,收率不高,生产成本较高等方面存在许多不足,因此,研究寻找一条反应条件温和,操作过程简便,产品收率高、纯度高,生产成本低的适合工业化生产唑吡坦的反应路线仍是目前需要解决的问题。
技术实现要素:
[0014]
针对目前现有唑吡坦制备技术存在的问题,本发明提供了一种新的唑吡坦的制备方法。通过该方法所制得的目标产品具有较高的纯度、收率。
[0015]
本发明的具体技术方案如下:
[0016][0017]
一种唑吡坦的制备方法,具体包括以下步骤:
[0018]
惰性气体保护下,将sm-1、二甲胺、甲基丙烯酸甲酯(mma)加入反应溶剂中,控温加入预冷的催化剂,保温搅拌均匀后,升温至室温,继续反应至结束后,经后处理制得唑吡坦。
[0019]
优选方案,所述的反应溶剂为乙醚、异丙醚、甲基叔丁基醚、四氢呋喃、2-甲基四氢呋喃中的一种或其组合,其中特别优选2-甲基四氢呋喃。
[0020]
优选方案,所述的催化剂为rh(trop2n)(pph3),其化学结构如下所示,可通过商业获得,也可以依据现有技术自行制备,可将其直接加入反应体系中,也可以经反应溶剂稀释后加入。
[0021]
[0022]
优选方案,所述的sm-1与二甲胺、mma、催化剂的投料摩尔比为1:1.2~2.0:1.5~4.0:0.1%~0.5%,其中特别优选1:1.6:2.8:0.2%。
[0023]
优选方案,所述的二甲胺可以是气体通入,或者是二甲胺的有机溶剂溶液,本发明优选2mol/l二甲胺的四氢呋喃溶液,只要加入配比量的二甲胺即可。
[0024]
优选方案,所述的控温、催化剂预冷的温度为-20~0℃。
[0025]
优选方案,所述的后处理步骤为:将反应液过滤,滤液减压浓缩至干后,用1~5mol/l盐酸溶解,过滤,滤液二氯甲烷洗涤,酸水层用1~5mol/l氢氧化钠溶液调节ph至9~11,继续搅拌析晶后,过滤,纯化水洗至中性,所得滤饼减压干燥后即为唑吡坦。
[0026]
本发明中,所述的惰性气体通常选择氮气、氩气,其中特别优选氩气。
[0027]
本发明的有益效果:
[0028]
1.本发明提供了一种新的唑吡坦的制备方法,该制备工艺后处理操作简便,只需通过简单的酸溶碱析精制即可得到纯度较高的产品;
[0029]
2.本发明的唑吡坦的制备工艺,相较于现有技术可明显缩短工艺路线,适合工业化生产。
附图说明
[0030]
图1是实施例1得到的唑吡坦纯度液相分析图谱。
[0031]
图2是实施例4得到的唑吡坦纯度液相分析图谱。
具体实施方式
[0032]
下面通过实施例来进一步说明本发明,应该正确理解的是:本发明的实施例仅仅是用于说明本发明,而不是对本发明的限制,所以,在本发明的方法前提下对本发明的简单改进均属于本发明要求保护的范围。
[0033]
对本发明得到的唑吡坦化合物结构确证如下:
[0034]
mp.:195.3~196.0℃;esi-hrms(m/z):308.1758[m+h]
+
;1h nmr(400mhz,cd3od-d4)δ9.56(s,1h),7.61~7.85(m,2h),7.42(d,j=7.4hz,2h),7.30(d,j=7.4hz,2h),4.50(d,j=5.4hz,2h),2.80(s,3h),2.48(s,3h),2.43(s,3h),2.41(s,3h);
13
c nmr(101mhz,cd3od-d4)δ176.73,140.55,138.15,137.99,133.35,129.89,129.57,129.33,126.69,124.78,119.91,119.00,36.93,36.73,21.13,15.47。
[0035]
本发明采用hplc测定唑吡坦的纯度,色谱条件如下:
[0036]
色谱柱:ymc triart-c
18
柱(4.6mm
×
250mm,5μm)或效能相当的色谱柱;
[0037]
流动相:流动相a:硫酸钠水溶液(取无水硫酸钠2.84g与三氟乙酸1ml,加水溶解并稀释至1000ml),流动相b:乙腈,梯度洗脱;
[0038]
柱温:35℃;
[0039]
检测波长:248nm;
[0040]
流速:1.0ml/min;
[0041]
进样量:20μl;
[0042]
洗脱梯度如表1所示:
[0043]
表1洗脱梯度表
[0044][0045]
以下各实施例中,未详细描述的各种过程和方法是本领域中公知的常规方法。
[0046]
实施例1
[0047]
氩气保护,将sm-1(26.63g,0.1mol)、二甲胺的四氢呋喃溶液(2mol/l,80ml,0.16mol)、甲基丙烯酸甲酯(mma,28.03g,0.28mol)加入2-甲基四氢呋喃(260ml)中,控温-15~-10℃,加入提前预冷-15~-10℃的rh(trop2n)(pph3)(0.15g,0.2mmol)的2-甲基四氢呋喃(10ml)溶液,保温-15~-10℃搅拌均匀后,将反应液升温至室温,经检测反应完毕后,过滤,滤液减压浓缩至干后,用盐酸(3mol/l,200ml)溶解,过滤,滤液用二氯甲烷(100ml)洗涤,酸水层用4mol/l氢氧化钠溶液调节ph至9~11,有大量淡黄色固体析出,继续搅拌析晶1~2h后过滤,纯化水洗至中性,所得滤饼减压干燥后即为目标产品,收率96.5%,纯度99.931%。
[0048]
实施例2
[0049]
氩气保护,将sm-1(26.63g,0.1mol)、二甲胺的四氢呋喃溶液(2mol/l,60ml,0.12mol)、mma(28.03g,0.28mol)加入四氢呋喃(260ml)中,控温-5~0℃,加入提前预冷-5~0℃的rh(trop2n)(pph3)(0.15g,0.2mmol)的四氢呋喃(10ml)溶液,控温-5~0℃搅拌均匀后,将反应液升温至室温,经检测反应完毕后,过滤,滤液减压浓缩至干后,用盐酸(2mol/l,300ml)溶解,过滤,滤液用二氯甲烷(150ml)洗涤,酸水层用5mol/l氢氧化钠溶液调节ph至9~11,有大量淡黄色固体析出,继续搅拌析晶1~2h后过滤,纯化水洗至中性,所得滤饼减压干燥后即为目标产品,收率93.2%,纯度99.882%。
[0050]
实施例3
[0051]
氩气保护,将sm-1(26.63g,0.1mol)、二甲胺的四氢呋喃溶液(2mol/l,55ml,0.11mol)、mma(28.03g,0.28mol)加入乙醚(260ml)中,控温-5~0℃,加入提前预冷-5~0℃的rh(trop2n)(pph3)(0.15g,0.2mmol)的乙醚(10ml)溶液,保温-5~0℃搅拌均匀后,将反应液升温至室温,经检测反应完毕后,过滤,滤液减压浓缩至干后,用盐酸(2mol/l,300ml)溶解,过滤,滤液用二氯甲烷(150ml)洗涤,酸水层用5mol/l氢氧化钠溶液调节ph至9~11,有大量淡黄色固体析出,继续搅拌析晶1~2h后过滤,纯化水洗至中性,所得滤饼减压干燥后即为目标产品,收率90.5%,纯度99.876%。
[0052]
实施例4
[0053]
氩气保护,将sm-1(26.63g,0.1mol)、二甲胺的四氢呋喃溶液(2mol/l,100ml,0.20mol)、mma(28.03g,0.28mol)加入甲基叔丁基醚(260ml)中,控温-15~-10℃,加入提前预冷-15~-10℃的rh(trop2n)(pph3)(0.15g,0.2mmol)的甲基叔丁基醚(10ml)溶液,保温-15~-10℃搅拌均匀后,将反应液升温至室温,经检测反应完毕后,过滤,滤液减压浓缩至干后,用盐酸(4mol/l,150ml)溶解,过滤,滤液用二氯甲烷(75ml)洗涤,酸水层用2.5mol/l氢氧化钠溶液调节ph至9~11,有大量淡黄色固体析出,继续搅拌析晶1~2h后过滤,纯化水洗
至中性,所得滤饼减压干燥后即为目标产品,收率96.2%,纯度99.897%。
[0054]
实施例5
[0055]
氩气保护,将sm-1(26.63g,0.1mol)、二甲胺的四氢呋喃溶液(2mol/l,105ml,0.21mol)、mma(28.03g,0.28mol)加入异丙醚(260ml)中,控温-15~-10℃,加入提前预冷-15~-10℃的rh(trop2n)(pph3)(0.15g,0.2mmol)的异丙醚(10ml)溶液,保温-15~-10℃搅拌均匀后,将反应液升温至室温,经检测反应完毕后,过滤,滤液减压浓缩至干后,用盐酸(5mol/l,120ml)溶解,过滤,滤液用二氯甲烷(60ml)洗涤,酸水层用2mol/l氢氧化钠溶液调节ph至9~11,有大量淡黄色固体析出,继续搅拌析晶1~2h后过滤,纯化水洗至中性,所得滤饼减压干燥后即为目标产品,收率95.6%,纯度99.872%。
[0056]
实施例6
[0057]
氩气保护,将sm-1(26.63g,0.1mol)、二甲胺的四氢呋喃溶液(2mol/l,80ml,0.16mol)、mma(15.02g,0.15mol)加入2-甲基四氢呋喃(260ml)中,控温-10~-5℃,加入提前预冷-10~-5℃的rh(trop2n)(pph3)(0.15g,0.2mmol)的2-甲基四氢呋喃(10ml)溶液,保温-10~-5℃搅拌均匀后,将反应液升温至室温,经检测反应完毕后,过滤,滤液减压浓缩至干后,用盐酸(4mol/l,150ml)溶解,过滤,滤液用二氯甲烷(75ml)洗涤,酸水层用3mol/l氢氧化钠溶液调节ph至9~11,有大量淡黄色固体析出,继续搅拌析晶1~2h后过滤,纯化水洗至中性,所得滤饼减压干燥后即为目标产品,收率93.5%,纯度99.853%。
[0058]
实施例7
[0059]
氩气保护,将sm-1(26.63g,0.1mol)、二甲胺的四氢呋喃溶液(2mol/l,80ml,0.16mol)、mma(14.02g,0.14mol)加入2-甲基四氢呋喃(260ml)中,控温-10~-5℃,加入提前预冷-10~-5℃的rh(trop2n)(pph3)(0.15g,0.2mmol)的2-甲基四氢呋喃(10ml)溶液,保温-10~-5℃搅拌均匀后,将反应液升温至室温,经检测反应完毕后,过滤,滤液减压浓缩至干后,用盐酸(2.5mol/l,240ml)溶解,过滤,滤液用二氯甲烷(120ml)洗涤,酸水层用4mol/l氢氧化钠溶液调节ph至9~11,有大量淡黄色固体析出,继续搅拌析晶1~2h后过滤,纯化水洗至中性,所得滤饼减压干燥后即为目标产品,收率91.5%,纯度99.786%。
[0060]
实施例8
[0061]
氩气保护,将sm-1(26.63g,0.1mol)、二甲胺的四氢呋喃溶液(2mol/l,80ml,0.16mol)、mma(40.05g,0.40mol)加入2-甲基四氢呋喃(260ml)中,控温-15~-10℃,加入提前预冷-15~-10℃的rh(trop2n)(pph3)(0.15g,0.2mmol)的2-甲基四氢呋喃(10ml)溶液,保温-15~-10℃搅拌均匀后,将反应液升温至室温,经检测反应完毕后,过滤,滤液减压浓缩至干后,用盐酸(3mol/l,200ml)溶解,过滤,滤液用二氯甲烷(100ml)洗涤,酸水层用2.5mol/l氢氧化钠溶液调节ph至9~11,有大量淡黄色固体析出,继续搅拌析晶1~2h后过滤,纯化水洗至中性,所得滤饼减压干燥后即为目标产品,收率94.2%,纯度99.812%。
[0062]
实施例9
[0063]
氩气保护,将sm-1(26.63g,0.1mol)、二甲胺的四氢呋喃溶液(2mol/l,80ml,0.16mol)、mma(41.05g,0.41mol)加入2-甲基四氢呋喃(260ml)中,控温-15~-10℃,加入提前预冷-15~-10℃的rh(trop2n)(pph3)(0.15g,0.2mmol)的2-甲基四氢呋喃(10ml)溶液,保温-15~-10℃搅拌均匀后,将反应液升温至室温,经检测反应完毕后,过滤,滤液减压浓缩至干后,用盐酸(2mol/l,300ml)溶解,过滤,滤液用二氯甲烷(150ml)洗涤,酸水层用
2.5mol/l氢氧化钠溶液调节ph至9~11,有大量淡黄色固体析出,继续搅拌析晶1~2h后过滤,纯化水洗至中性,所得滤饼减压干燥后即为目标产品,收率93.1%,纯度99.759%。
[0064]
实施例10
[0065]
氩气保护,将sm-1(26.63g,0.1mol)、二甲胺的四氢呋喃溶液(2mol/l,80ml,0.16mol)、mma(28.03g,0.28mol)加入四氢呋喃(260ml)中,控温-5~0℃,加入提前预冷-5~0℃的rh(trop2n)(pph3)(76mg,0.1mmol)的四氢呋喃(10ml)溶液,保温-5~0℃搅拌均匀后,将反应液升温至室温,经检测反应完毕后,过滤,滤液减压浓缩至干后,用盐酸(3mol/l,200ml)溶解,过滤,滤液用二氯甲烷(100ml)洗涤,酸水层用4mol/l氢氧化钠溶液调节ph至9~11,有大量淡黄色固体析出,继续搅拌析晶1~2h后过滤,纯化水洗至中性,所得滤饼减压干燥后即为目标产品,收率92.5%,纯度99.868%。
[0066]
实施例11
[0067]
氮气保护,将sm-1(26.63g,0.1mol)、二甲胺的四氢呋喃溶液(2mol/l,80ml,0.16mol)、mma(28.03g,0.28mol)加入2-甲基四氢呋喃(260ml)中,控温-15~-10℃,加入提前预冷-15~-10℃的rh(trop2n)(pph3)(0.38g,0.5mmol)的2-甲基四氢呋喃(10ml)溶液,保温-15~-10℃搅拌均匀后,将反应液升温至室温,经检测反应完毕后,过滤,滤液减压浓缩至干后,用盐酸(3mol/l,200ml)溶解,过滤,滤液用二氯甲烷(100ml)洗涤,酸水层用4mol/l氢氧化钠溶液调节ph至9~11,有大量淡黄色固体析出,继续搅拌析晶1~2h后过滤,纯化水洗至中性,所得滤饼减压干燥后即为目标产品,收率95.4%,纯度99.867%。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1