一种米库氯铵对照品的制备方法与流程
本发明属于大极性季铵盐类化合物的分离纯化领域,尤其涉及一种米库氯铵对照品的制备方法。
背景技术:
米库氯铵,又名美维松,是苄异喹啉类短效非去极化神经肌肉阻滞药,主要用于气管插管和维持肌松。由于米库氯铵结构中含有两个手性碳和两个手性氮,其中手性碳的构型为固定的r型,且分子又是对称结构,故最终合成的米库氯铵由三种未分离的非对映异构体组成,目前市售的成药中三种非对映异构体的比例为顺-顺:顺-反:反-反≈5:40:55。由于成品是未经分离的混合物,不同的工艺操作会使三者比例出现小的偏差,故对其进行含量标定时选择其水解产物950u77(反式水解产物)作为其对照品,来增加含量测定的准确度。
米库氯铵结构式如下:
米库氯铵对照品950u77结构式如下:
专利us4761418使用水溶解950u77粗品后,冷冻到0℃,抽除固体,再用大孔树脂吸附后,用dmf去除剩余的异构体,即得950u77,实际操作发现无法实现两个异构体的分离。现有技术一般采用普通柱层析分离方法分离得到米库氯铵对照品950u77,由于季铵盐类化合物极性大且950u77与反应过程中生成的顺式非对映异构体极性接近,使得分离过程费时费力,且柱层析分离的固定相硅胶对样品有不可逆吸附作用,样品长时间与硅胶接触会产生一部分的降解,使得最终收率不高。高速逆流色谱(hsccc)是近年发展起来的一种连续的无需固体支撑物的高效、快速的液液分配色谱分离技术,具有样品无损失、无污染、高效、快速等诸多优点,已经广泛应用于生物、医药等领域化学物质的制备分离和纯化。
技术实现要素:
本发明的目的是提供一种利用高速逆流色谱分离纯化制备米库氯铵对照品的方法,该方法能够快速高效的分离纯化得到米库氯铵对照品。
为实现上述目的,本发明所采取的技术方案为:一种米库氯铵对照品的制备方法,所述制备方法包括以下步骤:
(1)将r-(+)-5’-甲氧基劳丹素、3-氯-1-丙醇、碘化钠和碳酸钠在有机溶剂中回流反应后,过滤,滤液蒸干,得到固体a;
(2)米库氯铵对照品粗品的制备:将步骤(1)所得的固体a用水溶解后,加入阴离子交换树脂,搅拌、抽滤,滤液再用阴离子交换树脂交换一次,向水中加入氯化钠将产品析出,再用萃取剂萃取,蒸干,得到所述米库氯铵对照品粗品;
(3)米库氯铵对照品粗品的纯化:以溶剂a、溶剂b和溶剂c组成高速逆流溶剂体系,将所述高速逆流溶剂体系混合充分后静置,按上下两相分开,取上相为固定相,下相为流动相,将固定相充满于高速逆流色谱仪的多层线圈分离柱,然后注入流动相;将步骤(2)所得的米库氯铵对照品粗品用下相溶解后进样,收集流出液中第一个峰,向收集液中加入氯化钠将产品析出,再用萃取剂萃取,蒸干后加入乙醚,析出固体,倾去上清液后,固体干燥,得到米库氯铵对照品;
优选地,所述溶剂a为乙酸乙酯或乙酸丙酯或乙酸丁酯,所述溶剂b为正丁醇或异丁醇,所述溶剂c为水。
优选地,所述溶剂a为乙酸乙酯,溶剂b为正丁醇。
优选地,所述溶剂a、溶剂b、溶剂c的体积比为:溶剂a:溶剂b:溶剂c=(1-5):(1-10):(1-10)。
优选地,所述溶剂a、溶剂b、溶剂c的体积比为:溶剂a:溶剂b:溶剂c=1:4:6。
优选地,所述步骤(1)中,r-(+)-5’-甲氧基劳丹素、3-氯-1-丙醇、碘化钠、碳酸钠的摩尔比为:r-(+)-5’-甲氧基劳丹素:3-氯-1-丙醇:碘化钠:碳酸钠=1:(1-2.5):(1-2.5):(0.1-1),有机溶剂体积与r-(+)-5’-甲氧基劳丹素的质量的比值为(5-20):1ml/g,有机溶剂为丙酮、乙腈、乙酸乙酯中的至少一种。
优选地,所述步骤(2)中,阴离子交换树脂为强碱性阴离子交换树脂。
优选地,所述步骤(2)中,强碱性阴离子交换树脂为201×7强碱性阴离子交换树脂。
优选地,所述步骤(3)中,设定高速逆流色谱仪紫外检测器的波长为254~300nm,在500~1000r/min转速下,以2~10ml/min的流速注入流动相。
优选地,所述步骤(3)中,高速逆流色谱仪中紫外检测器的波长为279nm。
优选地,所述步骤(3)中,转速为800r/min。
相比于现有技术,本发明的有益效果为:本发明的分离过程可连续进行,操作简便,效率高;并且采用高速逆流色谱制备,不存在不可逆吸附,避免了样品的损耗,具有分离效果好、溶剂用量少、无污染、高效和快速的特点。
附图说明
图1为实施例1制得的950u77粗品液相色谱图;
图2为实施例1制得的950u77高速逆流色谱分离图;
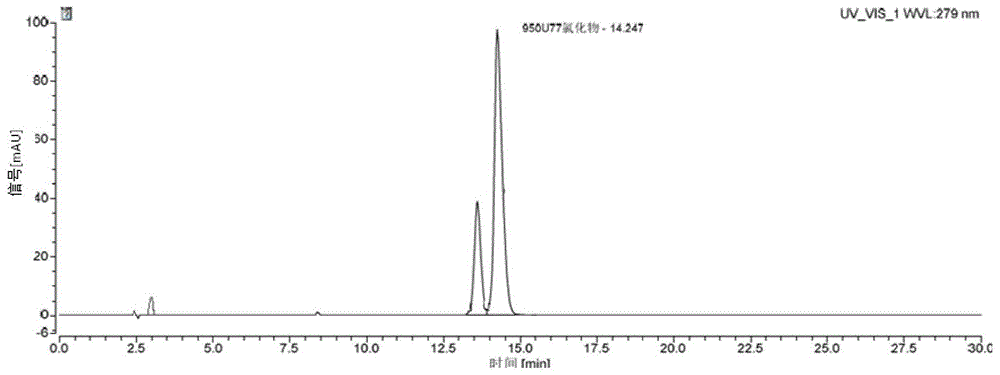
图3为实施例1制得的纯化的950u77液相色谱图。
具体实施方式
为更好地说明本发明的目的、技术方案和优点,下面将结合附图和具体实施例对本发明作进一步说明。
实施例1
本发明所述制备方法的一种实施例,本实施例所述制备方法包括如下步骤:
1)于100ml单口瓶中加入5.0gr-(+)-5’-甲氧基劳丹素,加入75ml丙酮使其溶解,再依次加入2.8g碘化钠、0.25g碳酸钠、2.2g3-氯-1-丙醇,加热回流反应96h,薄层色谱(tlc)检测反应完全,停止反应,反应液过滤,滤液蒸干后得浅黄色泡沫状固体7.7g,加入38ml去离子水溶解固体,再加入15.4g201×7强碱性阴离子交换树脂,室温搅拌30min,抽滤,滤液再用15.4g201×7强碱性阴离子交换树脂交换一次,向水中加入14.0g氯化钠将产品析出,再用二氯甲烷(30ml×3次)萃取,二氯甲烷层干燥,蒸干得浅黄色泡沫状固体6.8g,即为米库氯铵对照品950u77粗品。
使用高效液相色谱仪对得到的粗品进行纯度的检测。微粒多孔硅胶为填充剂(partisil5silica)(5μm,250mm×4.6mm);以乙腈-水(乙腈:水=70:30,v/v)为流动相;柱温35℃,流速1ml/min,检测波长279nm。其高效液相色谱图如图1所示,两个峰面积比约为1:3,与阳性对照品比较,从左至右第2个峰为950u77,液相含量为72.72%。
2)应用高速逆流色谱分离950u77:取乙酸乙酯、正丁醇、纯化水,按体积比1:4:6混合,混合充分后静置,按上下两相分开,上相为固定相,下相为流动相,先将固定相泵入高速逆流色谱仪中,待完全充满多层线圈分离柱后,开启高速逆流色谱仪,设定高速逆流色谱仪紫外检测器的波长为279nm,调整转速800r/min(正转),泵入流动相,换量筒接收,当接收处流动相上相不增加后表示系统已平衡好。取实施例1所得950u77粗品100mg,用6ml下相溶解后进样,进样后每隔25min再进样一次,所测紫外检测器光谱图为图2,对双峰中的第一个峰进行收集合并(收集4次共87ml),向收集液中加入31g氯化钠,二氯甲烷(50ml×3次)萃取,无水硫酸钠干燥,蒸干得油状物1.2g,加入20ml乙醚析出白色固体,静置后倾去上清液,干燥得固体162mg。
3)使用高效液相色谱仪对得到的纯品进行纯度的检测。微粒多孔硅胶为填充剂(partisil5silica)(5μm,250mm×4.6mm);以乙腈-水(70:30,v/v)为流动相;柱温35℃,流速1ml/min,检测波长279nm。其高效液相色谱图如图3所示,测得950u77液相纯度为99.67%,异构体含量为0.28%。
实施例2
本发明所述制备方法的一种实施例,所述实施例的制备方法与实施例1相比,仅存在如下区别:步骤(1)中,碘化钠为1.94g、碳酸钠为0.14g、3-氯-1-丙醇为1.22g,丙酮的用量为25ml。
使用高效液相色谱仪对实施例2获得的纯品进行纯度的检测,检测方法与实施例1相同,测得950u77液相纯度为99.23%。
实施例3
本发明所述制备方法的一种实施例,所述实施例的制备方法与实施例1相比,仅存在如下区别:步骤(1)中,碘化钠为4.83g、碳酸钠为1.37g、3-氯-1-丙醇为3.05g,丙酮的用量为100ml。
使用高效液相色谱仪对实施例3获得的纯品进行纯度的检测,检测方法与实施例1相同,测得950u77液相纯度为99.35%。
实施例4
本发明所述制备方法的一种实施例,所述实施例的制备方法与实施例1相比,仅存在如下区别:步骤(2)中,阴离子交换树脂为202型强碱性树脂。
使用高效液相色谱仪对实施例4获得的纯品进行纯度的检测,检测方法与实施例1相同,测得950u77液相纯度为97.50%。
最后所应当说明的是,以上实施例仅用以说明本发明的技术方案而非对本发明保护范围的限制,尽管参照较佳实施例对本发明作了详细说明,本领域的普通技术人员应当理解,可以对本发明的技术方案进行修改或者等同替换,但并不脱离本发明技术方案的实质和范围。
- 还没有人留言评论。精彩留言会获得点赞!