联苯类化合物及其制备方法和医药用途与流程
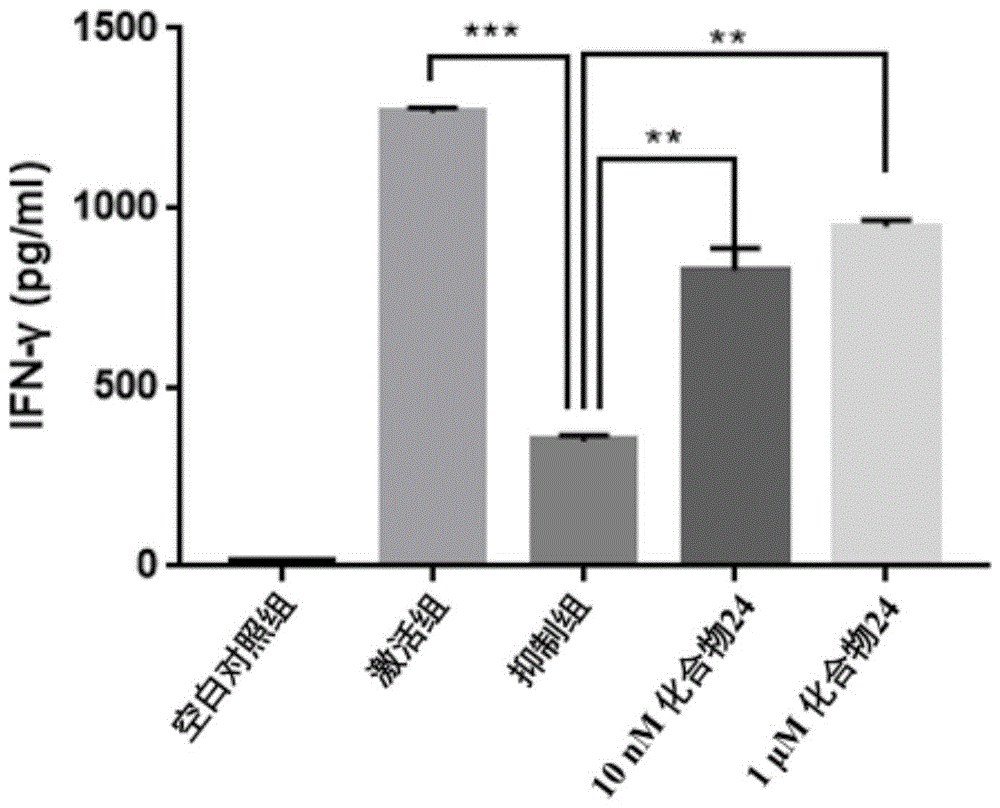
本发明属于生物医药领域,具体涉及一种具有pd-l1抑制活性的联苯类化合物,本发明还涉及该类化合物的制备方法及其作为pd-l1抑制剂的医药用途及用于免疫调节治疗的药物组合物。
背景技术:
:程序性死亡受体1(pd-1)主要表达于t细胞表面,是机体免疫检查点通路中的重要因子。而肿瘤细胞表面高表达pd-1的配体,即程序性死亡配体1(pd-l1),当其与t细胞表面的pd-1结合后,pd-1的itim(tyr223)和itsm(tyr248)酪氨酸基序分别被磷酸化,随后募集蛋白酪氨酸磷酸酶shp-2和shp-1,通过去磷酸化信号传导中间体(例如cd3ζ和plcγ1等),导致t细胞受体(tcr)信号的下调,使肿瘤细胞逃脱机体的免疫监控(immunity.2016,44(5):955-972)。pd-1与pd-l1结合后还可以通过减少与效应细胞功能相关的细胞因子(如ifn-γ、il-2和tnfα)和转录因子(如gata-3、t-bet和eomes)的表达来抑制t细胞的免疫功能(sciencesignaling.2012,5(230):ra46)。研究表明,抑制pd-1/pd-l1介导的免疫检查点通路,可以阻断肿瘤对t细胞的免疫抑制作用,从而激活t细胞对肿瘤的杀伤作用。目前上市的pd-1/pd-l1抑制剂都是单克隆抗体类药物,在研的pd-1/pd-l1抗体更是多达几十种。小分子药物相对于生物大分子药物具有明显的优势,由于其分子量较小,透膜性强,可以对一些实体瘤产生更好的疗效。此外,小分子药物适合口服给药,患者依从性好,且生产成本低。目前,pd-1/pd-l1小分子抑制剂还处于前期研发阶段。curis和aurigene公司联合开发的ca-170目前已完成i期临床研究(wo2016142833)。bms公司公开了一类苄基苯基醚类小分子pd-l1抑制剂(wo2015160641;wo2015034820)。中国专利申请2019102477713公开了一种苯并噁二唑类pd-l1抑制剂,然而,该类化合物的化学稳定性很差,这严重影响其成药性。综上所述,临床上亟需开发活性高、毒副作用小且成药性好的新型pd-l1小分子抑制剂。技术实现要素:发明目的:针对现有技术存在的问题,本发明提供一种新型联苯类化合物。本发明人发现,中国专利申请2019102477713公开的一种苯并噁二唑类pd-l1抑制剂尽管具有较好的pd-l1抑制活性,但该类化合物的化学稳定性和代谢稳定性不够理想,因而影响其成药性。而本发明的新型联苯类化合物是强效的pd-l1抑制剂,且具有非常好的化学稳定性和代谢稳定性,因而可应用于预防或治疗肿瘤、自身免疫性疾病、器官移植排斥、感染性疾病和炎症性疾病。本发明还提供所述联苯类化合物及其中间体的制备方法、药物组合物及其医药用途。技术方案:为了实现上述目的,本发明提供如下式(i)和式(ii)所示的联苯类化合物:r1选自:h、f、cl、br、i、cn、no2、nh2、oh、cf3、cf2cf3、ocf3、ocf2cf3、c1-c3烷基、c1-c4烷基磺酰基、c(o)oh、c(o)nh2、烯基、炔基、烷氧基、烷硫基、环烷基、卤代烷基、卤代烷氧基、卤代烷硫基、卤代环烷基或杂环烷基;r2选自:-(ch2)mcho、-(ch2)moh、-ch=ch-coor13或-(ch2)mnr9r9’;m是0、1、2、3或4;r9选自:h、c1-c4烷基或苄基;r9’选自以下任意一种:p是0、1、2、3或4;r10选自:h或c1-c3烷基;r11选自:h、甲基或苄基;r11’选自:h、或羟基;r12选自:h或c1-c3烷基;r12’选自:h或c1-c3烷基;r13选自:h、c1-c3烷基或苄基;r14选自:h、c1-c3烷基或苄基;r15选自:h、c1-c6烷基或c1-c6烷氧羰基;r16选自:h或c1-c4烷基;或者,r9和r9’与它们所连接的n原子一起形成一个环,选自以下任意一种:s是0、1或2;t是1、2或3;q选自:s、o、nh、nch3、n(ch2)2oh或chr18a;其中,r18a选自:h、oh、羟基取代的c1-c3烷基或c(o)oh;r17选自:h、c(o)oh、羟基取代的c1-c4烷基或c(o)nhso2r20;r18选自:h、c(o)oh、羟基取代的c1-c4烷基、oh、c(o)或c(o)nhso2r20;r19选自:c1-c4烷氧羰基、c1-c6烷基、c(o)oh、f、cl、br、i、oh、羟基取代的c1-c4烷基、nrarb或苯氧基羰基;其中,苯氧基羰基的苯基任选被f、cl、br、i、oh、cn、no2、nh2、cf3、cf2cf3、ocf3、ocf2cf3、so2nh2、c(o)oh、c(o)nh2或nhc(o)nh2取代;ra和rb各自独立地选自:h、c1-c4烷氧羰基或c1-c4烷基羰基;r20选自:cf3、环丙基、c1-c4烷基、二甲基氨基或甲基取代的咪唑基;r3和r4各自独立地选自:h、f、cl、br或i;r5、r6、r7和r8各自独立地选自h、f、cl、br、i、cn、no2、nh2、oh、cf3、orc、cf2cf3、ocf3、ocf2cf3、so2ch3、c(o)nhch3、c1-c4烷基、c3-c4环烷基、ch2c(o)n(rd)2或ch2ch2c(o)re;rc选自:取代或非取代的c1-c4烷基,所述取代或非取代的c1-c4烷基是未取代的或被一个或两个或三个独立地各自选自下列的取代基所取代:oh、(o)、c(o)oh、nhrf、n(rd)2、c(o)n(rd)2、吡咯烷-1-基、哌啶-1-基、4-羟基哌啶-1-基、4-羟基-4-羧基哌啶-1-基、4-羟基-4-羧酸酯哌啶-1-基、4-羟基-4-腈基哌啶-1-基、哌嗪-1-基、4-甲基-哌嗪-1-基、吗啉-4-基、硫代吗啉-1,1-二氧代-4-基、氨基酸或氨基酸酯;rd选自:c1-c3烷基或羟基取代的c2-c3烷基;re选自:4-羟基-4-羧基哌啶-1-基或4-羟基-4-腈基哌啶-1-基;rf选自:c1-c3烷基、羟基取代的c2-c3烷基或5-氟-2-氧代-2,3-二氢嘧啶-4-基;或者,r5、r6、r7和r8之中每两个与它们所连接到的原子一起形成取代或非取代的苯环、取代或非取代的杂芳环、取代或非取代的环烷烃环、取代或非取代的杂环烷烃环或取代或非取代的杂环烯烃环;w选自:h、f、cl、br、i、cn、no2、nh2、oh、cf3、cf2cf3、ocf3、ocf2cf3、c1-c4烷基、烯基、炔基、烷氧基、烷硫基、环烷基、卤代烷基、卤代烷氧基、卤代烷硫基、卤代环烷基或杂环烷基;z选自:o或s。在某些实施方案中,所述联苯类化合物不仅包括化合物本身还包括其药学上可接受的盐、互变异构体、内消旋体、外消旋体、立体异构体、代谢产物、代谢前体、前药或溶剂化物。在某些优选的实施方案中,所述联苯类化合物中的r1选自:h、f、cl、br、i、cn、no2、cf3、cf2cf3、ocf3、ocf2cf3、c1-c3烷基、c1-c4烷基磺酰基、c(o)oh或c(o)nh2;r2为-(ch2)mnr9r9’,其中,m是1;r9选自:h或c1-c4烷基;r9’选自以下任意一种:p是1或2;r10选自:h;r11是h;r12选自:h或c1-c3烷基;r13选自:h或c1-c3烷基;r14选自:h或c1-c3烷基;r15选自:h、c1-c6烷基或c1-c6烷氧羰基;或者,r9和r9’与它们所连接的n原子一起形成一个环,选自以下任意一种:s是0或1;t是2或3;q选自:s、o、nh、nch3、n(ch2)2oh或chr18a;其中,r18a选自:h、oh、羟基取代的c1-c3烷基或c(o)oh;r17选自:h、c(o)oh、羟基取代的c1-c4烷基或c(o)nhso2r20;r18选自:h、c(o)oh、羟基取代的c1-c4烷基、oh、c(o)或c(o)nhso2r20;r19选自:c1-c4烷氧羰基、c1-c6烷基、c(o)oh、f、cl、br、i、oh、羟基取代的c1-c4烷基、-nrarb或苯氧基羰基;其中,苯氧基羰基的苯基任选被f、cl、br、i、oh、cn、no2、nh2、cf3、cf2cf3、ocf3、ocf2cf3、so2nh2、c(o)oh、c(o)nh2或nhc(o)nh2取代;ra和rb各自独立地选自:h、c1-c4烷氧羰基或c1-c4烷基羰基;r20选自:cf3、环丙基、c1-c4烷基、二甲基氨基或甲基取代的咪唑基;r3和r4各自独立地选自:h、f、cl、br或i;r5、r6、r7和r8各自独立地选自h、f、cl、br、i、cn、no2、nh2、oh、cf3、orc、cf2cf3、ocf3、ocf2cf3、so2ch3、c(o)nhch3、c1-c4烷基、c3-c4环烷基、ch2c(o)n(rd)2或ch2ch2c(o)re;rc选自:取代或非取代的c1-c4烷基,所述取代或非取代的c1-c4烷基是未取代的或被一个或两个或三个独立地各自选自下列的取代基所取代:oh、(o)、c(o)oh、nhrf、n(rd)2、c(o)n(rd)2、吡咯烷-1-基、哌啶-1-基、4-羟基哌啶-1-基、4-羟基-4-羧基哌啶-1-基、4-羟基-4-羧酸酯哌啶-1-基、4-羟基-4-腈基哌啶-1-基、哌嗪-1-基、4-甲基-哌嗪-1-基、吗啉-4-基、硫代吗啉-1,1-二氧代-4-基、氨基酸或氨基酸酯;rd选自:c1-c3烷基或羟基取代的c2-c3烷基;re选自:4-羟基-4-羧基哌啶-1-基或4-羟基-4-腈基哌啶-1-基;rf选自:c1-c3烷基、羟基取代的c2-c3烷基或5-氟-2-氧代-2,3-二氢嘧啶-4-基;或者,r5、r6、r7和r8之中每两个与它们所连接到的原子一起形成取代或非取代的苯环、取代或非取代的杂芳环、取代或非取代的环烷烃环、取代或非取代的杂环烷烃环或取代或非取代的杂环烯烃环;w选自:h、f、cl、br、i、cn、no2、nh2、oh、cf3、cf2cf3、ocf3、ocf2cf3、c1-c4烷基、烯基、炔基、烷氧基、烷硫基、环烷基、卤代烷基、卤代烷氧基、卤代烷硫基、卤代环烷基或杂环烷基;z选自:o或s。作为更优选的实施方案,本发明所述的联苯类化合物或其药学上可接受的盐、互变异构体、内消旋体、外消旋体、立体异构体、代谢产物、代谢前体、前药或溶剂化物选自如下表1所示的化合物:表1化合物的结构与命名本发明的联苯类化合物也可作为药用盐使用。该盐可为下列酸中的至少一种的酸盐:半乳糖二酸、d-葡糖醛酸、甘油磷酸、马尿酸、羟乙磺酸、乳糖酸、马来酸、1,5-萘二磺酸、萘-2-磺酸、新戊酸、对苯二甲酸、硫氰酸、胆酸、正十二烷基硫酸、苯磺酸、柠檬酸、d-葡萄糖,乙醇酸、乳酸、苹果酸、丙二酸、扁桃酸、磷酸、丙酸、盐酸、硫酸、酒石酸、琥珀酸、甲酸、氢碘酸、氢溴酸、甲烷磺酸、烟酸、硝酸、乳清酸、草酸、苦味酸、l-焦谷氨酸、糖精酸、水杨酸、龙胆酸、对甲苯磺酸、戊酸、棕榈酸、葵二酸、硬脂酸、月桂酸、乙酸、己二酸、碳酸、苯磺酸、乙烷二磺酸、乙基琥珀酸、富马酸、3-羟基萘-2-甲酸、1-羟基萘-2-甲酸、油酸、十一碳烯酸、抗坏血酸、樟脑酸、樟脑磺酸、二氯乙酸、乙烷磺酸。另一方面,该盐也可以是本发明的化合物与金属(包括钠、钾、钙等)离子或药学上可接受的胺(包括乙二胺、氨丁三醇等)或铵离子形成的盐。本发明还提供了所述联苯类化合物或其药学上可接受的盐、互变异构体、内消旋体、外消旋体、立体异构体、代谢产物、代谢前体、前药或溶剂化物在制备pd-l1抑制剂中的应用。本发明发现,式(i)或式(ii)所示的联苯类化合物或其药学上可接受的盐、互变异构体、内消旋体、外消旋体、立体异构体、代谢产物、代谢前体、前药或溶剂化物是pd-l1抑制剂,可显著抑制pd-1和pd-l1蛋白-蛋白相互作用,且能增加t细胞的增殖能力,促进免疫因子的生成,并激活t细胞的抗肿瘤免疫活性,因而可用于肿瘤的免疫治疗。另一方面,在某些实施方案中,式(i)或式(ii)化合物或其药学上可接受的盐、互变异构体、内消旋体、外消旋体、立体异构体、代谢产物、代谢前体、前药或溶剂化物还具有抑制免疫炎症反应的功效。因此,本发明的联苯类化合物,包括其药学上可接受的盐、互变异构体、内消旋体、外消旋体、立体异构体、代谢产物、代谢前体、前药或溶剂化物可用于制备免疫调节剂类药物。所述免疫调节剂类药物可用于预防或治疗肿瘤、自身免疫性疾病、器官移植排斥、感染性疾病和炎症性疾病。本发明的联苯类化合物,包括其药学上可接受的盐、互变异构体、内消旋体、外消旋体、立体异构体、代谢产物、代谢前体、前药或溶剂化物可用于预防或治疗肿瘤。所述肿瘤包括但不限于:骨癌、急性骨髓性白血病、慢性骨髓性白血病、急性淋巴细胞系白血病、慢性淋巴细胞系白血病、骨髓增生性疾病、多发性骨髓瘤、骨髓增生异常综合征、霍奇金氏淋巴瘤、非霍奇金氏淋巴瘤、血管瘤、肉芽瘤、黄瘤、脑膜肉瘤、神经胶质瘤、神经母细胞瘤、星形细胞瘤、成神经管细胞瘤、室管膜瘤、生殖细胞瘤(松果体瘤)、多形性成胶质细胞瘤、少突神经胶质细胞瘤、神经鞘瘤、成视网膜细胞瘤、纤维神经瘤、肉瘤、食道癌、胃癌、胰腺癌、大肠癌、结肠癌、直肠癌、肾癌、前列腺癌、淋巴癌、睾丸癌、间质细胞癌、肺癌、肝癌、皮肤癌、恶性黑素瘤和基底细胞癌等。本发明的联苯类化合物,包括其药学上可接受的盐、互变异构体、内消旋体、外消旋体、立体异构体、代谢产物、代谢前体、前药或溶剂化物可用于预防或治疗自身免疫性疾病。所述自身免疫性疾病包括但不限于:溃疡性结肠炎、克罗恩病、系统性红斑狼疮、家族性冻疮狼疮、查加斯病、类风湿关节炎、银屑病、多发性硬化症、硬皮症、白塞氏病、自身免疫性肝炎和aicardi-goutières综合征等。本发明的联苯类化合物,包括其药学上可接受的盐、互变异构体、内消旋体、外消旋体、立体异构体、代谢产物、代谢前体、前药或溶剂化物可用于预防或治疗器官移植排斥。所述器官移植包括但不限于:骨髓移植、肝脏移植、肺脏移植、心脏移植和肾脏移植等。本发明的联苯类化合物,包括其药学上可接受的盐、互变异构体、内消旋体、外消旋体、立体异构体、代谢产物、代谢前体、前药或溶剂化物可用于预防或治疗感染性疾病。所述感染性疾病包括但不限于:乙肝病毒感染、丙肝病毒感染、hiv病毒感染、新冠病毒感染、疱疹病毒感染和细菌感染等。本发明的联苯类化合物,包括其药学上可接受的盐、互变异构体、内消旋体、外消旋体、立体异构体、代谢产物、代谢前体、前药或溶剂化物可用于预防或治疗炎症性疾病。所述炎症性疾病包括但不限于:非酒精性脂肪性肝炎、原发性胆汁性胆管炎、原发性硬化性胆管炎、哮喘、气管炎、支气管炎、肺炎、呼吸窘迫综合征、肺气肿、慢性阻塞性肺病、特发性肺纤维化、囊性纤维化肺病、鼻炎、心肌炎、肾炎和神经性皮炎等。在某些实施方案中,本发明的式(i)或式(ii)化合物可单独使用。而在某些实施方案中,本发明的式(i)或式(ii)化合物可与一种或多种其他药物联合使用。可选择与本发明的联苯类化合物联合使用的抗肿瘤药物包括但不限于:化疗药物,如多西他赛、紫杉醇、abraxane、多柔比星、奥沙利铂、卡铂、顺铂、伊立替康、吉西他滨、环磷酰胺、ast-1001和ast-3424等;免疫检查点抑制剂,如ctla-4抑制剂(如yervoy)、pd-1抑制剂(如keytruda和opdivo)、pd-l1抑制剂(如tecentriq)、lag-3抑制剂(如bms986016、regn3767和lag525)、tim-3抑制剂(如tsr-022和mbg-453)、tigit抑制剂(如mtig7192a)、b7h4抑制剂和vista抑制剂(如jnj-61610588)等;抗体药物缀合物(adc),如kadcyla等;激酶抑制剂,如shp-2抑制剂、b-raf抑制剂(如vemurafenib)、mek抑制剂(如cobimetinib)和btk抑制剂(如ibrutinib)等;ido抑制剂(如epacadostat);usp7抑制剂(如p5091);tgfβr抑制剂(如galunisertib和vactosertib);mdm2抑制剂(如idasanutlin和apg-115);hdac抑制剂(如saha);lsd1抑制剂(如og-l002)和pcaf(p300/cbp-associatedfactor)抑制剂(如gsk4027)等。可选择与本发明的联苯类化合物联合使用的抗自身免疫性疾病药物包括但不限于:抗银屑病药物(如卡泊三醇、他卡西醇、糖皮质激素、他扎罗汀、维a酸、他克莫司和吡美莫司等)、抗溃疡性结肠炎药物(如柳氮磺胺吡啶、美沙拉嗪、强的松和地塞米松等)、抗克罗恩病药物(如柳氮磺胺吡啶、5-氨基水杨酸、氢化可的松、甲基强的松龙、英夫利西单抗、阿达木单抗和赛妥珠单抗等)、抗系统性红斑狼疮药(如氯喹、羟基氯喹、环孢素a、甲氨蝶呤和硫唑嘌呤等)、抗类风湿关节炎药(如jak抑制剂、非甾体抗炎药、依那西普和来氟米特等)和抗多发性硬化症药(如干扰素、米托蒽醌、芬戈利德、ozanimod、特立氟胺、富马酸二甲酯和ocrelizumab等)等。可选择与本发明的联苯类化合物联合使用的抗器官移植排斥药物包括但不限于:环孢素、西罗莫司、吗替麦考酚酯和他克莫司等。可选择与本发明的联苯类化合物联合使用的抗感染性疾病药物包括但不限于:抗乙肝病毒药(如恩替卡韦和阿德福韦等)、抗丙肝病毒药(如索磷布韦和维帕他韦等)、抗hiv病毒药(如替诺福韦和恩曲他滨等)和抗生素(如头孢类药物等)等。可选择与本发明的联苯类化合物联合使用的抗炎症性疾病药物包括但不限于:甾体类抗炎药(如布地奈德和地塞米松等)和非甾体类抗炎药(如水杨酸和塞来昔布等)等。本发明还提供了一种用于免疫调节治疗的药物组合物,其中含有如本发明所述的治疗有效量的式(i)或式(ii)所示联苯类化合物或其药学上可接受的盐、互变异构体、内消旋体、外消旋体、立体异构体、代谢产物、代谢前体、前药或溶剂化物作为活性成分和药学上可接受的载体。可任意混合的载体根据剂型、给药形式等可以改变。载体的例子包括赋形剂、粘合剂、崩解剂、润滑剂、矫味剂、香味剂、着色剂和甜味剂等。所述药物组合物可以是胶囊剂、散剂、片剂、颗粒剂、丸剂、注射剂、糖浆剂、口服液、吸入剂、软膏剂、栓剂和贴剂等制剂学上常规的制剂形式。进一步地,所述药物组合物是胶囊剂、散剂、片剂、颗粒剂、丸剂、注射剂、糖浆剂、口服液、吸入剂、软膏剂、栓剂或贴剂。为了使片剂形式的药物组合物成形,可使用本领域任何已知并广泛使用的赋形剂。例如,载体,如乳糖、白糖、氯化钠、葡萄糖、尿素、淀粉、碳酸钙、高岭土、结晶纤维素和硅酸等;粘合剂,如水、乙醇、丙醇、普通糖浆、葡萄糖溶液、淀粉溶液、明胶溶液,羧甲基纤维素、紫胶、甲基纤维素和磷酸钾、聚乙烯吡咯烷酮等;崩解剂,如干淀粉、藻酸钠、琼脂粉和海带粉,碳酸氢钠、碳酸钙、聚乙烯脱水山梨醇的脂肪酸酯、十二烷基硫酸钠、单硬脂酸甘油酯、淀粉和乳糖等;崩解抑制剂,如白糖、甘油三硬脂酸酯、椰子油和氢化油;吸附促进剂,如季胺碱和十二烷基硫酸钠等;润湿剂,如甘油、淀粉等;吸附剂,如淀粉、乳糖、高岭土、膨润土和胶体硅酸等;以及润滑剂,如纯净的滑石,硬脂酸盐、硼酸粉和聚乙二醇等。还可以根据需要选用通常的涂渍材料制成糖衣片剂、涂明胶膜片剂、肠衣片剂、涂膜片剂、双层膜片剂及多层片剂。为了使丸剂形式的药物组合物成形,可使用本领域任何已知的并广泛使用的赋形剂,例如,载体,如乳糖,淀粉,椰子油,硬化植物油,高岭土和滑石粉等;粘合剂,如阿拉伯树胶粉,黄蓍胶粉,明胶和乙醇等;崩解剂,如琼脂和海带粉等。为了使栓剂形式的药物组合物成形,可使用本领域任何已知并广泛使用的赋性剂,例如,聚乙二醇,椰子油,高级醇,高级醇的酯,明胶和半合成的甘油酯等。为了制备针剂形式的药物组合物,可将溶液或悬浮液消毒后,制成与血液等渗压的针剂。在制备针剂时,也可使用本领域内任何常用的载体。例如,水,乙醇,丙二醇,乙氧基化的异硬脂醇,聚氧基化的异硬脂醇和聚乙烯脱水山梨醇的脂肪酸酯等。此外,还可加入通常的溶解剂、缓冲剂和止痛剂等。所述药物组合物中,所述稀释剂可为本领域中常规的稀释剂。所述的药物组合物可以是口服的形式,也可以是无菌注射水溶液形式,可按照本领域任何已知制备药用组合物的方法制备口服或注射组合物。本发明中制备式(i)或式(ii)所示的联苯类化合物的方法,其合成路线如下所示:在上述合成路线中,t和y是溴、氯、碘、对甲苯磺酸酯、甲磺酸酯或三氟甲磺酸酯;r1、r3、r4、r5、r6、r7、r8、r9、r9’和w的定义与所述式(i)或式(ii)化合物中的定义一致;所述化合物的合成具体包括以下步骤:(1)由化合物n-1与化合物n-2在碱的作用下反应得到化合物n-3;(2)由化合物n-3与la-7或lc-4在碱的作用下反应得到化合物n-4a或n7a;(3)由化合物n-4a或n7a与nhr9r9’进行还原胺化反应得到化合物ia’或iia’;(4)或者,由化合物n-4a或n-7a经还原反应得到化合物n-5a或n-8a;(5)由n-5a或n-8a经卤化或磺酸酯化得到化合物n-6a或n-9a;(6)n-6a或n-9a在碱的作用下与nhr9r9’进行烷基化反应得到化合物ia’或iia’。本发明的联苯类化合物的制备可参照实施例中的方法进行。特别地,当所述式(i)或式(ii)化合物中的r2为-(ch2)mnr9r10,且m是1时,本发明的联苯类化合物的制备可参照以下合成路线或改进的方法进行。苯并噁二唑边链的制备方法(一):苯并噻二唑边链的制备方法(二):苯并噁二唑边链的制备方法(三):苯并噻二唑边链的制备方法(四):目标化合物的合成路线(一):目标化合物的合成路线(二):在上述合成路线中,t和y是溴、氯、碘、对甲苯磺酸酯、甲磺酸酯或三氟甲磺酸酯;r1、r3、r4、r5、r6、r7、r8、r9、r9’、w和z的定义与所述式(i)或式(ii)化合物中的定义一致。所述化合物的合成具体包括以下步骤:(1)由la-1经硝化反应得到la-2,所采用的溶剂包括但不限于:硫酸、乙酸、三氟乙酸、乙酸酐、三氟乙酸酐或者用这些溶剂任选组成的混合溶剂;所采用的硝化试剂包括但不限于:硝酸、硝酸钾、硝酸钠、硝酸铵、四氟硼酸硝或硝酰氯;反应温度为0℃至100℃,优选温度为20℃至60℃;(2)由化合物la-2与叠氮化物反应得到化合物la-3,所采用的溶剂包括但不限于:苯、甲苯、氯仿、正己烷、环己烷、二氯甲烷、1,2-二氯乙烷、甲基叔丁基醚、乙酸乙酯、乙酸丙酯、乙酸丁酯、甲醇、乙醇、丙酮、四氢呋喃、乙醚、乙腈、n,n-二甲基甲酰胺、二甲亚砜或者用这些溶剂任选组成的混合溶剂,优选丙酮、n,n-二甲基甲酰胺、四氢呋喃或乙腈;所采用的叠氮化物包括但不限于:叠氮化钠或叠氮化钾;反应温度为0℃至150℃,优选温度为50℃至100℃;(3)由化合物la-3经环合反应得到化合物la-4,所采用的溶剂包括但不限于:苯、甲苯、氯仿、正己烷、环己烷、二氯甲烷、1,2-二氯乙烷、甲基叔丁基醚、乙酸乙酯、乙酸丙酯、乙酸丁酯、甲醇、乙醇、丙酮、四氢呋喃、乙腈、n,n-二甲基甲酰胺、二甲亚砜或者用这些溶剂任选组成的混合溶剂,优选丙酮、n,n-二甲基甲酰胺、四氢呋喃、甲苯或乙腈;反应温度为室温至200℃,优选温度为50℃至150℃;(4)由化合物la-4在脱氧剂的作用下反应得到化合物la-5,所采用的溶剂包括但不限于:苯、甲苯、氯仿、正己烷、环己烷、二氯甲烷、1,2-二氯乙烷、甲基叔丁基醚、乙酸乙酯、乙酸丙酯、乙酸丁酯、丙酮、甲醇、乙醇、四氢呋喃、乙腈、n,n-二甲基甲酰胺、二甲亚砜或者用这些溶剂任选组成的混合溶剂,优选n,n-二甲基甲酰胺、四氢呋喃或乙腈;所采用的脱氧剂包括但不限于:三苯基膦、三乙基膦、三丁基膦或三环己基膦;反应温度为0℃至150℃,优选温度为50℃至120℃;(5)由化合物la-5经还原反应得到化合物la-6,所采用的溶剂包括但不限于:苯、甲苯、氯仿、正己烷、环己烷、二氯甲烷、1,2-二氯乙烷、甲基叔丁基醚、乙酸乙酯、乙酸丙酯、乙酸丁酯、丙酮、四氢呋喃、乙腈、甲醇、乙醇、n,n-二甲基甲酰胺、二甲亚砜或者用这些溶剂任选组成的混合溶剂,优选甲醇、乙醇或四氢呋喃;所采用的还原剂包括但不限于:硼氢化钠或硼氢化钾;反应温度为-20℃至100℃,优选温度为室温至70℃;(6)由化合物la-6经卤化或磺酸酯化反应得到化合物la-7,所采用的溶剂包括但不限于:苯、甲苯、氯仿、正己烷、环己烷、二氯甲烷、1,2-二氯乙烷、甲基叔丁基醚、乙酸乙酯、乙酸丙酯、乙酸丁酯、丙酮、四氢呋喃、乙腈、n,n-二甲基甲酰胺、二甲亚砜或者用这些溶剂任选组成的混合溶剂;所采用的卤化剂包括但不限于:pph3/cbr4、pbr3、pobr3、hbr、pcl3、pocl3、hcl或hi;所采用的磺酸酯化试剂(在碱的作用下)包括但不限于:对甲苯磺酰氯、甲磺酰氯、甲磺酸酐、三氟甲磺酰氯或三氟甲磺酸酐;所采用的碱包括但不限于:三乙胺、二异丙基乙胺、1,8-二氮杂环[5,4,0]十一烯-7、碳酸钾、碳酸钠、碳酸氢钾或碳酸氢钠;反应温度为-20℃至100℃,优选温度为0℃至70℃;(7)由化合物lb-1在碱的作用下与二氯亚砜环化得到化合物lb-2,所采用的溶剂包括但不限于:苯、甲苯、氯仿、正己烷、环己烷、二氯甲烷、1,2-二氯乙烷、甲基叔丁基醚、乙酸乙酯、乙酸丙酯、乙酸丁酯、丙酮、四氢呋喃、乙腈、n,n-二甲基甲酰胺、二甲亚砜或者用这些溶剂任选组成的混合溶剂,优选二氯甲烷、甲苯或氯仿;所采用的碱包括但不限于:三乙胺、二异丙基乙胺、1,8-二氮杂环[5,4,0]十一烯-7、碳酸钾、碳酸钠、碳酸氢钾或碳酸氢钠;反应温度为0℃至150℃,优选温度为20℃至80℃;(8)由化合物lb-2经卤化反应得到化合物lb-3,所采用的溶剂包括但不限于:苯、甲苯、氯仿、正己烷、环己烷、二氯甲烷、1,2-二氯乙烷、甲基叔丁基醚、乙酸乙酯、乙酸丙酯、乙酸丁酯、丙酮、四氢呋喃、乙腈、n,n-二甲基甲酰胺、二甲亚砜或者用这些溶剂任选组成的混合溶剂;所采用的卤化剂包括但不限于:cl2、br2或nbs;反应温度为0℃至120℃,优选温度为20℃至80℃;(9)由化合物lc-1在碱的存在下与氧化剂反应得到化合物lc-2,所采用的溶剂包括但不限于:苯、甲苯、氯仿、正己烷、环己烷、二氯甲烷、1,2-二氯乙烷、甲基叔丁基醚、乙酸乙酯、乙酸丙酯、乙酸丁酯、丙酮、四氢呋喃、乙腈、甲醇、乙醇、n,n-二甲基甲酰胺、二甲亚砜或者用这些溶剂任选组成的混合溶剂,优选甲醇、乙醇或四氢呋喃;所采用的碱包括但不限于:氢氧化钠或氢氧化钾;所采用的氧化剂包括但不限于:次氯酸钠、次氯酸钾、双氧水或间氯过氧苯甲酸;反应温度为-20℃至100℃,优选温度为0℃至70℃;(10)由化合物lc-2在脱氧剂的作用下反应得到化合物lc-3,所采用的溶剂包括但不限于:苯、甲苯、氯仿、正己烷、环己烷、二氯甲烷、1,2-二氯乙烷、甲基叔丁基醚、乙酸乙酯、乙酸丙酯、乙酸丁酯、丙酮、甲醇、乙醇、四氢呋喃、乙腈、n,n-二甲基甲酰胺、二甲亚砜或者用这些溶剂任选组成的混合溶剂,优选n,n-二甲基甲酰胺、四氢呋喃或乙腈;所采用的脱氧剂包括但不限于:三苯基膦、三乙基膦、三丁基膦或三环己基膦;反应温度为0℃至150℃,优选温度为50℃至120℃;(11)由化合物lc-3经卤化反应得到化合物lc-4,所采用的溶剂包括但不限于:苯、甲苯、氯仿、正己烷、环己烷、二氯甲烷、1,2-二氯乙烷、甲基叔丁基醚、乙酸乙酯、乙酸丙酯、乙酸丁酯、丙酮、四氢呋喃、乙腈、n,n-二甲基甲酰胺、二甲亚砜或者用这些溶剂任选组成的混合溶剂;所采用的卤化剂包括但不限于:cl2、br2或nbs;反应温度为0℃至120℃,优选温度为20℃至80℃;(12)由化合物ld-1在碱的作用下与二氯亚砜环化得到化合物ld-2,所采用的溶剂包括但不限于:苯、甲苯、氯仿、正己烷、环己烷、二氯甲烷、1,2-二氯乙烷、甲基叔丁基醚、乙酸乙酯、乙酸丙酯、乙酸丁酯、丙酮、四氢呋喃、乙腈、n,n-二甲基甲酰胺、二甲亚砜或者用这些溶剂任选组成的混合溶剂,优选二氯甲烷、甲苯或氯仿;所采用的碱包括但不限于:三乙胺、二异丙基乙胺、1,8-二氮杂环[5,4,0]十一烯-7、碳酸钾、碳酸钠、碳酸氢钾或碳酸氢钠;反应温度为0℃至150℃,优选温度为20℃至80℃;(13)由化合物ld-2经卤化反应得到化合物ld-3,所采用的溶剂包括但不限于:苯、甲苯、氯仿、正己烷、环己烷、二氯甲烷、1,2-二氯乙烷、甲基叔丁基醚、乙酸乙酯、乙酸丙酯、乙酸丁酯、丙酮、四氢呋喃、乙腈、n,n-二甲基甲酰胺、二甲亚砜或者用这些溶剂任选组成的混合溶剂;所采用的卤化剂包括但不限于:cl2、br2或nbs;反应温度为0℃至120℃,优选温度为20℃至80℃;(14)由化合物n-1与化合物n-2在碱的作用下反应得到化合物n-3,所采用的溶剂包括但不限于:苯、甲苯、氯仿、正己烷、环己烷、二氯甲烷、1,2-二氯乙烷、甲基叔丁基醚、乙酸乙酯、乙酸丙酯、乙酸丁酯、甲醇、乙醇、丙酮、四氢呋喃、乙醚、乙腈、n,n-二甲基甲酰胺、二甲亚砜或者用这些溶剂任选组成的混合溶剂,优选丙酮、n,n-二甲基甲酰胺、四氢呋喃、乙腈或二氯甲烷;所采用的碱包括但不限于:三乙胺、二异丙基乙胺、1,8-二氮杂环[5,4,0]十一烯-7、碳酸钾、碳酸钠、碳酸氢钾或碳酸氢钠;反应温度为0℃至150℃,优选温度为50℃至100℃;(15)由化合物n-3与la-7或lb-3在碱的作用下反应得到化合物n-4,所采用的溶剂包括但不限于:苯、甲苯、氯仿、正己烷、环己烷、二氯甲烷、1,2-二氯乙烷、甲基叔丁基醚、乙酸乙酯、乙酸丙酯、乙酸丁酯、丙酮、四氢呋喃、乙腈、n,n-二甲基甲酰胺、二甲亚砜或者用这些溶剂任选组成的混合溶剂,优选丙酮、n,n-二甲基甲酰胺、四氢呋喃或乙腈;所采用的碱包括但不限于:三乙胺、二异丙基乙胺、1,8-二氮杂环[5,4,0]十一烯-7、碳酸钾、碳酸钠、碳酸铯、碳酸氢钾或碳酸氢钠;反应温度为-20℃至100℃,优选温度为室温至50℃;(16)由化合物n-4与nhr9r9’进行还原胺化反应得到化合物ia,所采用的溶剂包括但不限于:乙酸、苯、甲苯、氯仿、正己烷、环己烷、二氯甲烷、1,2-二氯乙烷、甲基叔丁基醚、乙酸乙酯、乙酸丙酯、乙酸丁酯、甲醇、乙醇、四氢呋喃、乙腈、n,n-二甲基甲酰胺、二甲亚砜或者用这些溶剂任选组成的混合溶剂;所采用的还原剂包括但不限于:硼氢化钠、三乙酰氧基硼氢化钠或氰基硼氢化钠;反应温度为-20℃至80℃,优选温度为0℃至50℃;(17)或者,由化合物n-4经还原反应得到化合物n-5,所采用的还原剂包括但不限于:硼氢化钠或硼氢化钾;n-5再经卤化或磺酸酯化得到化合物n-6,所采用的卤化剂包括但不限于:pph3/cbr4、pbr3、pobr3、hbr、pcl3、pocl3、hcl或hi,所采用的磺酸酯化试剂(在碱的作用下)包括但不限于:对甲苯磺酰氯、甲磺酰氯、甲磺酸酐、三氟甲磺酰氯或三氟甲磺酸酐;n-6在碱的作用下与nhr9r9’进行烷基化反应得到化合物(ia),所采用的碱包括但不限于:三乙胺、二异丙基乙胺、1,8-二氮杂环[5,4,0]十一烯-7、碳酸钾、碳酸钠、碳酸氢钾或碳酸氢钠。(18)由化合物n-3与lc-4或ld-3在碱的作用下反应得到化合物n-7,所采用的溶剂包括但不限于:苯、甲苯、氯仿、正己烷、环己烷、二氯甲烷、1,2-二氯乙烷、甲基叔丁基醚、乙酸乙酯、乙酸丙酯、乙酸丁酯、丙酮、四氢呋喃、乙腈、n,n-二甲基甲酰胺、二甲亚砜或者用这些溶剂任选组成的混合溶剂,优选丙酮、n,n-二甲基甲酰胺、四氢呋喃或乙腈;所采用的碱包括但不限于:三乙胺、二异丙基乙胺、1,8-二氮杂环[5,4,0]十一烯-7、碳酸钾、碳酸钠、碳酸铯、碳酸氢钾或碳酸氢钠;反应温度为-20℃至100℃,优选温度为室温至50℃;(19)由化合物n-7与nhr9r9’进行还原胺化反应得到化合物iia,所采用的溶剂包括但不限于:乙酸、苯、甲苯、氯仿、正己烷、环己烷、二氯甲烷、1,2-二氯乙烷、甲基叔丁基醚、乙酸乙酯、乙酸丙酯、乙酸丁酯、甲醇、乙醇、四氢呋喃、乙腈、n,n-二甲基甲酰胺、二甲亚砜或者用这些溶剂任选组成的混合溶剂;所采用的还原剂包括但不限于:硼氢化钠、三乙酰氧基硼氢化钠或氰基硼氢化钠;反应温度为-20℃至80℃,优选温度为0℃至50℃;(20)或者,由化合物n-7经还原反应得到化合物n-8,所采用的还原剂包括但不限于:硼氢化钠或硼氢化钾;n-8再经卤化或磺酸酯化得到化合物n-9,所采用的卤化剂包括但不限于:pph3/cbr4、pbr3、pobr3、hbr、pcl3、pocl3、hcl或hi,所采用的磺酸酯化试剂(在碱的作用下)包括但不限于:对甲苯磺酰氯、甲磺酰氯、甲磺酸酐、三氟甲磺酰氯或三氟甲磺酸酐;n-9在碱的作用下与nhr9r9’进行烷基化反应得到化合物iia:所采用的碱包括但不限于:三乙胺、二异丙基乙胺、1,8-二氮杂环[5,4,0]十一烯-7、碳酸钾、碳酸钠、碳酸氢钾或碳酸氢钠。本发明的式(i)或式(ii)化合物或其药学上可接受的盐、互变异构体、内消旋体、外消旋体、立体异构体、代谢产物、代谢前体、前药或溶剂化物的用量可根据患者年龄、体重、症状和给药途径等而适当改变。对成人而言,在口服给药时,一次给药量的下限是0.1mg(优选1mg),上限是1000mg(优选500mg)。在静脉给药时,一次给药量的下限是0.01mg(优选0.1mg),上限是500mg(优选250mg)。也可根据疾病程度的不同和剂型的不同而偏离此剂量范围。有益效果:与现有技术相比,本发明具有如下优点:(1)本发明的联苯类化合物是新型pd-l1抑制剂,可以显著抑制pd-1和pd-l1蛋白-蛋白相互作用,且活性显著优于已知的pd-l1抑制剂bms-1016。本发明的联苯类化合物可以显著地阻断pd-l1对t细胞的抑制作用,并可阻断肿瘤细胞抑制t细胞增殖和分泌ifn-γ的效应,因而具有增强t细胞抗肿瘤免疫的功效。尤其重要的是,本发明的联苯类化合物在荷瘤动物体内显示了显著的抗肿瘤功效。因此,本发明的联苯类化合物作为pd-l1抑制剂可用于制备肿瘤免疫治疗的药物。(2)本发明人意外地发现,本发明的联苯类化合物可抑制银屑病动物模型中炎症因子的释放,且对银屑病具有显著疗效,这提示本发明的联苯类化合物可用于预防和治疗自身免疫性疾病、器官移植排斥、感染性疾病和炎症性疾病。因此本发明的化合物可以制备免疫调节剂类药物用于预防或治疗肿瘤、自身免疫性疾病、器官移植排斥、感染性疾病和炎症性疾病等。(3)本发明的联苯类化合物的合成路线设计巧妙、简便易行,原料便宜易得,合成工艺安全、环保,易于规模化生产。尤其重要的是,本发明的新型联苯类化合物不仅对pd-l1具有很高的抑制活性,同时还具有非常好的化学稳定性和代谢稳定性,因而具有很好的成药性。与中国专利申请2019102477713公开的一种苯并噁二唑类pd-l1抑制剂相比,本发明的联苯类化合物的化学稳定性和代谢稳定性有了极大的提高,且具有非常好的安全性。因此,本发明的联苯类化合物具有非常好的成药性。(4)本发明的联苯类化合物作为新型pd-l1小分子抑制剂,比现有的pd-1/pd-l1抗体类药物具有成本低廉、易于制备及免疫反应副作用小等优点。其即可单独使用,也可与化疗药或其他抗肿瘤药物联用,有望成为新型肿瘤免疫治疗药物。(5)通过分子对接手段,将本发明的化合物与已报道的hpd-l1的共晶结构(pdb编号:6r3k)进行对接,结果显示,本发明化合物优势结构中的苯并噁二唑杂环可以很好地与hpd-l1蛋白发生相互作用,其中杂环的氧原子与hpd-l1蛋白中125位的精氨酸上的胍基产生关键的氢键相互作用;苯并噁二唑杂环与123位酪氨酸上的苯环产生关键的π-π相互作用,并且苯并噁二唑中的噁二唑环的强吸电子作用会加强π-π相互作用的强度,使该侧链与蛋白结合的强度更高,从而得到与hpd-l1蛋白结合能力更好的分子。通过打分函数打分结果,本发明如化合物9的打分为-11.627,而若将化合物9中的苯并噁二唑替换为苯环所得到的相应化合物的打分为-9.87,其打分显著低于化合物9,且本发明的其他化合物中苯并噁二唑均比替换成苯环得分更高。以上分子对接结果说明本发明化合物中的苯并噁二唑侧链对于化合物活性提升至关重要,即通过引入苯并噁二唑侧链,该类化合物阻断pd-1和pd-l1蛋白-蛋白相互作用的能力显著增加。附图说明图1为联苯类化合物阻断pd-l1抑制人外周血单个核细胞(pbmc)分泌ifn-γ的效应图:空白对照组、激活组、抑制组以及不同浓度的化合物组的人pbmc在48小时后ifn-γ的分泌情况(n=3,**p<0.01,***p<0.001);图2为联苯类化合物阻断肿瘤细胞抑制人t细胞增殖的效应图:空白对照组、激活组、抑制组以及不同浓度的化合物组的t细胞在48小时后的增殖情况;图3为联苯类化合物阻断肿瘤细胞抑制人t细胞分泌ifn-γ的效应图:空白对照组、激活组、抑制组以及不同浓度的化合物组的t细胞在48小时后ifn-γ的分泌情况(n=3,*p<0.05,**p<0.01,***p<0.001)。图4为联苯类化合物阻断肿瘤细胞抑制jurkat细胞分泌il-2的效应图:空白对照组、激活组、抑制组以及不同浓度的化合物组的jurkat细胞在48小时后il-2的分泌情况(n=3,*p<0.05,**p<0.01,***p<0.001vs抑制组);图5为不同浓度的化合物9与hpd-l1蛋白结合的传感图;图6为化合物8影响亚急毒实验小鼠体重变化的效应图;图7为化合物8影响亚急毒实验小鼠体内脏器重量的效应图;图8为化合物8影响亚急毒实验小鼠血浆指标变化的效应图;图9为化合物8影响小鼠黑色素瘤高转移细胞(b16f10)皮下植瘤模型的肿瘤体积随时间变化的效应图;图10为化合物8影响mc38-hpd-l1结肠癌细胞移植c57bl/6-hpd-l1荷瘤模型中肿瘤体积变化的效应图;图11为化合物8影响mc38-hpd-l1结肠癌细胞移植c57bl/6-hpd-l1荷瘤模型中小鼠体重变化的效应图;图12为化合物8影响银屑病模型小鼠体重变化的效应图;图13为化合物8影响银屑病模型pasi打分结果的效应图;图14为化合物8影响银屑病模型小鼠耳厚度的效应图;图15为elisa测定化合物8影响银屑病模型小鼠血浆il-17a含量的效应图;图16为qpcr测定化合物8影响银屑病模型小鼠背部组织炎症相关基因表达的效应图;图17为qpcr测定化合物8影响银屑病模型小鼠耳朵组织炎症相关基因表达的效应图。具体实施方式下面通过实施例具体说明本发明的内容。在本发明中,以下所述的实施例是为了更好的阐述本发明,并不是用来限制本发明的范围。在不背离本发明的精神和范围的前提下,可以对本发明进行各种变化和修饰。实施例1(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((2-氟-[1,1'-联苯]-3-基)甲氧基)苄基)-d-丝氨酸(化合物1)合成路线:化合物xa-2的合成取化合物xa-1(3.71g)溶于浓硫酸(30ml)中,冰浴下滴加发烟硝酸(1.4ml)。约3小时后停止反应。将反应液倒入100ml冰水中,有淡黄色固体析出,抽滤,得淡黄色色固体化合物xa-2(3.0g)。化合物xa-3的合成取化合物xa-2(3.0g)溶于dmso(50ml)中,缓慢加入nan3(1.5g)。约1小时后停止反应。将反应液倒入50ml水中,有固体析出,抽滤,得淡黄绿色固体化合物xa-3(3.1g)。化合物xa-4的合成取化合物xa-3(1.5g)溶于甲苯(20ml)中,油浴加热110℃。约5小时后停止加热,减压蒸除溶剂,得暗黄色固体化合物xa-4(1.2g)。化合物xa-5的合成取化合物xa-4(1.2g)溶于乙醇(20ml)中,加入三苯基膦(1.5g),油浴加热70℃。约1小时后停止加热,减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=15:1)纯化,得淡黄色固体化合物xa-5(883mg)。化合物xa-6的合成取化合物xa-5(883mg)溶于甲醇(3ml)和四氢呋喃(1ml)中,冰浴下缓慢加入硼氢化钠(250mg),约1小时后停止反应,减压蒸除溶剂,加入乙酸乙酯(25ml)稀释,用水(10mlx3)和饱和食盐水(10mlx2)洗涤,无水硫酸钠干燥。减压蒸除溶剂,得淡黄色固体化合物xa-6(890mg)。化合物xa-7的合成取化合物xa-6(890mg)、和三苯基膦(3.2g)溶于无水四氢呋喃(30ml)中,于冰浴下缓慢加入四溴化碳(4g),室温下反应约4小时后停止反应。减压蒸除溶剂,用二氯甲烷(30ml)稀释后硅藻土抽滤,母液经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=10:1)纯化,得淡黄色固体化合物xa-7(752mg)。化合物i-2的合成取化合物i-1(7ml)溶于无水四氢呋喃(150ml)中,-78℃缓慢滴加2n的lda(二异丙基氨基锂)四氢呋喃溶液(36ml),反应1小时后滴加无水n,n-二甲基甲酰胺(5.568ml),约3小时后停止反应,冰浴下向反应液中加入饱和氯化铵水溶液(100ml),用乙酸乙酯(30mlx3)萃取,无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=40:1)纯化,得黄色固体化合物i-2(3.0g)。化合物i-3的合成取化合物i-2(3.0g)、苯硼酸(1.342g)、[1,1’-双(二苯基膦)二茂铁]二氯化钯二氯甲烷络合物(82mg)、无水碳酸钾(2.68g)和18-冠-6-醚(250mg)加入到100ml三颈瓶中,氩气保护,换气三次。室温下加入甲苯(16ml)、无水乙醇(4ml)和水(2ml)。90℃油浴加热。反应约5小时后停止加热,减压蒸除溶剂,用乙酸乙酯25ml稀释,硅藻土抽滤,母液用乙酸乙酯(45mlx3)萃取,饱和食盐水(30mlx2)洗涤,无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=4:1)纯化,得淡黄色油状化合物i-3(2.0g)。化合物i-4的合成取化合物i-3(2.0g)溶于甲醇(5ml)和四氢呋喃(15ml)中,冰浴下缓慢加入硼氢化钠(400mg),约1小时后停止反应,减压蒸除溶剂,加入乙酸乙酯(25ml)稀释,用水(10mlx3)和饱和食盐水(10mlx2)洗涤,无水硫酸钠干燥。减压蒸除溶剂,得淡黄色固体化合物i-4(1.76g)。化合物i-5的合成取化合物i-4(1.76g)和三苯基膦(4.15g)溶于无水四氢呋喃(30ml)中,于冰浴下缓慢加入四溴化碳(5.24g),室温下反应约4小时后停止反应。减压蒸除溶剂,用二氯甲烷(30ml)稀释后硅藻土抽滤,母液经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=10:1)纯化,得淡白色固体化合物i-5(2.02g)。化合物i-6的合成取化合物2,4-二羟基苯甲醛(714mg)溶于无水乙腈(20ml)中,加入无水碳酸氢钠(865mg),并缓慢加入xa-7(1.371g),70℃油浴加热。反应约20小时后停止加热,减压蒸除溶剂,加入乙酸乙酯(10ml)稀释,用水(10mlx3)和饱和食盐水(10mlx2)洗涤,无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/二氯甲烷=2:1)纯化,得灰白色固体化合物i-6(1.1g)。化合物i-7的合成取化合物i-6(200mg)和碳酸铯(377mg)加入n,n-二甲基甲酰胺(4ml)中,室温搅拌15分钟后缓慢加入xa-7(149mg),室温反应约1小时后停止反应。加入水(30ml),用乙酸乙酯(10mlx3)萃取,无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=2:1)纯化,得黄色固体化合物i-7(75mg)。化合物i-8的合成取d-丝氨酸乙酯盐酸盐(80mg)溶于二氯甲烷与甲醇的混合溶剂(3:1,4ml),加入n,n-二异丙基乙胺(dipea,59mg)室温搅拌20分钟,加入化合物i-7(75mg)和三乙酰氧基硼氢化钠(stab,133mg),室温反应12小时后停止反应。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=1:1)纯化,得黄色油状化合物i-8(70mg)。化合物1的合成取化合物i-8(70mg)溶于甲醇与四氢呋喃的混合溶剂(2:1,3ml),加入一水合氢氧化锂(24mg),室温反应5小时后,减压蒸除溶剂,加入水(5ml),用1n盐酸调节ph=5~6,抽滤,得淡黄色固体化合物1(32mg):1hnmr(300mhz,dmso-d6)δ8.14(s,1h),8.05(d,j=9.3hz,1h),7.72(d,j=9.4hz,1h),7.61–7.37(m,7h),7.30(d,j=8.1hz,2h),6.83(d,j=2.3hz,1h),6.69(d,j=8.3hz,1h),5.31(s,2h),5.20(s,2h),3.95(d,j=13.6hz,1h),3.88(d,j=13.5hz,1h),3.54(t,j=5.5hz,2h),2.96(t,j=6.3hz,1h).hrms(esi):m/z544.1869[m+h]+.实施例22-((2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((2-氟-[1,1'-联苯]-3-基)甲氧基)苄基)氨基)乙-1-醇(化合物2)参照实施例1的方法,将实施例1中的d-丝氨酸乙酯盐酸盐替换成乙醇胺,制得化合物2:1hnmr(300mhz,dmso-d6)δ8.16(s,1h),8.10(d,j=9.3hz,1h),7.72(d,j=9.4hz,1h),7.65–7.39(m,8h),7.30(t,j=7.6hz,1h),6.91(d,j=2.3hz,1h),6.79(d,j=8.3hz,1h),5.35(s,2h),5.25(s,3h),4.18(s,2h),3.70(d,j=5.3hz,1h),3.66(d,j=5.3hz,1h),2.97(t,j=5.3hz,2h).hrms(esi):m/z500.1982[m+h]+.实施例3(2-((2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((2-氟-[1,1'-联苯]-3-基)甲氧基)苄基)氨基)乙基)乙酰胺(化合物3)参照实施例1的方法,将实施例1中的d-丝氨酸乙酯盐酸盐替换成n-乙酰基乙二胺,制得化合物3:1hnmr(300mhz,dmso-d6)δ8.08(d,j=8.8hz,2h),7.96(s,1h),7.67(d,j=9.5hz,1h),7.60–7.39(m,6h),7.38–7.22(m,2h),6.85(d,j=2.4hz,1h),6.72(dd,j=8.4,2.3hz,1h),5.32(s,2h),5.21(s,2h),3.96(s,2h),3.24(d,j=6.0hz,2h),2.77(t,j=6.5hz,2h),1.77(s,3h).hrms(esi):m/z541.2245[m+h]+.实施例4(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-5-氯-4-((2-氯-[1,1'-联苯]-3-基)甲氧基)苄基)-d-丝氨酸乙酯盐酸盐(化合物4)合成路线:化合物h-1的合成取化合物2,4-二羟基苯甲醛(5g)溶于氯仿(100ml)中,加入n-氯代丁二酰亚胺(ncs,5.32g),油浴加热70℃。7小时后停止加热,减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=15:1)纯化,得白色固体化合物h-1(2.6g)。化合物a-2的合成取化合物a-1(6.15g)溶于无水四氢呋喃(50ml)中,冰盐浴下缓慢滴加硼烷四氢呋喃复合物(60ml),室温下反应约5小时停止反应,冰浴下加水淬灭反应。用乙酸乙酯(50mlx4)萃取,和饱和食盐水(30mlx2)洗涤,无水硫酸钠干燥。减压蒸除溶剂,得白色固体化合物a-2(5.18g)。化合物a-3的合成取化合物a-2(2.5g)、苯硼酸(1.6g)、[1,1’-双(二苯基膦)二茂铁]二氯化钯二氯甲烷络合物(180mg)、无水醋酸钾(2.1g)和18-冠-6-醚(581mg)加入到100ml三颈瓶中,氩气保护,换气三次。室温下加入甲苯(21ml)、无水乙醇(7ml)和水(3.5ml)。90℃油浴加热,反应约5小时后停止加热,减压蒸除溶剂,用乙酸乙酯25ml稀释,硅藻土抽滤,母液用乙酸乙酯(45mlx3)萃取,饱和食盐水(30mlx2)洗涤,无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=4:1)纯化,得无色油状化合物a-3(2.2g)。化合物a-4的合成取化合物a-3(2.2g),三苯基膦(5.3g),溶于无水四氢呋喃(30ml)中,于冰浴下缓慢加入四溴化碳(6.6g),室温下反应约4小时后停止反应。减压蒸除溶剂,用二氯甲烷(25ml)稀释后硅藻土抽滤,母液经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=5:1)纯化,得无色油状化合物a-4(2.4g)。化合物ii-1的合成取化合物a-4(600mg)溶于无水乙腈(25ml)中,加入无水碳酸氢钠(587mg),缓慢加入h-1(997mg)的n,n-二甲基甲酰胺溶液(2ml),70℃油浴加热。反应约6小时后停止加热,减压蒸除溶剂,加入乙酸乙酯(15ml)稀释,用水(10mlx3)和饱和食盐水(10mlx2)洗涤,无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=5:1)纯化,得白色固体化合物ii-1(590mg)。化合物4的合成参照实施例1的方法,将实施例1中的i-6替换成ii-1,不经水解直接制得化合物4的游离碱,再将化合物4的游离碱加入盐酸乙醇溶液中,搅拌过夜,抽滤,得白色固体化合物4:1hnmr(300mhz,dmso-d6)δ9.42(s,2h),8.17(s,1h),8.10(d,j=9.3hz,1h),7.74–7.60(m,3h),7.53–7.30(m,7h),7.14(s,1h),5.62(s,1h),5.41(s,2h),5.37(s,2h),4.26(s,2h),4.16–4.02(m,3h),3.89(q,j=11.7hz,2h),1.15(t,j=7.1hz,3h).hrms(esi):m/z622.1500[m+h]+.实施例5(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-5-氯-4-((2-氯-[1,1'-联苯]-3-基)甲氧基)苄基)-d-丝氨酸(化合物5)以化合物4的游离碱为关键中间体,参照实施例1的方法水解制得化合物5:1hnmr(300mhz,dmso-d6)δ8.14(s,1h),8.06(d,j=9.3hz,1h),7.70(d,j=9.4hz,1h),7.62(s,1h),7.56–7.31(m,8h),7.11(s,1h),5.39(s,2h),5.33(s,2h),4.00(s,3h),3.79–3.55(m,3h),3.19(t,j=5.5hz,1h).hrms(esi):m/z594.1192[m+h]+.实施例62-((2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-5-氯-4-((2-氯-[1,1'-联苯]-3-基)甲氧基)苄基)氨基)乙-1-醇(化合物6)参照实施例5的方法,将实施例5中的d-丝氨酸乙酯盐酸盐替换成乙醇胺,制得化合物6:1hnmr(300mhz,dmso-d6)δ8.13–8.03(m,2h),7.71–7.59(m,2h),7.53–7.31(m,8h),7.07(s,1h),5.36(s,2h),5.31(s,2h),3.72(s,2h),3.47(t,j=5.8hz,2h),2.58(t,j=5.7hz,2h).hrms(esi):m/z550.1293[m+h]+.实施例7(s)-2-((2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-5-氯-4-((2-氯-[1,1'-联苯]-3-基)甲氧基)苄基)氨基)-2-苯基乙酸(化合物7)参照实施例5的方法,将实施例5中的d-丝氨酸乙酯盐酸盐替换成l-苯甘氨酸甲酯盐酸盐,制得化合物7:1hnmr(300mhz,dmso-d6)δ8.09–7.94(m,2h),7.68–7.53(m,2h),7.54–7.27(m,10h),7.23–7.11(m,2h),7.11–6.99(m,2h),5.30(s,4h),3.79(s,1h),3.57(s,2h).hrms(esi):m/z640.1392[m+h]+.实施例8(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((2-溴-[1,1'-联苯]-3-基)甲氧基)-5-氯苄基乙基)-d-丝氨酸乙酯盐酸盐(化合物8)合成路线:化合物b-2的合成取化合物b-1(4.2g,2.8ml)溶于水(10ml)中,冰盐浴下缓慢加入盐酸(6ml),搅拌5分钟,缓慢滴加亚硝酸钠(1.88g)的水溶液,20分钟加毕,冰盐浴下搅拌30分钟后,向反应液中缓慢滴加碘化钾(7.5g)的水溶液,室温下反应8小时后停止反应。加入硫代硫酸钠的饱和水溶液(35ml),用乙酸乙酯(50mlx3)萃取,饱和食盐水(25mlx3)洗涤,无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚)纯化,得淡黄色油状化合物b-2(5.2g)。化合物b-3的合成取苯硼酸(1.08g),四(三苯基膦)钯(465mg),无水碳酸钠(1.28g)置于三颈瓶中,氩气保护。将化合物b-2(2.39g)溶于甲苯(8ml)中,加入反应瓶,加入甲苯(22ml),水(3ml),80℃油浴加热。反应约4小时后停止加热,减压蒸除溶剂,用乙酸乙酯(15ml)稀释,硅藻土抽滤,母液经硅胶柱层析(洗脱剂:石油醚)纯化,得淡黄色油状化合物b-3(1.78g)。化合物b-4的合成取化合物b-3(1.6g),溶于四氯化碳(50ml)中,加入n-溴代丁二酰亚胺(nbs,1.21g),分批加入过氧化苯甲酰(bpo,10mg),90℃油浴加热。反应约6小时后停止加热,减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚)纯化,得无色油状化合物b-4(1.43g)。化合物iii-1的合成取实施例4中化合物h-1(405mg)溶于无水乙腈(25ml)中,加入无水碳酸氢钠(587mg),缓慢加入b-4(765mg)的n,n-二甲基甲酰胺溶液(2ml),70℃油浴加热。反应约6小时后停止加热,减压蒸除溶剂,加入乙酸乙酯(15ml)稀释,用水(10mlx3)和饱和食盐水(10mlx2)洗涤,无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=5:1)纯化,得淡黄色固体化合物iii-1(300mg)。化合物8的合成参照实施例1的方法,将实施例1中的i-6替换成iii-1,不经水解直接制得化合物8的游离碱,再将化合物8的游离碱加入盐酸乙醇溶液中,搅拌过夜,抽滤,得化合物8:1hnmr(300mhz,dmso-d6)δ9.46(s,2h),8.18(s,1h),8.10(d,j=9.3hz,1h),7.77–7.65(m,2h),7.61(dd,j=7.6,1.8hz,1h),7.54–7.29(m,7h),7.12(s,1h),5.63(s,1h),5.41(s,2h),5.33(s,2h),4.26(s,2h),4.19–4.01(m,3h),4.01–3.77(m,2h),1.15(t,j=7.1hz,3h).hrms(esi):m/z668.0984[m+h]+.实施例9(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((2-溴-[1,1'-联苯]-3-基)甲氧基)-5-氯苄基)-d-丝氨酸(化合物9)以化合物8的游离碱为关键中间体,参照实施例1的方法进行水解制得化合物9:1hnmr(300mhz,dmso-d6)δ8.14(s,1h),8.06(d,j=9.3hz,1h),7.71(d,j=9.3hz,1h),7.67–7.56(m,1h),7.56–7.26(m,8h),7.08(s,1h),5.39(s,2h),5.29(s,2h),3.98(s,2h),3.64(qd,j=11.0,5.6hz,2h),3.16(t,j=5.5hz,1h).hrms(esi):m/z640.0658[m+h]+.实施例10(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((2-溴-[1,1'-联苯]-3-基)甲氧基)-5-氯苄基)-l-丝氨酸(化合物10)参照实施例9的方法,将实施例9中的d-丝氨酸乙酯盐酸盐替换成l-丝氨酸乙酯盐酸盐,制得化合物10:1hnmr(300mhz,dmso-d6)δ8.10(s,1h),8.05(d,j=9.2hz,1h),7.72(d,j=9.4hz,1h),7.61(d,j=7.7hz,1h),7.53–7.26(m,8h),7.03(s,1h),5.35(s,2h),5.26(s,2h),3.68(s,2h),3.43(q,j=5.7hz,1h),3.21(q,j=9.9hz,1h),2.62(dd,j=9.5,5.5hz,1h).hrms(esi):m/z640.0659[m+h]+.实施例112-((2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((2-溴-[1,1'-联苯]-3-基)甲氧基)-5-氯苄基)氨基)乙-1-醇(化合物11)参照实施例9的方法,将实施例5中的d-丝氨酸乙酯盐酸盐替换成乙醇胺,制得化合物11:1hnmr(300mhz,dmso-d6)δ8.18–8.00(m,2h),7.77–7.55(m,2h),7.56–7.22(m,8h),7.05(s,1h),5.36(s,2h),5.27(s,2h),3.72(s,2h),3.47(t,j=5.7hz,2h),2.57(t,j=5.7hz,2h).hrms(esi):m/z596.0769[m+h]+.实施例12(s)-2-((2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((2-溴-[1,1'-联苯]-3-基)甲氧基)-5-氯苄基)氨基)-2-苯乙酸甲酯(化合物12)参照实施例8的方法,将实施例5中的d-丝氨酸乙酯盐酸盐替换成l-苯甘氨酸甲酯盐酸盐,制得化合物12:1hnmr(300mhz,dmso-d6)δ8.04(d,j=9.3hz,1h),8.00(s,1h),7.66–7.53(m,2h),7.53–7.19(m,12h),7.04(s,1h),5.33(s,2h),5.28(s,2h),4.39(d,j=8.1hz,1h),3.65(d,j=6.4hz,2h),3.54(s,3h),2.98(q,j=7.2hz,1h).hrms(esi):m/z700.1032[m+h]+.实施例13(s)-2-((2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((2-溴-[1,1'-联苯]-3-基)甲氧基)-5-氯苄基)氨基)-2-苯乙酸(化合物13)以化合物12为关键中间体,参照实施例1的方法水解,制得化合物13:1hnmr(300mhz,dmso-d6)δ8.11–7.94(m,2h),7.69–7.56(m,2h),7.54–7.32(m,10h),7.27–7.11(m,3h),7.04(s,1h),5.32(s,2h),5.28(s,2h),4.02(s,1h),3.67(s,2h).hrms(esi):m/z686.0867[m+h]+.实施例14(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((3-(2,3-二氢苯并[b][1,4]二噁英-6-基)-2-(三氟甲基)苄基)氧基)苄基)-d-丝氨酸(化合物14)合成路线:化合物c-2的合成取化合物c-1(10g)溶于甲醇(75ml)中,于室温下缓慢滴加浓硫酸(20ml),约15分钟加毕,85℃油浴加热。反应约15小时后停止加热,减压蒸除溶剂,用乙酸乙酯(60mlx4),饱和碳酸钠(50mlx3)和饱和食盐水(100mlx4)萃取,无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=50:1)纯化,得淡黄色油状化合物c-2(9.0g)。化合物c-3的合成取化合物c-2(9.0g)溶于水(50ml)中,冰盐浴下缓慢加入盐酸(20ml),搅拌10分钟,缓慢滴加亚硝酸钠(3.2g)的水溶液,约30分钟加毕,冰盐浴下搅拌约45分钟后,向反应液中缓慢滴加碘化钾(8.4g)的水溶液,室温下反应8小时后停止反应。用硫代硫酸钠的饱和水溶液(60ml),乙酸乙酯(100mlx3)和饱和食盐水(80mlx3)萃取,无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=50:1)纯化,得淡黄色油状化合物c-3(11g)。化合物c-4的合成取化合物c-3(8.645g)、碘化亚铜(10.6g)和氟磺酰基二氟乙酸甲酯(8.031g)溶于n,n-二甲基甲酰胺(60ml)中,66℃油浴加热。反应约7小时后停止反应,加水150ml,用乙酸乙酯(50mlx4)萃取,饱和食盐水(50ml),减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/二氯甲烷=30:1)纯化,得无色油状化合物c-4(7.120g)。化合物c-5的合成取化合物c-4(7.120g)溶于无水二氯甲烷(100ml),-78℃滴加二异丁基氢化铝(1.5m,40ml),加毕自然升至室温。反应两小时后停止反应,冰浴下缓慢滴加饱和氯化铵溶液(50ml),待反应淬灭完毕,二氯甲烷(40mlx3)萃取,无水硫酸钠干燥。减压蒸除溶剂,得白色透明固体化合物c-5(5.3g)。化合物c-6的合成取化合物c-5(2.55g)、苯并二氧六环-4-硼酸酯(2.88g)、[1,1’-双(二苯基膦)二茂铁]二氯化钯二氯甲烷络合物(82mg)、无水碳酸钾(2.68g)和18-冠-6-醚(250mg)加入到50ml三颈瓶中,氩气保护,换气三次。室温下加入甲苯(18ml)、无水乙醇(6ml)和水(3ml),90℃油浴加热。反应约5小时后停止加热,减压蒸除溶剂,用乙酸乙酯25ml稀释,硅藻土抽滤,母液用乙酸乙酯(40mlx3)和饱和食盐水(40mlx3)萃取,无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/二氯甲烷=30:1)纯化,得无色油状化合物c-6(2.85g)。化合物c-7的合成取化合物c-6(2.85g)和三苯基膦(4.82g)溶于无水四氢呋喃(30ml)中,于冰浴下缓慢加入四溴化碳(6.1g),室温下反应约4小时后停止反应。减压蒸除溶剂,用二氯甲烷(25ml)稀释后硅藻土抽滤,母液经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=5:1)纯化,得无色油状化合物c-7(2.824g)。化合物iv-1的合成取化合物2,4-二羟基苯甲醛(342mg)溶于无水乙腈(25ml)中,加入无水碳酸氢钠(420mg),缓慢加入c-7(840mg)的n,n-二甲基甲酰胺溶液(2ml),70℃油浴加热。反应约6小时后停止加热,减压蒸除溶剂,加入乙酸乙酯(15ml)稀释,用水(10mlx3)和饱和食盐水(10mlx2)洗涤,无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=5:1)纯化,得淡黄色固体化合物iv-1(710mg)。化合物14的合成参照实施例1的方法,将实施例1中的i-6替换成iv-1,制得化合物14:1hnmr(300mhz,dmso-d6)δ8.12(s,1h),8.05(d,j=9.3hz,1h),7.78–7.67(m,2h),7.62(t,j=7.7hz,1h),7.37–7.22(m,2h),6.89(d,j=8.2hz,1h),6.84–6.74(m,2h),6.70(dd,j=8.2,2.2hz,1h),6.62(d,j=8.1hz,1h),5.29(s,2h),5.25(s,2h),4.27(s,4h),3.89(s,2h),3.58–3.49(m,2h),2.94(t,j=6.3hz,1h).hrms(esi):m/z674.1718[m+h]+.实施例152-((2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((3-(2,3-二氢苯并[b][1,4]二噁英-6-基)-2-(三氟甲基)苄基)氧基)苄基)氨基)乙-1-醇(化合物15)参照实施例14的方法,将实施例14中的d-丝氨酸乙酯盐酸盐替换成乙醇胺,制得化合物15:1hnmr(300mhz,dmso-d6)δ8.09(d,j=8.5hz,2h),7.82–7.54(m,3h),7.31(d,j=7.6hz,1h),7.25(d,j=8.3hz,1h),6.90(d,j=8.2hz,1h),6.78(s,2h),6.72(d,j=8.1hz,1h),6.61(d,j=8.3hz,1h),5.30(s,2h),5.25(s,2h),4.29(s,4h),3.73(s,2h),3.47(t,j=5.7hz,2h),3.37(t,j=6.1hz,1h),2.59(t,j=5.7hz,2h).hrms(esi):m/z608.2004[m+h]+.实施例16甲基(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((3-(2,3-二氢苯并[b][1,4]二噁英-6-基)-2-(三氟甲基)苄基)氧基)苄基)-l-亮氨酸甲酯盐酸盐(化合物16)参照实施例14的方法,将实施例14中的d-丝氨酸乙酯盐酸盐替换成l-亮氨酸甲酯盐酸盐,不经水解直接制得化合物16的游离碱,再将化合物4的游离碱加入盐酸乙醇溶液中,搅拌过夜,抽滤,得化合物16:1hnmr(300mhz,dmso-d6)δ9.44(d,j=48.7hz,2h),8.17(s,1h),8.11(d,j=9.3hz,1h),7.77–7.67(m,2h),7.63(t,j=7.6hz,1h),7.48(d,j=8.5hz,1h),7.32(d,j=7.5hz,1h),6.97–6.80(m,2h),6.79–6.61(m,3h),5.31(d,j=7.5hz,4h),4.27(s,4h),4.20(s,2h),3.95(s,1h),3.66(s,3h),1.79–1.66(m,2h),1.66–1.53(m,1h),0.79(t,j=7.0hz,6h).hrms(esi):m/z692.2573[m+na]+.实施例172-((2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((2-甲基-[1,1'-联苯]-3-基)甲氧基)苄基)氨基)乙-1-醇(化合物17)参照实施例1的方法,将实施例1中的i-4替换为2-甲基-3-苯基苯甲醇,将d-丝氨酸乙酯盐酸盐替换成乙醇胺,制得化合物17:1hnmr(300mhz,dmso-d6)δ8.17(s,1h),8.11(d,j=9.3hz,1h),7.73(d,j=9.3hz,1h),7.52–7.35(m,5h),7.34–7.26(m,2h),7.27–7.15(m,2h),6.92(d,j=2.3hz,1h),6.79(d,j=8.3hz,1h),5.36(s,2h),5.24(t,j=5.1hz,1h),5.19(s,2h),4.19(s,2h),3.68(q,j=5.3hz,2h),2.98(t,j=5.2hz,3h),2.18(s,3h).hrms(esi):m/z496.2238[m+h]+.实施例183-((2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((2-甲基-[1,1'-联苯]-3-基)甲氧基)苄基)氨基)丙酸(化合物18)参照实施例17的方法,将实施例17中的乙醇胺替换成3-氨基丙酸甲酯,经水解后制得化合物18:1hnmr(300mhz,dmso-d6)δ8.09(s,1h),8.07(d,j=9.6hz,1h),7.70(d,j=9.3hz,1h),7.49–7.34(m,4h),7.32–7.19(m,4h),7.16(d,j=7.5hz,1h),6.80(d,j=2.3hz,1h),6.66(dd,j=8.3,2.2hz,1h),5.29(s,2h),5.11(s,2h),3.77(s,2h),2.70(t,j=6.4hz,2h),2.24–2.08(m,5h).hrms(esi):m/z524.2184[m+h]+.实施例19(s)-2-((2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((2-甲基-[1,1'-联苯]-3-基)甲氧基)苄基)氨基)-2-苯乙酸(化合物19)参照实施例18的方法,将实施例18中的3-氨基丙酸甲酯替换成l-苯甘氨酸甲酯盐酸盐,再经过水解,制得化合物19:1hnmr(300mhz,dmso-d6)δ8.04(d,j=9.8hz,2h),7.60(d,j=9.3hz,1h),7.48–7.37(m,5h),7.37–7.21(m,7h),7.16(d,j=7.7hz,1h),6.83(d,j=2.3hz,1h),6.72(d,j=8.6hz,1h),5.25(s,2h),5.15(s,2h),4.41(s,1h),3.90(s,2h),2.16(s,3h).hrms(esi):m/z586.2336[m+h]+.实施例20(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((2-甲基-[1,1'-联苯]-3-基)甲氧基)苄基)-d-丝氨酸(化合物20)参照实施例18的方法,将实施例18中的3-氨基丙酸甲酯替换成d-丝氨酸乙酯盐酸盐,再经过水解,制得化合物20:1hnmr(300mhz,dmso-d6)δ8.18(s,1h),8.07(d,j=9.3hz,1h),7.73(d,j=9.3hz,1h),7.50–7.41(m,3h),7.38(t,j=7.9hz,2h),7.33–7.27(m,2h),7.23(d,j=7.4hz,1h),7.18(d,j=7.5hz,1h),6.88(d,j=2.2hz,1h),6.74(dd,j=8.4,2.3hz,1h),5.34(s,2h),5.16(s,2h),4.19–4.03(m,2h),3.76(dd,j=11.3,4.6hz,1h),3.67(d,j=6.6hz,1h),3.21(t,j=5.6hz,1h),2.18(s,3h).hrms(esi):m/z562.1924[m+na]+.实施例21(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-5-氯-4-((2-甲基-[1,1'-联苯]-3-基)甲氧基)苄基)-d-丝氨酸(化合物21)合成路线:化合物k-2的合成取化合物k-1(5.95g)和三苯基膦(15.7g)溶于无水四氢呋喃(100ml)中,于冰浴下缓慢加入四溴化碳(19.9g),室温下反应约4小时后停止反应。减压蒸除溶剂,用二氯甲烷(100ml)稀释后硅藻土抽滤,母液经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=10:1)纯化,得淡白色固体化合物k-2(6.5g)。化合物k-3的合成取实施例4中化合物h-1(1.7g)溶于无水乙腈(20ml)中,加入无水碳酸氢钠(1.6g),并缓慢加入k-2(3.9g)的n,n-二甲基甲酰胺溶液(3ml),70℃油浴加热。反应约20小时后停止加热,减压蒸除溶剂,加入乙酸乙酯(10ml)稀释,用水(10mlx3)和饱和食盐水(10mlx2)洗涤,无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/二氯甲烷=2:1)纯化,得灰白色固体化合物k-3(1.9g)。化合物21的合成参照实施例1的方法,将实施例1中的i-6替换为k-3,制得化合物21:1hnmr(300mhz,dmso-d6)δ8.15(s,1h),8.08(d,j=9.2hz,1h),7.73(d,j=9.4hz,1h),7.54–7.43(m,4h),7.39(d,j=6.9hz,1h),7.32(s,1h),7.29(s,1h),7.25–7.10(m,3h),5.40(s,2h),5.27(s,2h),3.93(s,2h),3.59(d,j=5.7hz,2h),3.06(s,1h),2.22(s,3h).hrms(esi):m/z574.1753[m+h]+.实施例22(s)-2-((2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-5-氯-4-((2-甲基-[1,1'-联苯]-3-基)甲氧基)苄基)氨基)-2-苯基乙酸甲酯(化合物22)参照实施例21的方法,将实施例21中的d-丝氨酸乙酯盐酸盐替换为l-苯甘氨酸甲酯盐酸盐,不经水解直接制得化合物22:1hnmr(300mhz,dmso-d6)δ8.05(d,j=9.3hz,1h),8.00(s,1h),7.58(d,j=9.3hz,1h),7.50–7.41(m,3h),7.40–7.33(m,4h),7.32–7.15(m,7h),7.10(s,1h),5.34(s,2h),5.25(s,2h),4.38(s,1h),3.65(s,2h),3.53(s,3h),2.21(s,3h).hrms(esi):m/z634.2102[m+h]+.实施例232-((2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-5-氯-4-((2-甲基-[1,1'-联苯]-3-基)甲氧基)苄基)氨基)乙-1-醇(化合物23)参照实施例21的方法,将实施例21中的d-丝氨酸乙酯盐酸盐替换为乙醇胺,制得化合物23:1hnmr(300mhz,dmso-d6)δ8.13–8.06(m,2h),7.66(d,j=9.5hz,1h),7.51–7.42(m,3h),7.41–7.35(m,2h),7.33–7.26(m,2h),7.25–7.13(m,2h),7.11(s,1h),5.37(s,2h),5.24(s,2h),3.71(s,2h),3.46(t,j=5.7hz,2h),2.57(t,j=5.7hz,2h),2.21(s,3h).hrms(esi):m/z530.1844[m+h]+.实施例24(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((2-甲基-[1,1'-联苯]-3-基)甲氧基)-5-硝基苄基)-d-丝氨酸(化合物24)合成路线:化合物d-1的合成取化合物2,4-二羟基苯甲醛(1.3g)溶于无水乙腈(20ml)中,加入无水碳酸氢钠(1.6g),并缓慢加入g-2(2.6g)的n,n-二甲基甲酰胺溶液(3ml),70℃油浴加热。反应约20小时后停止加热,减压蒸除溶剂,加入乙酸乙酯(10ml)稀释,用水(10mlx3)和饱和食盐水(10mlx2)洗涤,无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/二氯甲烷=2:1)纯化,得灰白色固体化合物d-1(3.0g)。化合物v-1的合成取化合物d-1(200mg)溶于冰醋酸(2ml)中,冰浴下加入发烟硝酸(0.14ml),室温反应5小时后停止反应,将反应液倒入冰水中,有黄色固体析出,抽滤,得黄色固体,用乙醇(4ml)打浆得黄色固体化合物v-1(203mg)。化合物24的合成参照实施例1的方法,将实施例1中的i-6替换为v-1,制得化合物24:1hnmr(300mhz,dmso-d6)δ8.17(s,1h),8.13(s,1h),8.10(d,j=9.3hz,1h),7.71(d,j=9.4hz,1h),7.52–7.41(m,3h),7.37(t,j=7.1hz,1h),7.32–7.25(m,2h),7.25–7.12(m,3h),5.52(s,2h),5.39(s,2h),4.00(d,j=14.2hz,1h),3.92(d,j=14.3hz,1h),3.74–3.65(m,1h),3.65–3.56(m,1h),3.21(d,j=5.3hz,1h),2.20(s,3h).ms(esi):m/z585.2[m+h]+.实施例25(2r,4r)-1-(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-5-氯-4-((2-氯-[1,1'-联苯]-3-基)甲氧基)苄基)-4-羟基脯氨酸(化合物25)参照实施例5的方法,将实施例5中的d-丝氨酸乙酯盐酸盐替换成(2r,4r)-4-羟基吡咯烷-2-羧酸甲酯盐酸盐,制得化合物25:ms(esi):m/z620.5[m+h]+.实施例26(2r,4r)-1-(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-5-氯-4-((2-溴-[1,1'-联苯]-3-基)甲氧基)苄基)-4-羟基脯氨酸(化合物26)合成路线:化合物j-1的合成取化合物iii-1(410mg)和碳酸铯(478mg)加入n,n-二甲基甲酰胺(4ml)中,室温搅拌15分钟后缓慢加入xa-7(209mg),室温反应约1小时后停止反应。加入水(30ml),用乙酸乙酯(10mlx3)萃取,无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=2:1)纯化,得白色固体化合物j-1(538mg)。化合物j-2的合成取化合物j-1(300mg)溶于无水乙醇(10ml)中,冰浴下加入硼氢化钠(11mg),反应约3小时后停止反应,加入水(10ml),用乙酸乙酯(10mlx3)萃取,无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=1:1)纯化,得白色固体化合物j-2(280mg)。化合物j-3的合成取化合物j-2(280mg)和三苯基膦(262mg)溶于无水四氢呋喃(10ml)中,于冰浴下缓慢加入四溴化碳(336mg),室温下反应约4小时后停止反应。减压蒸除溶剂,用二氯甲烷(10ml)稀释后硅藻土抽滤,母液经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=2:1)纯化,得淡黄色固体化合物j-3(250mg)。化合物j-4的合成取(2r,4r)-4-羟基吡咯烷-2-羧酸甲酯盐酸盐(55mg)溶于n,n-二甲基甲酰胺(5ml),加入无水碳酸钾(80mg),室温搅拌20分钟,加入化合物g-5(125mg),70℃油浴加热。反应12小时后停止反应。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=1:1)纯化,得透明油状化合物j-4(93mg)。化合物26的合成取化合物j-4(50mg)溶于甲醇与四氢呋喃的混合溶剂(2:1,3ml),加入一水合氢氧化锂(6mg),室温反应5小时后,减压蒸除溶剂,加入水(5ml),用1n盐酸调节ph=5~6,抽滤,固体用乙醚打浆,得白色固体化合物26:1hnmr(300mhz,dmso-d6)δ8.16(s,1h),8.07(d,j=9.3hz,1h),7.70(d,j=9.4hz,1h),7.67–7.54(m,2h),7.54–7.28(m,7h),7.13(s,1h),5.42(s,2h),5.31(s,2h),4.36–4.21(m,2h),4.12(d,j=13.3hz,1h),3.90(d,j=8.4hz,1h),3.23(d,j=11.0hz,1h),3.05(dd,j=11.3,4.5hz,1h),2.44(d,j=13.0hz,1h),2.03–1.84(m,1h).ms(esi):m/z664.2[m+h]+.实施例27(s)-1-(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-5-氯-4-((2-溴-[1,1'-联苯]-3-基)甲氧基)苄基)哌啶-2-羧酸(化合物27)参照实施例26的方法,将实施例26中的(2r,4r)-4-羟基吡咯烷-2-羧酸甲酯盐酸盐替换成(s)-哌啶-2-甲酸甲酯盐酸盐,制得化合物27:1hnmr(300mhz,dmso-d6)δ8.20–8.04(m,2h),7.72–7.58(m,2h),7.55–7.27(m,8h),7.07(s,1h),5.38(s,2h),5.29(s,2h),3.83(d,j=14.0hz,1h),3.69(d,j=13.9hz,1h),3.22(t,j=5.7hz,1h),3.03–2.86(m,1h),2.43–2.27(m,1h),1.78(s,2h),1.50(s,3h),0.93–0.76(m,1h).ms(esi):m/z662.8[m+h]+.实施例28(2-(苯并[c][1,2,5]噁二唑-4-基甲氧基)-5-氯-4-((2-氯-[1,1'-联苯]-3-基)甲氧基)苄基)-d-丝氨酸(化合物28)合成路线:化合物xb-2的合成取化合物xb-1(1.0g)溶于koh(600mg)的乙醇溶液(10ml)中,冰浴下滴加14%的次氯酸钠水溶液(10ml),反应2小时后,向反应液中加入水(50ml),有大量黄色固体析出,减压抽滤,真空干燥后得黄色固体化合物xb-2(900mg)。化合物xb-3的合成取化合物xb-2(890mg)溶于乙醇(20ml)中,加入三苯基膦(1.5g),油浴70℃反应2小时后停止反应,减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=20:1)纯化,得淡黄色油状化合物xb-3(480mg)。化合物xb-4的合成取化合物xb-3(416mg)溶于二氯甲烷(12ml),加入n-溴代琥珀酰亚胺(nbs,552mg)和偶氮二异丁腈(aibn,26mg),加热回流反应,48小时后停止反应,减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=100:1)纯化,得淡黄色固体化合物xb-4(420mg)。化合物vi-1的合成取化合物ii-1(300mg),碳酸铯(520mg)加入n,n-二甲基甲酰胺(5ml)中,室温搅拌15分钟后缓慢加入xb-4(257mg),室温反应约1小时后停止反应。加入水(30ml),用乙酸乙酯(10mlx3)萃取,无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=2:1)纯化,得黄色固体化合物v-1(390mg)。化合物28的合成参照实施例1的方法,将实施例1中的i-7替换成vi-1,制得化合物28:1hnmr(300mhz,dmso-d6)δ8.00(d,j=9.0hz,1h),7.77(d,j=6.9hz,1h),7.69–7.54(m,3h),7.54–7.29(m,7h),7.18(d,j=14.6hz,1h),5.63(s,2h),5.36(s,2h),4.09(s,2h),3.73(dd,j=30.6,11.5hz,2h),3.50(s,1h).ms(esi):m/z592.1[m-h]-.实施例292-((2-(苯并[c][1,2,5]噁二唑-4-基甲氧基)-5-氯-4-((2-氯-[1,1'-联苯]-3-基)甲氧基)苄基)氨基)乙-1-醇(化合物29)参照实施例28的方法,将实施例28中的d-丝氨酸乙酯盐酸盐替换成乙醇胺,制得化合物29:1hnmr(300mhz,dmso-d6)δ8.03(d,j=8.9hz,1h),7.75–7.57(m,3h),7.55–7.35(m,8h),7.19(s,1h),5.62(s,2h),5.36(s,2h),3.82(s,2h),3.48(t,j=5.6hz,2h),2.66(t,j=5.6hz,2h).ms(esi):m/z550.2[m+h]+.实施例30(2-(苯并[c][1,2,5]噻二唑-5-基甲氧基)-5-氯-4-((3-(2,3-二氢苯并[b][1,4]二噁英-6-基)基)-2-甲基苄基)氧基)苄基)-d-丝氨酸乙酯盐酸盐(化合物30)合成路线:化合物xc-2的合成取化合物xc-1(2.26g)溶于dcm(60ml)中,加入三乙胺(et3n,10.3ml),室温搅拌至固体完全溶解,冰浴下将二氯亚砜(socl2,2.7ml)缓慢滴入,油浴加热45℃反应7小时后停止加热,减压蒸除溶剂,加入水(70ml),用1n盐酸调ph至1,用乙酸乙酯(30mlx3)萃取,无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=10:1)纯化,得黄色油状化合物xc-2(2.4g)。化合物xc-3的合成取化合物xc-2(1.54g)溶于二氯甲烷(50ml),加入n-溴代琥珀酰亚胺(nbs,2.2g)和偶氮二异丁腈(aibn,85mg),加热回流反应,12小时后停止反应,减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=100:1)纯化,得白色固体化合物xc-3(977mg)。化合物e-2的合成取化合物e-1(5g)溶于甲醇(60ml)中,于室温下缓慢滴加浓硫酸(10ml),约15分钟加毕,85℃油浴加热。反应约10小时后停止加热,减压蒸除溶剂,用乙酸乙酯(50mlx3)萃取,饱和碳酸钠(30mlx3)和饱和食盐水(25mlx2)洗涤,无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=50:1)纯化,得淡黄色油状化合物e-2(5.1g)。化合物e-3的合成取化合物e-2(3g)、苯并二氧六环-4-硼酸酯(3.8g)、[1,1’-双(二苯基膦)二茂铁]二氯化钯二氯甲烷络合物(530mg)、无水碳酸钾(3.5g)和18-冠-6-醚(345mg),加入到100ml三颈瓶中,氩气保护,换气三次。室温下加入甲苯(21ml)、无水乙醇(7ml)和水(3.5ml)。90℃油浴加热,反应约5小时后停止加热,减压蒸除溶剂,用乙酸乙酯25ml稀释,硅藻土抽滤,母液用乙酸乙酯(45mlx3)萃取,饱和食盐水(30mlx2)洗涤,无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=8:1)纯化,得淡黄色油状化合物e-3(3.96g)。化合物e-4的合成取化合物e-3(3.96g)溶于甲醇(15ml)和四氢呋喃(15ml)的混合溶剂中,于冰浴下缓慢加入一水合氢氧化锂(1.5g),室温下反应约6小时后停止反应,减压蒸除溶剂。1n盐酸酸化调ph=2~3,析出白色固体,抽滤,得白色固体化合物e-4(3.3g)。化合物e-5的合成取化合物e-4(3.96g)溶于无水四氢呋喃(45ml)中,冰盐浴下缓慢滴加硼烷四氢呋喃复合物(29ml),室温下反应约5小时停止反应,冰浴下加水淬灭反应。用乙酸乙酯(50mlx3)萃取,和饱和食盐水(25mlx2)洗涤,无水硫酸钠干燥。减压蒸除溶剂,得白色固体化合物e-5(3.0g)。化合物e-6的合成取化合物e-5(3.0g)和三苯基膦(6.2g)溶于无水四氢呋喃(30ml)中,于冰浴下缓慢加入四溴化碳(7.8g),室温下反应约4小时后停止反应。减压蒸除溶剂,用二氯甲烷(30ml)稀释后硅藻土抽滤,母液经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=5:1)纯化,得淡黄色油状化合物e-6(3.6g)。化合物e-7的合成取实施例4中化合物h-1(351mg)溶于无水丙酮(10ml)中,加入无水碳酸氢钠(395mg),室温搅拌20分钟,缓慢加入e-6(600mg),70℃油浴加热。反应约6小时后停止加热,减压蒸除溶剂,加入乙酸乙酯(10ml)稀释,用水(10mlx3)和饱和食盐水(10mlx2)洗涤,无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=3:1)纯化,得白色固体化合物e-7(626mg)。化合物vii-1的合成取化合物e-7(360mg)和碳酸铯(559mg)加入n,n-二甲基甲酰胺(4ml)中,室温搅拌15分钟后缓慢加入xc-3(229mg),室温反应约4小时后停止反应。加入水(30ml),用乙酸乙酯(10mlx3)萃取,无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=2:1)纯化,得黄色固体化合物vii-1(224mg)。化合物30的合成参照实施例1的方法,将实施例1中的化合物i-7替换成化合物vii-1,不经水解直接制得化合物30的游离碱,再将化合物30的游离碱加入盐酸乙醇溶液中,搅拌过夜,抽滤,得化合物30:1hnmr(300mhz,dmso-d6)δ9.66–9.28(m,2h),8.24(s,1h),8.13(d,j=9.0hz,1h),7.87(dd,j=9.1,1.6hz,1h),7.67(s,1h),7.39(dd,j=4.6hz,1h),7.20(s,1h),7.13(d,j=4.5hz,2h),6.92(d,j=8.1hz,1h),6.79–6.68(m,2h),5.49(s,2h),5.30(s,2h),4.36–4.16(m,6h),4.13–4.00(m,3h),3.95(d,j=12.4hz,1h),3.84(dd,j=12.0,3.9hz,1h),2.22(s,3h),1.13(t,j=7.1hz,3h).ms(esi):m/z676.3[m+h]+.实施例31(2-(苯并[c][1,2,5]噻二唑-5-基甲氧基)-5-氯-4-((3-(2,3-二氢苯并[b][1,4]二噁英-6-基)基)-2-甲基苄基)氧基)苄基)-d-丝氨酸乙酯盐酸盐(化合物31)以化合物30的游离碱为关键中间体,参照实施例1的方法水解,制得化合物31:1hnmr(300mhz,dmso-d6)δ8.24(s,1h),8.12(d,j=9.0hz,1h),7.88(dd,j=9.1,1.6hz,1h),7.61(s,1h),7.39(dd,j=4.6hz,1h),7.19(s,1h),7.13(d,j=4.5hz,2h),6.92(d,j=8.1hz,1h),6.80–6.69(m,2h),5.50(s,2h),5.28(s,2h),4.28(s,4h),4.17(s,2h),3.88(dd,j=11.8,3.6hz,1h),3.78(dd,j=11.7,4.8hz,1h),3.68–3.62(m,1h),2.22(s,3h).ms(esi):688.3[m+h]+.实施例32(2r,4r)-1-(2-(苯并[c][1,2,5]噻二唑-5-基甲氧基)-5-氯-4-((3-(2,3-二氢苯并[b]甲基)[1,4]二氧杂环己烯-6-基)-2-甲基苄基)氧基)苄基)-4-羟基吡咯烷-2-羧酸甲酯盐酸盐(化合物32)参照实施例30的方法,将实施例30中的d-丝氨酸乙酯盐酸盐替换成(2r,4r)-4-羟基吡咯烷-2-羧酸甲酯盐酸盐,制得化合物32:1hnmr(300mhz,dmso-d6)δ10.20(s,1h),8.25(s,1h),8.16(d,j=9.1hz,1h),7.86(dd,j=9.0,1.6hz,1h),7.70(s,1h),7.46–7.35(m,1h),7.25(s,1h),7.15(d,j=4.4hz,2h),6.93(d,j=8.1hz,1h),6.80–6.70(m,2h),5.53(s,2h),5.32(s,2h),4.56–4.46(m,2h),4.44–4.35(m,2h),4.29(s,4h),3.56(s,3h),3.50–3.41(m,1h),3.41–3.31(m,1h),2.67–2.55(m,1h),2.23(s,3h),2.06–1.93(m,1h).ms(esi):674.3[m+h]+.实施例33(2r,4r)-1-(2-(苯并[c][1,2,5]噻二唑-5-基甲氧基)-5-氯-4-((3-(2,3-二氢苯并[b]甲基)[1,4]二氧杂环己烯-6-基)-2-甲基苄基)氧基)苄基)-4-羟基吡咯烷-2-羧酸(化合物33)以化合物32的游离碱为关键中间体,参照实施例1的方法水解制得化合物33:1hnmr(300mhz,dmso-d6)δ8.22(s,1h),8.12(d,j=9.0hz,1h),7.87(d,j=9.1hz,1h),7.51(s,1h),7.41(dd,j=5.9,2.4hz,1h),7.19(s,1h),7.17–7.11(m,2h),6.93(d,j=8.1hz,1h),6.81–6.70(m,2h),5.49(s,2h),5.25(s,2h),4.29(s,4h),4.23(s,1h),4.06(d,j=13.5hz,1h),3.91(d,j=13.2hz,1h),3.65–3.47(m,2h),3.04(d,j=10.4hz,1h),2.86–2.75(m,1h),2.47–2.32(m,1h),2.22(s,3h),1.87(d,j=18.1hz,1h).ms(esi):674.3[m+h]+.实施例34(2r,4r)-1-(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-5-氯-4-((2-甲基-[1,1'-联苯]-3-基)甲氧基)苄基)-4-羟基吡咯烷-2-羧酸(化合物34)参照实施例21的方法,将实施例21中的d-丝氨酸乙酯盐酸盐替换成(2r,4r)-4-羟基吡咯烷-2-羧酸甲酯盐酸盐,制得化合物34:1hnmr(300mhz,chloroform-d)δ8.17(s,1h),8.10(d,j=9.4hz,1h),7.72(d,j=9.2hz,1h),7.60(s,1h),7.52–7.43(m,3h),7.40(d,j=6.9hz,1h),7.31(d,j=7.1hz,2h),7.26–7.14(m,4h),5.44(s,2h),5.30(s,2h),4.36–4.22(m,2h),4.18–4.05(m,1h),3.97–3.83(m,1h),3.29–3.22(m,1h),3.22–3.16(m,1h),3.11–2.98(m,1h),2.23(s,3h),2.00–1.84(m,1h).ms(esi):600.4[m+h]+.实施例35(2-(苯并[c][1,2,5]噻二唑-5-基甲氧基)-4-((2-氯-3-(2,3-二氢苯并[b][1,4]二氧杂环己烯-6-基)苄基)氧基)苄基)-d-丝氨酸(化合物35)合成路线:化合物f-1的合成取化合物a-2(3.5g)、苯并二氧六环-4-硼酸酯(4.56g)、[1,1’-双(二苯基膦)二茂铁]二氯化钯二氯甲烷络合物(636mg)、无水醋酸钾(4.2g)和18-冠-6-醚(412mg)加入到100ml三颈瓶中,氩气保护,换气三次。室温下加入甲苯(21ml)、无水乙醇(7ml)和水(3.5ml)。90℃油浴加热,反应约5小时后停止加热,减压蒸除溶剂,用乙酸乙酯25ml稀释,硅藻土抽滤,母液用乙酸乙酯(45mlx3)萃取,饱和食盐水(30mlx2)洗涤,无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=4:1)纯化,得无色油状化合物f-1(3.87g)。化合物f-2的合成取化合物f-1(3.87g)和三苯基膦(7.3g)溶于无水四氢呋喃(30ml)中,于冰浴下缓慢加入四溴化碳(9.3g),室温下反应约4小时后停止反应。减压蒸除溶剂,用二氯甲烷(25ml)稀释后硅藻土抽滤,母液经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=5:1)纯化,得无色油状化合物f-2(4.3g)。化合物f-3的合成取化合物2,4-二羟基苯甲醛(407mg)溶于无水乙腈(25ml)中,加入无水碳酸氢钠(495mg),缓慢加入f-2(1.2g)的n,n-二甲基甲酰胺溶液(2ml),70℃油浴加热。反应约6小时后停止加热,减压蒸除溶剂,加入乙酸乙酯(15ml)稀释,用水(10mlx3)和饱和食盐水(10mlx2)洗涤,无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=5:1)纯化,得白色固体化合物f-3(607mg)。化合物viii-1的合成取化合物e-7(607mg),碳酸铯(1.50g)加入n,n-二甲基甲酰胺(4ml)中,室温搅拌15分钟后缓慢加入z-3(526mg),室温反应约4小时后停止反应。加入水(30ml),用乙酸乙酯(10mlx3)萃取,无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=2:1)纯化,得白色固体化合物viii-1(471mg)。化合物35的合成参照实施例1的方法,将实施例1中的i-7替换成viii-1,制得化合物35:1hnmr(300mhz,chloroform-d)δ8.19(s,1h),8.08(d,j=9.0hz,1h),7.88(d,j=8.9hz,1h),7.54(d,j=7.1hz,1h),7.35(d,j=11.3hz,3h),7.04–6.76(m,4h),6.66(d,j=8.5hz,1h),5.39(s,2h),5.19(s,2h),4.29(s,4h),4.13–3.95(m,2h),3.76–3.61(m,2h),3.23–3.11(m,1h).ms(esi):634.3[m+h]+.实施例36(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((2-溴-[1,1'-联苯]-3-基)甲氧基)-5-硝基苄基)-d-丝氨酸乙酯盐酸盐(化合物36)合成路线:化合物p-1的合成取化合物2,4-二羟基苯甲醛(925mg)溶于无水乙腈(20ml)中,加入无水碳酸氢钠(1.0g),并缓慢加入b-4(2.0g)的n,n-二甲基甲酰胺溶液(3ml),70℃油浴加热。反应约20小时后停止加热,减压蒸除溶剂,加入乙酸乙酯(10ml)稀释,用水(10mlx3)和饱和食盐水(10mlx2)洗涤,无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/二氯甲烷=2:1)纯化,得灰白色固体化合物p-1(1.6g)。化合物p-2的合成取化合物p-1(1.6g)溶于冰醋酸(20ml)中,冰浴下加入发烟硝酸(0.23ml),室温反应5小时后停止反应,将反应液倒入冰水中,有黄色固体析出,抽滤,得黄色固体,用乙醇(4ml)打浆得黄色固体化合物p-2(620mg)。化合物p-3的合成取化合物p-2(200mg)和碳酸铯(304mg)加入n,n-二甲基甲酰胺(4ml)中,室温搅拌15分钟后缓慢加入xa-7(119mg),室温反应约1小时后停止反应。加入水(30ml),用乙酸乙酯(10mlx3)萃取,无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=2:1)纯化,得黄色固体化合物p-3(200mg)。化合物36的合成参照实施例1的方法,将实施例1中的i-7替换成p-3,不经水解直接制得化合物36的游离碱,再将化合物36的游离碱加入盐酸乙醇溶液中,搅拌过夜,抽滤,得化合物36:ms(esi):m/z677.4[m+h]+.实施例37(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((2-溴-[1,1'-联苯]-3-基)甲氧基)-5-硝基苄基)-d-丝氨酸(化合物37)以化合物36的游离碱为关键中间体,参照实施例1的方法进行水解制得化合物37:1hnmr(300mhz,dmso-d6)δ8.26(s,1h),8.21(s,1h),8.10(d,j=9.2hz,1h),7.74(d,j=9.2hz,1h),7.68(d,j=7.1hz,1h),7.53–7.42(m,4h),7.41–7.32(m,3h),7.18(s,1h),5.54(s,2h),5.44(s,2h),4.22–4.11(m,2h),3.88–3.80(m,1h),3.79–3.72(m,1h),3.63(s,1h).ms(esi):m/z649.2[m+h]+.实施例382-((2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((2-溴-[1,1'-联苯]-3-基)甲氧基)-5-硝基苄基)氨基)乙-1-醇(化合物38)参照实施例36的方法,将实施例36中的d-丝氨酸乙酯盐酸盐替换成乙醇胺,制得化合物38:1hnmr(300mhz,dmso-d6)δ8.12(dd,j=13.4,3.2hz,3h),7.69(d,j=9.3hz,2h),7.56–7.41(m,4h),7.41–7.31(m,3h),7.15(s,1h),5.53(s,2h),5.41(s,2h),3.84(s,2h),3.52(t,j=5.6hz,2h),2.66(t,j=5.5hz,2h).ms(esi):m/z605.4[m+h]+.实施例392-((2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((2-溴-[1,1'-联苯]-3-基)甲氧基)-5-硝基苄基)氨基)乙烷-1-磺酸(化合物39)参照实施例36的方法,将实施例36中的d-丝氨酸乙酯盐酸盐替换成牛磺酸,制得化合物39:ms(esi):m/z669.5[m+h]+.实施例40(2-((2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((2-溴-[1,1'-联苯]-3-基)甲氧基)-5-硝基苄基)氨基)乙基)甲磺酰胺(化合物40)参照实施例36的方法,将实施例36中的d-丝氨酸乙酯盐酸盐替换成n-(2-氨基乙基)甲烷磺酰胺,制得化合物40:ms(esi):m/z682.2[m+h]+.实施例41(s)-2-((2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((2-溴-[1,1'-联苯]-3-基)甲氧基)-5-硝基苄基)氨基)-3-羟基-2-甲基丙酸(化合物41)参照实施例36的方法,将实施例36中的d-丝氨酸乙酯盐酸盐替换成2-甲基-l-丝氨酸乙酯盐酸盐,制得化合物41:ms(esi):m/z663.6[m+h]+.实施例42(s)-1-(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((2-溴-[1,1'-联苯]-3-基)甲氧基)-5-硝基苄基)哌啶-2-羧酸甲酯盐酸盐(化合物42)合成路线:化合物g-4的合成取化合物p-3(150mg)溶于无水乙醇(10ml)中,冰浴下加入硼氢化钠(38mg),反应约3小时后停止反应,加入水(10ml),用乙酸乙酯(10mlx3)萃取,无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=1:1)纯化,得黄色固体化合物g-4(125mg)。化合物g-5的合成取化合物g-4(125mg)和三苯基膦(115mg)溶于无水四氢呋喃(10ml)中,于冰浴下缓慢加入四溴化碳(146mg),室温下反应约4小时后停止反应。减压蒸除溶剂,用二氯甲烷(10ml)稀释后硅藻土抽滤,母液经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=2:1)纯化,得淡黄色固体化合物g-5(90mg)。化合物42的合成取(s)-哌啶-2-甲酸甲酯盐酸盐(38mg)溶于n,n-二甲基甲酰胺(5ml),加入无水碳酸钾(50mg),室温搅拌20分钟,加入化合物g-5(90mg),70℃油浴加热。反应12小时后停止反应。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=1:1)纯化,得黄色油状化合物42的游离碱(42mg),再将化合物42的游离碱加入盐酸乙醇溶液中,搅拌过夜,抽滤,得化合物42:ms(esi):m/z687.5[m+h]+.实施例43(s)-1-(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((2-溴-[1,1'-联苯]-3-基)甲氧基)-5-硝基苄基)哌啶-2-羧酸(化合物43)以化合物42的游离碱为关键中间体,参照实施例1的方法水解制得化合物43:ms(esi):m/z673.4[m+h]+.实施例44(2r,4r)-1-(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((2-溴-[1,1'-联苯]-3-基)甲氧基)-5-硝基苄基)-4-羟基吡咯烷-2-羧酸甲酯盐酸盐(化合物44)参照实施例42的方法,将实施例42中的(s)-哌啶-2-甲酸甲酯盐酸盐替换成(2r,4r)-4-羟基吡咯烷-2-羧酸甲酯盐酸盐,制得化合物44:ms(esi):m/z689.7[m+h]+.实施例45(2r,4r)-1-(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((2-溴-[1,1'-联苯]-3-基)甲氧基)-5-硝基苄基)-4-羟基吡咯烷-2-羧酸(化合物45)以化合物44的游离碱为关键中间体,参照实施例1的方法水解制得化合物45:1hnmr(300mhz,dmso-d6)δ8.23–8.02(m,3h),7.79–7.63(m,2h),7.55–7.41(m,4h),7.41–7.29(m,3h),7.17(s,1h),5.53(s,2h),5.41(s,2h),4.89(s,1h),4.23(s,1h),4.01(d,j=13.8hz,1h),3.85(d,j=13.8hz,1h),3.44–3.40(m,1h),2.97(d,j=10.2hz,1h),2.69(dd,j=10.3,5.4hz,1h),2.45–2.30(m,1h),1.83(d,j=13.5hz,1h).ms(esi):m/z675.5[m+h]+.实施例46(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((2-氯-[1,1'-联苯]-3-基)甲氧基)-5-硝基苄基)-d-丝氨酸乙酯盐酸盐(化合物46)参照实施例24的方法,将实施例24中的d-1替换成a-4,不经水解直接制得化合物46的游离碱,再将化合物46的游离碱加入盐酸乙醇溶液中,搅拌过夜,抽滤,得化合物46:ms(esi):m/z633.2[m+h]+.实施例47(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((2-氯-[1,1'-联苯]-3-基)甲氧基)-5-硝基苄基)-d-丝氨酸(化合物47)以化合物46的游离碱为关键中间体,参照实施例1的方法进行水解制得化合物47:ms(esi):m/z605.0[m+h]+.实施例482-((2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((2-氯-[1,1'-联苯]-3-基)甲氧基)-5-硝基苄基)氨基)乙-1-醇(化合物48)参照实施例46的方法,将实施例46中的d-丝氨酸乙酯盐酸盐替换成乙醇胺,制得化合物48:ms(esi):m/z561.2[m+h]+.实施例492-((2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((2-氯-[1,1'-联苯]-3-基)甲氧基)-5-硝基苄基)氨基)乙烷-1-磺酸(化合物49)参照实施例46的方法,将实施例46中的d-丝氨酸乙酯盐酸盐替换成牛磺酸,制得化合物49:ms(esi):m/z623.0[m+h]+.实施例50(2-((2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((2-氯-[1,1'-联苯]-3-基)甲氧基)-5-硝基苄基)氨基)乙基)甲磺酰胺(化合物50)参照实施例46的方法,将实施例46中的d-丝氨酸乙酯盐酸盐替换成n-(2-氨基乙基)甲烷磺酰胺,制得化合物50:ms(esi):m/z638.2[m+h]+.实施例51(s)-2-((2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((2-氯-[1,1'-联苯]-3-基)甲氧基)-5-硝基苄基)氨基)-3-羟基-2-甲基丙酸(化合物51)参照实施例46的方法,将实施例46中的d-丝氨酸乙酯盐酸盐替换成2-甲基-l-丝氨酸乙酯盐酸盐,再经过水解,制得化合物51:ms(esi):m/z619.6[m+h]+.实施例52(s)-1-(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((2-氯-[1,1'-联苯]-3-基)甲氧基)-5-硝基苄基)哌啶-2-羧酸甲酯盐酸盐(化合物52)合成路线:化合物q-1的合成取化合物2,4-二羟基苯甲醛(860mg)溶于无水乙腈(20ml)中,加入无水碳酸氢钠(1.05g),并缓慢加入a-4(1.76g)的n,n-二甲基甲酰胺溶液(3ml),70℃油浴加热。反应约20小时后停止加热,减压蒸除溶剂,加入乙酸乙酯(10ml)稀释,用水(10mlx3)和饱和食盐水(10mlx2)洗涤,无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/二氯甲烷=2:1)纯化,得灰白色固体化合物q-1(1.48g)。化合物q-2的合成取化合物q-1(1.48g)溶于冰醋酸(20ml)中,冰浴下加入发烟硝酸(0.23ml),室温反应5小时后停止反应,将反应液倒入冰水中,有黄色固体析出,抽滤,得黄色固体,用乙醇(4ml)打浆得黄色固体化合物q-2(783mg)。化合物q-3的合成取化合物q-2(400mg)和碳酸铯(605mg)加入n,n-二甲基甲酰胺(4ml)中,室温搅拌15分钟后缓慢加入xa-7(254mg),室温反应约1小时后停止反应。加入水(30ml),用乙酸乙酯(10mlx3)萃取,无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=2:1)纯化,得黄色固体化合物q-3(175mg)。化合物q-4的合成取化合物q-3(200mg)溶于无水乙醇(10ml)中,冰浴下加入硼氢化钠(15mg),反应约3小时后停止反应,加入水(10ml),用乙酸乙酯(10mlx3)萃取,无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=1:1)纯化,得黄色固体化合物q-4(175mg)。化合物q-5的合成取化合物q-4(175mg)和三苯基膦(168mg)溶于无水四氢呋喃(10ml)中,于冰浴下缓慢加入四溴化碳(221mg),室温下反应约4小时后停止反应。减压蒸除溶剂,用二氯甲烷(10ml)稀释后硅藻土抽滤,母液经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=2:1)纯化,得淡黄色固体化合物q-5(90mg)。化合物52的合成取(s)-哌啶-2-甲酸甲酯盐酸盐(38mg)溶于n,n-二甲基甲酰胺(5ml),加入无水碳酸钾(50mg),室温搅拌20分钟,加入化合物q-5(90mg),70℃油浴加热。反应12小时后停止反应。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=1:1)纯化,得黄色油状化合物42的游离碱(42mg),再将化合物52的游离碱加入盐酸乙醇溶液中,搅拌过夜,抽滤,得化合物52:ms(esi):m/z643.1[m+h]+.实施例53(s)-1-(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((2-氯-[1,1'-联苯]-3-基)甲氧基)-5-硝基苄基)哌啶-2-羧酸(化合物53)以化合物52的游离碱为关键中间体,参照实施例1的方法水解制得化合物53:ms(esi):m/z629.2[m+h]+.实施例54(2r,4r)-1-(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((2-氯-[1,1'-联苯]-3-基)甲氧基)-5-硝基苄基)-4-羟基吡咯烷-2-羧酸甲酯盐酸盐(化合物54)参照实施例52的方法,将实施例52中的(s)-哌啶-2-甲酸甲酯盐酸盐替换成(2r,4r)-4-羟基吡咯烷-2-羧酸甲酯盐酸盐,制得化合物54:ms(esi):m/z645.5[m+h]+.实施例55(2r,4r)-1-(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((2-氯-[1,1'-联苯]-3-基)甲氧基)-5-硝基苄基)-4-羟基吡咯烷-2-羧酸(化合物55)以化合物54的游离碱为关键中间体,参照实施例1的方法水解制得化合物55:ms(esi):m/z631.3[m+h]+.实施例56(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((2-氰基-[1,1'-联苯]-3-基)甲氧基)-5-硝基苄基-d-丝氨酸(化合物56)合成路线:化合物r-2的合成取化合物r-1(10g)溶于甲醇(75ml)中,于室温下缓慢滴加浓硫酸(20ml),约15分钟加毕,85℃油浴加热。反应约15小时后停止加热,减压蒸除溶剂,用乙酸乙酯(60mlx4)萃取,饱和碳酸钠(50mlx3)和饱和食盐水(100mlx4)洗涤,无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=50:1)纯化,得淡黄色油状化合物r-2(9.0g)。化合物r-3的合成取化合物r-2(9.0g)溶于水(50ml)中,冰盐浴下缓慢加入盐酸(20ml),搅拌10分钟,缓慢滴加亚硝酸钠(3.2g)的水溶液,约30分钟加毕,冰盐浴下搅拌约45分钟后,向反应液中缓慢滴加碘化钾(8.4g)的水溶液,室温下反应8小时后停止反应。用硫代硫酸钠的饱和水溶液(60ml),乙酸乙酯(100mlx3)萃取,饱和食盐水(80mlx3)洗涤,无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=50:1)纯化,得淡黄色油状化合物r-3(11g)。化合物r-4的合成取化合物r-3(11g)、氰化亚铜(11.5g)、三(二亚苄基丙酮)二钯(604mg)、双二苯基膦二茂铁(730mg)、无水醋酸钾(6.5g)和18-冠-6-醚(873mg)加入到100ml三颈瓶中,氩气保护,换气三次,加入无水1,4-二氧六环40ml,110℃油浴加热。反应约5小时后停止加热,减压蒸除溶剂,用乙酸乙酯(40ml)稀释,硅藻土抽滤,母液经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=3:1)纯化,得白色固体化合物r-4(3.47g)。化合物r-5的合成取化合物r-4(1.5g)、苯硼酸(914mg)、[1,1’-双(二苯基膦)二茂铁]二氯化钯二氯甲烷络合物(257mg)、无水醋酸钾(1.2g)和18-冠-6-醚(330mg)加入到50ml三颈瓶中,氩气保护,换气三次。室温下加入甲苯(18ml)、无水乙醇(6ml)和水(3ml)。90℃油浴加热。反应约5小时后停止加热,减压蒸除溶剂,用乙酸乙酯25ml稀释,硅藻土抽滤,母液用乙酸乙酯(40mlx3)萃取,饱和食盐水(40mlx3)洗涤,无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/二氯甲烷=30:1)纯化,得无色油状化合物r-5(1.4g)。化合物r-6的合成取化合物r-5(1.41g)溶于甲醇(10ml)和四氢呋喃(10ml)的混合溶剂中,于冰浴下缓慢加入一水合氢氧化锂(619mg),室温下反应约6小时后停止反应,减压蒸除溶剂。用1n盐酸酸化调ph=5~6,析出白色固体化合物r-6(1.36g)。化合物r-7的合成取化合物r-6(1.36g)溶于无水四氢呋喃(30ml)中,冰盐浴下缓慢滴加硼烷四氢呋喃复合物(12ml),室温下反应约5小时停止反应,冰浴下加水淬灭反应。用乙酸乙酯(30mlx3)萃取,和饱和食盐水(25mlx3)洗涤,无水硫酸钠干燥。减压蒸除溶剂,得白色固体化合物r-7(952mg)。化合物r-8的合成取化合物r-7(948mg)和三苯基膦(1.78g)溶于无水四氢呋喃(30ml)中,于冰浴下缓慢加入四溴化碳(2.2g),室温下反应约4小时后停止反应。减压蒸除溶剂,用二氯甲烷(20ml)稀释,硅藻土抽滤,母液经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=5:1)纯化,得黄色油状化合物r-8(1.2g)。化合物r-9的合成取化合物2,4-二羟基苯甲醛(1.07g)溶于无水乙腈(20ml)中,加入无水碳酸氢钠(1.09g),并缓慢加入r-8(1.78g)的n,n-二甲基甲酰胺溶液(3ml),70℃油浴加热。反应约20小时后停止加热,减压蒸除溶剂,加入乙酸乙酯(10ml)稀释,用水(10mlx3)和饱和食盐水(10mlx2)洗涤,无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/二氯甲烷=2:1)纯化,得白色固体化合物d-1(2.0g)。化合物r-10的合成取化合物r-9(2.0g)溶于冰醋酸(2ml)中,冰浴下加入发烟硝酸(0.38ml),室温反应5小时后停止反应,将反应液倒入冰水中,有黄色固体析出,抽滤,得黄色固体,用乙醇(4ml)打浆得黄色固体化合物r-10(1.0g)。化合物56的合成参照实施例1的方法,将实施例1中的i-6替换为r-10,制得化合物56:1hnmr(300mhz,dmso-d6)δ8.15(s,1h),8.13–8.04(m,2h),7.82–7.75(m,2h),7.65–7.48(m,7h),7.20(s,1h),5.56(s,2h),5.53(s,2h),3.93–3.75(m,3h),3.53–3.47(m,2h),2.90(s,1h).ms(esi):m/z595.5[m+h]+.实施例572-((2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((2-溴-[1,1'-联苯]-3-基)甲氧基)-5-氯苄基)氨基)乙烷-1-磺酸(化合物57)参照实施例8的方法,将实施例8中的d-丝氨酸乙酯盐酸盐替换成牛磺酸,制得化合物57:1hnmr(300mhz,dmso-d6)δ8.62(s,1h),8.16(s,1h),8.08(d,j=9.3hz,1h),7.74(d,j=9.4hz,1h),7.66–7.55(m,2h),7.52–7.28(m,7h),7.13(s,1h),5.44(s,2h),5.32(s,2h),4.23(s,2h),3.21(t,j=6.5hz,2h),2.82(t,j=6.4hz,2h).ms(esi):m/z565.1[m-h]-.实施例58(2-(苯并[c][1,2,5]噻二唑-5-基甲氧基)-4-((2-溴-[1,1'-联苯]-3-基)甲氧基)-5-氯苄基)-d-丝氨酸乙酯盐酸盐(化合物58)参照实施例8的方法,将实施例8中的xa-7替换成xc-3,制得化合物58:1hnmr(300mhz,dmso-d6)δ9.48(s,2h),8.22(s,1h),8.11(d,j=9.1hz,1h),7.84(d,j=9.1hz,1h),7.69(s,1h),7.58(d,j=7.5hz,1h),7.48(d,j=7.2hz,1h),7.51–7.37(m,3h),7.41–7.26(m,3h),7.13(s,1h),5.63(s,1h),5.47(s,2h),5.31(s,2h),4.24(s,2h),4.11–3.99(m,3h),3.94(d,j=13.0hz,1h),3.83(d,j=11.5hz,1h),1.13(t,j=7.1hz,1h).ms(esi):m/z684.2[m+h]+.实施例59(2-(苯并[c][1,2,5]噻二唑-5-基甲氧基)-4-((2-溴-[1,1'-联苯]-3-基)甲氧基)-5-氯苄基)-d-丝氨酸(化合物59)以化合物58的游离碱为关键中间体,参照实施例1的方法进行水解制得化合物59:1hnmr(300mhz,dmso-d6)δ8.01(d,j=9.0hz,1h),7.76(d,j=6.5hz,1h),7.62(d,j=7.4hz,2h),7.58–7.23(m,8h),7.16(s,1h),5.62(s,2h),5.31(s,2h),3.93(s,2h),3.61(dd,j=11.5,5.2hz,3h),3.16(s,1h).ms(esi):m/z652.1[m-h]-.实施例60(2-(苯并[c][1,2,5]噻二唑-4-基甲氧基)-4-((2-溴-[1,1'-联苯]-3-基)甲氧基)-5-氯苄基)-d-丝氨酸乙酯盐酸盐(化合物60)合成路线:化合物xd-2的合成取化合物xd-1(2.26g)溶于dcm(60ml)中,加入三乙胺(et3n,10.3ml),室温搅拌至固体完全溶解,冰浴下将二氯亚砜(socl2,2.7ml)缓慢滴入,油浴加热45℃反应7小时后停止加热,减压蒸除溶剂,加入水(70ml),用1n盐酸调ph至1,用乙酸乙酯(30mlx3)萃取,无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=10:1)纯化,得黄色油状化合物xd-2(2.1g)。化合物xd-3的合成取化合物xd-2(1.54g)溶于二氯甲烷(50ml),加入n-溴代琥珀酰亚胺(nbs,2.2g)和偶氮二异丁腈(aibn,85mg),加热回流反应,12小时后停止反应,减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=100:1)纯化,得白色固体化合物xd-3(977mg)。化合物60的合成参照实施例8的方法,将实施例8中的xa-7替换成xd-3,制得化合物60:1hnmr(300mhz,dmso-d6)δ9.43(d,j=78.7hz,2h),8.10(d,j=8.8hz,1h),7.89(d,j=6.8hz,1h),7.74(dd,j=8.8,6.8hz,1h),7.65(s,1h),7.61(d,j=7.5hz,1h),7.53–7.40(m,4h),7.36(d,j=7.4hz,3h),7.22(s,1h),5.72(s,2h),5.62(s,1h),5.33(s,2h),4.19(s,2h),4.04–3.91(m,3h),3.89(d,j=10.2hz,1h),3.77(d,j=12.1hz,1h),1.09(t,j=7.1hz,3h).ms(esi):m/z684.2[m+h]+.实施例61(2-(苯并[c][1,2,5]噻二唑-4-基甲氧基)-4-((2-溴-[1,1'-联苯]-3-基)甲氧基)-5-氯苄基)-d-丝氨酸(化合物61)以化合物60的游离碱为关键中间体,参照实施例1的方法进行水解制得化合物61:1hnmr(500mhz,chloroform-d)δ8.05(d,j=8.8hz,1h),7.86(d,j=6.8hz,1h),7.70(dd,j=8.8,6.8hz,1h),7.59(d,j=7.5hz,1h),7.49–7.42(m,4h),7.40(d,j=7.0hz,1h),7.37–7.31(m,3h),7.13(s,1h),5.73–5.64(m,2h),5.27(s,2h),3.91(q,j=13.7hz,2h),3.65–3.55(m,2h),3.15(t,j=5.4hz,1h).ms(esi):m/z652.1[m-h]-.实施例62(2-(苯并[c][1,2,5]噁二唑-4-基甲氧基)-4-((2-溴-[1,1'-联苯]-3-基)甲氧基)-5-氯苄基)-d-丝氨酸乙酯盐酸盐(化合物62)参照实施例8的方法,将实施例8中的xa-7替换成xb-4,制得化合物62:1hnmr(300mhz,dmso-d6)δ9.43(d,j=45.4hz,2h),8.04(d,j=9.1hz,1h),7.77(d,j=6.6hz,1h),7.70–7.58(m,3h),7.55–7.40(m,4h),7.36(d,j=7.3hz,3h),7.22(s,1h),5.64(s,2h),5.58(s,1h),5.35(s,2h),4.19(s,2h),4.09–3.92(m,3h),3.89(d,j=12.0hz,1h),3.78(d,j=12.4hz,1h),1.11(t,j=7.0hz,3h).ms(esi):m/z666.2[m+h]+.实施例63(2-(苯并[c][1,2,5]噁二唑-4-基甲氧基)-4-((2-溴-[1,1'-联苯]-3-基)甲氧基)-5-氯苄基)-d-丝氨酸(化合物63)以化合物62的游离碱为关键中间体,参照实施例1的方法进行水解制得化合物63:1hnmr(300mhz,dmso-d6)δ8.19(s,1h),8.08(d,j=9.0hz,1h),7.85(d,j=9.1hz,1h),7.58(d,j=7.4hz,1h),7.51(s,1h),7.50–7.23(m,7h),7.09(s,1h),5.45(s,2h),5.27(s,2h),3.99(s,2h),3.75–3.67(m,1h),3.63(dd,j=11.3,6.0hz,1h),3.20(t,j=5.3hz,1h).hrms(esi):m/z640.06742[m+h]+.实施例641-(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((2-溴-[1,1'-联苯]-3-基)甲氧基)-5-氯苄基)氮杂环丁烷-3-羧酸(化合物64)参照实施例26的方法,将实施例26中的(2r,4r)-4-羟基吡咯烷-2-羧酸甲酯盐酸盐替换成3-甲酸甲酯氮杂环丁烷盐酸盐,制得化合物64:1hnmr(300mhz,dmso-d6)δ8.13(s,1h),8.10(d,j=9.3hz,1h),7.71(d,j=9.3hz,1h),7.62(d,j=7.7hz,1h),7.46(dq,j=10.8,6.2,4.6hz,4h),7.41–7.31(m,4h),7.09(s,1h),5.40(s,2h),5.30(s,2h),3.79(s,2h),3.63(s,2h),3.49(s,2h),3.34–3.27(m,1h).ms(esi):m/z634.2[m+h]+.实施例65(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((2-溴-[1,1'-联苯]-3-基)甲氧基)-5-溴苄基)-d-丝氨酸乙酯盐酸盐(化合物65)合成路线:化合物h-2的合成取化合物2,4-二羟基苯甲醛(500mg),加入1,3-二丁基咪唑三溴化物中,室温反应0.5小时后停止反应,硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=5:1)纯化,得白色固体化合物h-2(330mg)。化合物65的合成参照实施例8的方法,将实施例8中的h-1替换成h-2,制得化合物65:1hnmr(300mhz,dmso-d6)δ9.43(s,2h),8.19(s,1h),8.11(d,j=9.4hz,1h),7.81(s,1h),7.71(dd,j=9.3,1.3hz,1h),7.64(dd,j=7.7,1.8hz,1h),7.53–7.41(m,4h),7.41–7.31(m,3h),7.09(s,1h),5.63(s,1h),5.42(s,2h),5.33(s,2h),4.27(s,2h),4.16–4.06(m,3h),3.91(q,j=12.3,11.5hz,2h),1.17(t,j=7.1hz,3h).ms(esi):m/z712.2[m+h]+.实施例66(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((2-溴-[1,1'-联苯]-3-基)甲氧基)-5-溴苄基)-d-丝氨酸(化合物66)以化合物65的游离碱为关键中间体,参照实施例1的方法进行水解制得化合物66:1hnmr(300mhz,dmso-d6)δ8.16(s,1h),8.07(d,j=9.3hz,1h),7.79–7.60(m,4h),7.54–7.30(m,8h),7.07(s,1h),5.41(s,2h),5.30(s,2h),4.00(s,2h),3.71–3.58(m,2h),3.16(t,j=5.6hz,1h).ms(esi):m/z680.1[m-h]-.实施例67(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((2-氟-[1,1'-联苯]-3-基)甲氧基)-5-硝基苄基)-d-丝氨酸乙酯盐酸盐(化合物67)合成路线:化合物o-1的合成取化合物i-6(1.48g)溶于冰醋酸(10ml)中,冰浴下加入发烟硝酸(0.287ml),室温反应5小时后停止反应,将反应液倒入冰水中,有黄色固体析出,抽滤,得黄色固体,用乙醇(4ml)打浆得黄色固体化合物o-1(1.3g)。化合物o-2的合成取化合物o-1(600mg)和碳酸铯(1.2g)加入n,n-二甲基甲酰胺(10ml)中,室温搅拌15分钟后缓慢加入xa-7(510mg),室温反应约1小时后停止反应。加入水(30ml),用乙酸乙酯(10mlx3)萃取,无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=2:1)纯化,得黄色固体化合物o-2(370mg)。化合物67的合成参照实施例1的方法,将实施例8中的i-7替换成o-2,制得化合物67:1hnmr(300mhz,dmso-d6)δ9.61(d,j=50.9hz,2h),8.33(s,1h),8.21(s,1h),8.10(d,j=9.3hz,1h),7.73(d,j=9.4hz,1h),7.66–7.38(m,7h),7.34–7.16(m,2h),5.53(d,j=7.7hz,4h),4.32(s,2h),4.22–4.05(m,3h),3.98(d,j=12.2hz,1h),3.86(d,j=12.9hz,1h),1.16(t,j=7.0hz,3h).ms(esi):m/z617.3[m+h]+.实施例68(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((2-氟-[1,1'-联苯]-3-基)甲氧基)-5-硝基苄基)-d-丝氨酸(化合物68)以化合物67的游离碱为关键中间体,参照实施例1的方法进行水解制得化合物68:1hnmr(300mhz,dmso-d6)δ8.18–8.01(m,3h),7.69(d,j=9.4hz,1h),7.62–7.36(m,7h),7.27(dd,j=7.6hz,1h),7.21(s,1h),5.51(s,2h),5.46(s,2h),3.95(q,j=14.2hz,2h),3.65–3.58(m,2h),3.19–3.12(m,1h).ms(esi):m/z587.2[m-h]-.实施例69(2r,4r)-1-(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((2-氟-[1,1'-联苯]-3-基)甲氧基)-5-硝基苄基)-4-羟基吡咯烷-2-羧酸甲酯盐酸盐(化合物69)合成路线:化合物o-3的合成取化合物o-2(220mg)溶于无水乙醇(10ml)中,冰浴下加入硼氢化钠(20mg),反应约3小时后停止反应,加入水(10ml),用乙酸乙酯(10mlx3)萃取,无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=1:1)纯化,得白色固体化合物o-3(228mg)。化合物o-4的合成取化合物o-3(228mg)和三苯基膦(262mg)溶于无水四氢呋喃(10ml)中,于冰浴下缓慢加入四溴化碳(331mg),室温下反应约4小时后停止反应。减压蒸除溶剂,用二氯甲烷(10ml)稀释后硅藻土抽滤,母液经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=2:1)纯化,得淡淡黄色固体化合物o-4(220mg)。化合物69的合成参照实施例26的方法,将实施例8中的j-3替换成o-4,制得化合物69:1hnmr(300mhz,dmso-d6)δ10.29(s,1h),8.33(d,j=10.1hz,1h),8.19(s,1h),8.10(d,j=9.3hz,1h),7.69(d,j=9.3hz,1h),7.63–7.35(m,8h),7.33–7.21(m,2h),5.54(s,2h),5.50(s,2h),4.66–4.30(m,4h),3.58(s,3h),2.66–2.51(m,1h),1.99(d,j=13.9hz,1h).ms(esi):m/z629.3[m+h]+.实施例70(2r,4r)-1-(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((2-氟-[1,1'-联苯]-3-基)甲氧基)-5-硝基苄基)-4-羟基吡咯烷-2-羧酸(化合物70)以化合物69的游离碱为关键中间体,参照实施例1的方法进行水解制得化合物70:1hnmr(500mhz,chloroform-d)δ8.13(s,1h),8.09(d,2h),7.71(d,j=9.3hz,1h),7.60(dd,j=7.1hz,1h),7.55(d,2h),7.54–7.47(m,3h),7.43(dd,j=7.3hz,1h),7.30(dd,j=7.6hz,1h),7.21(s,1h),5.55–5.47(m,2h),5.46(s,2h),4.19(s,1h),3.97(d,j=14.0hz,1h),3.82(d,j=13.9hz,1h),3.33(dd,j=9.0,5.6hz,3h),2.92(d,j=10.1hz,1h),2.68(dd,j=10.0,5.2hz,1h),2.37–2.26(m,1h),1.83–1.75(m,1h).ms(esi):m/z615.3[m+h]+.实施例71(2-(苯并[c][1,2,5]噻二唑-5-基甲氧基)-4-((2-氟-[1,1'-联苯]-3-基)甲氧基)-5-硝基苄基)-d-丝氨酸乙酯盐酸盐(化合物71)参照实施例67的方法,将实施例67中的xa-7替换成xc-3,制得化合物71:1hnmr(300mhz,dmso-d6)δ9.61(d,j=36.6hz,2h),8.31(d,j=18.6hz,2h),8.15(d,j=9.0hz,1h),7.89(d,j=9.2hz,1h),7.68–7.46(m,6h),7.43(dd,j=8.2,5.6hz,1h),7.35–7.19(m,2h),5.62(s,2h),5.53(s,2h),4.31(s,2h),4.17–4.03(m,3h),3.97(d,j=12.0hz,1h),3.85(d,j=12.6hz,1h),1.14(t,j=7.0hz,3h).ms(esi):m/z633.4[m+h]+.实施例72(2-(苯并[c][1,2,5]噻二唑-5-基甲氧基)-4-((2-氟-[1,1'-联苯]-3-基)甲氧基)-5-硝基苄基)-d-丝氨酸(化合物72)以化合物71游离碱为关键中间体,参照实施例1的方法进行水解制得化合物72:1hnmr(300mhz,dmso-d6)δ8.20(s,1h),8.15–8.06(m,2h),7.84(d,j=9.1hz,1h),7.62–7.44(m,6h),7.44–7.36(m,1h),7.29–7.19(m,2h),5.58(s,2h),5.45(s,2h),3.98(d,j=14.3hz,1h),3.90(d,j=14.4hz,1h),3.69–3.57(m,2h),3.17(t,j=5.4hz,1h).ms(esi):m/z604.5[m+h]+.实施例73(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-5-氯-4-((2-氟-3'-(3-(4-羟基哌啶-1-基))丙氧基)-[1,1'-联苯]-3-基)甲氧基)苄基)-d-丝氨酸乙酯(化合物73)合成路线:化合物s-1的合成取3-溴苯酚(5g)、1,3-二溴丙烷(15ml)和无水碳酸钾(8g)加入100ml茄形瓶中,加入n,n-二甲基甲酰胺(30ml),室温反应4小时后停止反应。加入水(100ml),用乙酸乙酯(40mlx3)萃取,无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=100:1)纯化,得无色油状化合物s-1(9.84g)。化合物s-2的合成取化合物s-1(3.4g)、联硼酸频那醇酯(1.2g)、[1,1’-双(二苯基膦)二茂铁]二氯化钯二氯甲烷络合物(139mg)和醋酸钾(834mg)加入到25ml三颈瓶中,氩气保护,换气三次。室温下加入1,4-二氧六环(15ml),100℃油浴加热。反应约5小时后停止加热,减压蒸除溶剂,用乙酸乙酯25ml稀释,硅藻土抽滤,母液中加入水(50ml),用乙酸乙酯(10mlx3)萃取,无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=30:1)纯化,得无色油状化合物s-2(560mg)。化合物m-2的合成取化合物m-1(1g)和三苯基膦(7g)溶于无水四氢呋喃(40ml)中,于冰浴下缓慢加入四溴化碳(8.9g),室温下反应约4小时后停止反应。减压蒸除溶剂,用二氯甲烷(10ml)稀释后硅藻土抽滤,母液经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=2:1)纯化,得淡黄色固体化合物m-2(3.7g)。化合物m-3的合成取实施例4中化合物h-1(1.6g)溶于无水乙腈(20ml)中,加入无水碳酸氢钠(1.5g),并缓慢加入m-2(3.7g)的n,n-二甲基甲酰胺溶液(3ml),70℃油浴加热。反应约20小时后停止加热,减压蒸除溶剂,加入乙酸乙酯(10ml)稀释,用水(10mlx3)和饱和食盐水(10mlx2)洗涤,无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/二氯甲烷=2:1)纯化,得灰白色固体化合物m-3(1.98g)。化合物m-4的合成取化合物m-3(200mg),化合物s-2(204mg),[1,1’-双(二苯基膦)二茂铁]二氯化钯二氯甲烷络合物(60mg),无水碳酸钾(134g),18-冠-6-醚(13mg)加入到25ml三颈瓶中,氩气保护,换气三次。室温下加入甲苯(4ml),无水乙醇(1ml),水(0.5ml),90℃油浴加热。反应约5小时后停止加热,减压蒸除溶剂,用乙酸乙酯25ml稀释,硅藻土抽滤,母液用乙酸乙酯(10mlx3)和饱和食盐水(10mlx3)萃取,无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/二氯甲烷=30:1)纯化,得无色油状化合物m-4(95mg)。化合物m-5的合成取化合物m-4(95mg),碳酸铯(125mg)加入n,n-二甲基甲酰胺(4ml)中,室温搅拌15分钟后缓慢加入xa-7(82mg),室温反应约1小时后停止反应。加入水(30ml),用乙酸乙酯(10mlx3)萃取,无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=2:1)纯化,得黄色固体化合物m-5(100mg)。化合物m-6的合成取化合物m-5(250mg),无水碳酸钾(66mg)和4-羟基哌啶(81mg)加入n,n-二甲基甲酰胺(4ml)中,60℃油浴加热。约4小时后停止反应。加入水(30ml),用乙酸乙酯(10mlx3)萃取,无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:二氯甲烷/甲醇=20:1)纯化,得黄色固体化合物m-6(250mg)。化合物73的合成取d-丝氨酸乙酯盐酸盐(105mg)溶于二氯甲烷与甲醇的混合溶剂(3:1,4ml),加入n,n-二异丙基乙胺(dipea,102mg)室温搅拌20分钟,加入化合物m-6(100mg)和三乙酰氧基硼氢化钠(stab,183mg),室温反应12小时后停止反应。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=1:1)纯化,得黄色油状化合物73(70mg):1hnmr(300mhz,methanol-d4)δ7.91(s,1h),7.85(d,j=9.3hz,1h),7.50(d,j=9.4hz,1h),7.45(d,j=6.9hz,1h),7.41–7.26(m,3h),7.16(dd,j=7.6hz,1h),7.12–7.01(m,2h),6.93(dd,j=8.4,2.5hz,1h),6.84(s,1h),5.27(s,2h),5.17(s,2h),4.23–3.96(m,4h),3.78(d,j=3.7hz,2h),3.74–3.65(m,2h),3.35(t,1h),3.32–3.30(m,1h),2.99(dt,j=10.9,4.5hz,2h),2.73(t,j=7.9hz,2h),2.46(s,2h),2.05(q,j=7.8,6.9hz,2h),1.91(t,j=8.3hz,2h),1.71–1.59(m,2h),1.17(t,j=7.1hz,3h).ms(esi):m/z763.6[m+h]+.实施例74(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-5-氯-4-((2-氟-3'-(3-(4-羟基哌啶-1-基))丙氧基)-[1,1'-联苯]-3-基)甲氧基)苄基)-d-丝氨酸(化合物74)以化合物73为关键中间体,参照实施例1的方法水解制得化合物74:1hnmr(300mhz,methanol-d4)δ7.94(s,1h),7.89(d,j=9.3hz,1h),7.59(dd,j=9.3,1.3hz,1h),7.50(s,1h),7.48–7.32(m,3h),7.18(dd,j=7.7hz,1h),7.14–7.03(m,2h),7.00–6.89(m,2h),5.34(s,2h),5.31(s,2h),4.25(q,j=13.2hz,2h),4.08(t,j=6.0hz,2h),3.96(dd,j=11.7,4.1hz,1h),3.82(dd,j=11.7,6.8hz,1h),3.70(d,j=5.7hz,1h),3.51(dd,j=6.8,4.1hz,1h),3.03–2.89(m,2h),2.69(t,2h),2.39(t,j=10.6hz,2h),2.11–1.97(m,2h),1.97–1.83(m,2h),1.70–1.55(m,2h).ms(esi):m/z736.4[m+h]+.实施例75(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-5-氯-4-((2-氟-3'-(3-(1,1-二氧代硫代吗啉代)丙氧基)-[1,1'-联苯]-3-基)甲氧基)苄基)-d-丝氨酸乙酯(化合物75)参照实施例73的方法,将实施例73中的4-羟基哌啶替换1,1-二氧化硫代吗啉,制得化合物75:1hnmr(300mhz,chloroform-d)δ7.93–7.79(m,2h),7.63–7.52(m,1h),7.47–7.37(m,3h),7.36(s,1h),7.24(dd,j=7.7hz,1h),7.16(d,j=7.9hz,1h),7.11(d,j=2.4hz,1h),6.97(dd,j=8.2,2.5hz,1h),6.67(s,1h),5.29(s,2h),5.13(s,2h),4.24–4.04(m,4h),3.94–3.78(m,3h),3.66(dd,j=10.9,6.2hz,1h),3.45(dd,j=6.1,4.2hz,1h),3.07(s,8h),2.75(t,j=7.0hz,2h),2.01(h,j=6.5,5.9hz,2h),1.26(t,j=7.1hz,3h).ms(esi):m/z797.4[m+h]+.实施例76(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-5-氯-4-((2-氟-3'-(3-(1,1-二氧代硫代吗啉代)丙氧基)-[1,1'-联苯]-3-基)甲氧基)苄基)-d-丝氨酸(化合物76)以化合物75为关键中间体,参照实施例1的方法水解制得化合物76:1hnmr(300mhz,methanol-d4)δ8.00(s,1h),7.91(d,j=9.3hz,1h),7.67–7.56(m,2h),7.54–7.34(m,3h),7.20(dd,j=7.6hz,1h),7.17–7.10(m,2h),7.09–6.97(m,2h),5.46–5.30(m,4h),4.49–4.33(m,2h),4.21(t,j=5.8hz,2h),4.10–4.01(m,3h),3.93–3.81(m,4h),3.63(t,j=5.3hz,4h),3.57–3.44(m,2h),2.35(t,j=7.8hz,2h).ms(esi):m/z767.3[m-h]-.实施例77(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((2-溴-[1,1'-联苯]-3-基)甲氧基)-5-氟苄基)-d-丝氨酸乙酯盐酸盐(化合物77)合成路线:化合物h-3的合成取二氯甲基甲醚(2.8ml)溶于无水二氯甲烷(20ml)中,滴加四氯化钛(ticl4,5.1ml),冰浴下加4-氟间苯二酚(2g),自然升温至室温反应过夜。冰浴下以10ml1nhcl与20ml水淬灭,二氯甲烷萃取(20mlx3),无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=5:1)纯化,得黄色固体化合物h-3(0.48g)。化合物ba-1的合成取化合物h-3(143mg)溶于无水乙腈(10ml)中,加入无水碳酸氢钠(103mg),并缓慢加入b-4(200mg),70℃油浴加热。反应约20小时后停止加热,减压蒸除溶剂,加入乙酸乙酯(10ml)稀释,用水(10mlx3)和饱和食盐水(10mlx2)洗涤,无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=10:1)纯化,得白色固体化合物ba-1(236mg)。化合物ba-2的合成取化合物ba-1(200mg)和碳酸铯(325mg)加入n,n-二甲基甲酰胺(6ml)中,室温搅拌15分钟后缓慢加入xa-7(127mg),室温反应约1小时后停止反应。加入水(30ml),用乙酸乙酯(20mlx3)萃取,无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=2:1)纯化,得白色固体化合物ba-2(170mg)。化合物77的合成参照实施例1的方法,将实施例1中的i-7替换成ba-2,不经水解直接制得化合物77的游离碱,再将化合物77的游离碱加入盐酸乙醇溶液中,搅拌过夜,抽滤,得化合物77:1hnmr(300mhz,dmso)δ9.56(s,1h),9.44(s,1h),8.17(s,1h),8.11(d,j=9.3hz,1h),7.70(d,j=9.3hz,1h),7.62–7.54(m,1h),7.52–7.41(m,4h),7.38–7.33(m,2h),7.17(d,j=7.2hz,1h),5.37(s,2h),5.33(s,2h),4.27(s,2h),4.15–4.03(m,3h),3.99–3.83(m,2h),1.16(t,j=7.1hz,3h).ms(esi):m/z650.2[m+h]+.实施例78(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((2-溴-[1,1'-联苯]-3-基)甲氧基)-5-氟苄基)-d-丝氨酸(化合物78)化合物78的合成取化合物77(80mg)溶于甲醇与四氢呋喃的混合溶剂(2:1,3ml),加入一水合氢氧化锂(15mg),室温反应5小时后,减压蒸除溶剂,加入水(5ml),用1n盐酸调节ph=5~6,抽滤,得淡黄色固体化合物78(50mg):1hnmr(300mhz,dmso)δ8.14(s,1h),8.07(d,j=9.3hz,1h),7.70(d,j=9.3hz,1h),7.59(d,j=7.3hz,1h),7.51–7.40(m,4h),7.41–7.31(m,4h),7.12(d,j=7.1hz,1h),5.35(s,2h),5.29(s,2h),3.98(s,2h),3.69–3.61(m,2h),3.17(t,1h).ms(esi):m/z622.2[m-h]-.实施例79(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((2-氟-[1,1'-联苯]-3-基)甲氧基)-5-氯苄基)-d-丝氨酸乙酯盐酸盐(化合物79)合成路线:化合物ia-1的合成取实施例4中化合物h-1(1g)溶于无水乙腈(15ml)中,加入无水碳酸氢钠(527mg),并缓慢加入i-5(830mg),70℃油浴加热。反应约10小时后停止加热,减压蒸除溶剂,加入乙酸乙酯(20ml)稀释,用饱和食盐水(10mlx2)与水(10mlx3)洗涤,无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=20:1)纯化,得白色固体化合物ia-1(760mg)。化合物ia-2的合成取化合物ia-1(120mg)和碳酸铯(220mg)加入n,n-二甲基甲酰胺(6ml)中,室温搅拌15分钟后缓慢加入xa-7(85mg),室温反应约1小时后停止反应。加入水(30ml),用乙酸乙酯(20mlx3)萃取,无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=2:1)纯化,得白色固体化合物ia-2(147mg)。化合物79的合成参照实施例1的方法,将实施例1中的i-7替换成ia-2,不经水解直接制得化合物79的游离碱,再将化合物79的游离碱加入盐酸乙醇溶液中,搅拌过夜,抽滤,得化合物79(149mg):1hnmr(300mhz,dmso)δ9.43(s,2h),8.17(s,1h),8.10(d,j=9.3hz,1h),7.69(d,j=8.0hz,2h),7.59–7.47(m,6h),7.44(d,j=6.8hz,1h),7.27(t,j=7.6hz,1h),7.19(s,1h),5.64(s,1h),5.42(s,2h),5.40(s,2h),4.27(s,2h),4.16–4.04(m,3h),3.99–3.82(m,2h),1.16(t,j=7.1hz,3h).ms(esi):m/z606.2[m+h]+.实施例80(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((2-氟-[1,1'-联苯]-3-基)甲氧基)-5-氯苄基)-d-丝氨酸(化合物80)化合物80的合成取化合物79(100mg)溶于甲醇与四氢呋喃的混合溶剂(2:1,3ml),加入一水合氢氧化锂(20mg),室温反应12小时后,减压蒸除溶剂,加入水(5ml),用1n盐酸调节ph=5~6,抽滤,得白色固体化合物80(17mg):1hnmr(300mhz,dmso)δ8.14(s,1h),8.07(d,j=9.3hz,1h),7.70(d,j=9.2hz,1h),7.59–7.40(m,8h),7.28(t,j=7.6hz,1h),7.15(s,1h),5.40(s,2h),5.36(s,2h),4.02(s,2h),3.72–3.63(m,2h),3.24(t,1h).ms(esi):m/z578.1[m+h]+.实施例81(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-5-氯-4-((2-氯-3'-(3-(4-羟基哌啶-1-基))丙氧基)-[1,1'-联苯]-3-基)甲氧基)苄基)-d-丝氨酸(化合物81)合成路线:化合物aa-1的合成取化合物a-2(2g)和三苯基膦(4.7g)溶于无水四氢呋喃(40ml)中,于冰浴下缓慢加入四溴化碳(5.9g),室温下反应约4小时后停止反应。减压蒸除溶剂,用二氯甲烷(10ml)稀释后硅藻土抽滤,母液经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=10:1)纯化,得无色透明液体化合物aa-1(2.7g)。化合物aa-2的合成取化合物2,4-二羟基苯甲醛(2.6g)溶于无水乙腈(20ml)中,加入无水碳酸氢钠(1.6g),并缓慢加入aa-1(2.7g),70℃油浴加热。反应约20小时后停止加热,减压蒸除溶剂,加入乙酸乙酯(10ml)稀释,用水(10mlx3)和饱和食盐水(10mlx2)洗涤,无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/二氯甲烷=2:1)纯化,得灰白色固体化合物aa-2(2.0g)。化合物aa-3的合成取化合物aa-2(1g)、化合物s-2(1.2g)、[1,1’-双(二苯基膦)二茂铁]二氯化钯二氯甲烷络合物(72mg)、无水碳酸钾(803mg)和18-冠-6-醚(13mg)加入到25ml三颈瓶中,氩气保护。室温下加入甲苯(4ml)、无水乙醇(1ml)和水(0.5ml),90℃油浴加热。反应约5小时后停止加热,减压蒸除溶剂,用乙酸乙酯25ml稀释,硅藻土抽滤,母液用乙酸乙酯(10mlx3)和饱和食盐水(10mlx3)萃取,无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/二氯甲烷=30:1)纯化,得白色固体化合物aa-3(670mg)。化合物aa-4的合成取化合物aa-3(450mg)溶于10ml无水乙醚中,冰浴下滴加磺酰氯(141mg),室温反应约12小时后,抽滤,并以乙醚洗涤,得白色固体化合物aa-4(190mg)。化合物aa-5的合成取化合物aa-4(150mg)和碳酸铯(144mg)加入n,n-二甲基甲酰胺(6ml)中,室温搅拌15分钟后缓慢加入xa-7(75mg),室温反应约1小时后停止反应。加入水(30ml),用乙酸乙酯(10mlx3)萃取,无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=2:1)纯化,得白色固体化合物aa-5(155mg)。化合物aa-6的合成取化合物aa-5(155mg)、无水碳酸钾(66mg)和4-羟基哌啶(36mg)加入n,n-二甲基甲酰胺(4ml)中,60℃油浴加热。约4小时后停止反应。加入水(30ml),用乙酸乙酯(10mlx3)萃取,无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:二氯甲烷/甲醇=20:1)纯化,得黄色固体化合物aa-6(120mg)。化合物aa-7的合成取d-丝氨酸乙酯盐酸盐(164mg)溶于二氯甲烷与甲醇的混合溶剂(3:1,4ml),加入n,n-二异丙基乙胺(dipea,125mg)室温搅拌20分钟,加入化合物aa-6(120mg)和三乙酰氧基硼氢化钠(stab,202mg),室温反应12小时后停止反应。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=1:1)纯化,得黄色油状化合物aa-7(65mg)化合物81的合成取化合物aa-7(65mg)溶于甲醇与四氢呋喃的混合溶剂(2:1,3ml),加入一水合氢氧化锂(10mg),室温反应12小时后,减压蒸除溶剂,加入水(5ml),用1n盐酸调节ph=5~6,抽滤,得黄色固体化合物81(24mg):1hnmr(300mhz,meod)δ7.99(s,1h),7.92(d,j=9.3hz,1h),7.66(d,j=9.3hz,1h),7.58–7.52(m,2h),7.39–7.28(m,3h),7.00–6.91(m,4h),5.37(s,2h),5.36(s,2h),4.37–4.22(m,2h),4.10(t,j=5.9hz,2h),3.99(dd,j=11.7,4.0hz,1h),3.85(dd,j=11.6,7.0hz,1h),3.71(s,1h),3.55(dd,j=6.8,4.1hz,1h),3.02–2.90(m,2h),2.71(t,2h),2.40(t,2h),2.04(t,j=7.2hz,2h),1.90(t,2h),1.69–1.57(m,2h).ms(esi):m/z751.3[m+h]+.实施例82(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((2-溴-3'-(3-羟基丙氧基)-[1,1'-联苯]-3-基)甲氧基)-5-氯苄基)-d-丝氨酸(化合物82)合成路线:化合物sa-1的合成取3-溴苯酚(5g)、3-溴-1-丙醇(15ml)和无水碳酸钾(8g)加入100ml茄形瓶中,加入n,n-二甲基甲酰胺(30ml),室温反应4小时后停止反应。加入水(100ml),用乙酸乙酯(40mlx3)萃取,无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=100:1)纯化,得无色油状化合物sa-1(9.84g)。化合物sa-2的合成取化合物sa-1(3.4g)、联硼酸频那醇酯(1.2g)、[1,1’-双(二苯基膦)二茂铁]二氯化钯二氯甲烷络合物(139mg)和醋酸钾(834mg)加入到25ml三颈瓶中,氩气保护,换气三次。室温下加入1,4-二氧六环(15ml),100℃油浴加热。反应约5小时后停止加热,减压蒸除溶剂,用乙酸乙酯25ml稀释,硅藻土抽滤,母液中加入水(50ml),用乙酸乙酯(10mlx3)萃取,无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=30:1)纯化,得无色油状化合物sa-2(560mg)。化合物bb-1的合成取化合物b-2(8.0g)溶于四氯化碳(50ml),加入n-溴代琥珀酰亚胺(nbs,5.3g)和过氧化二苯甲酰(bpo,654mg),加热回流反应,8小时后停止反应,减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚)纯化,淡黄色油状化合物bb-1(5.81g)。化合物bb-2的合成取实施例4中化合物h-1(1.35g)溶于无水乙腈(20ml)中,加入无水碳酸氢钠(1.08g),室温搅拌30分钟后缓慢加入bb-1(2.43g),70℃油浴加热。反应约20小时后停止加热,白色固体析出直接抽滤,水洗(60ml),烘干溶剂得灰白色固体化合物bb-2(2.47g)。化合物bb-3的合成取化合物bb-2(4.3g)、化合物sa-2(5.6g)、[1,1’-双(二苯基膦)二茂铁]二氯化钯二氯甲烷络合物(531mg)、无水碳酸钾(3.6g)和18-冠-6-醚(686mg)加入到250ml三颈瓶中,氩气保护,换气三次。室温下加入甲苯(30ml)、无水乙醇(10ml)和水(5ml),90℃油浴加热。反应约5小时后停止加热,减压蒸除溶剂,用乙酸乙酯80ml稀释,硅藻土抽滤,母液用乙酸乙酯(100mlx3)和饱和食盐水(50mlx3)萃取,无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/二氯甲烷=30:1)纯化,得淡黄色固体化合物bb-3(1.7g)。化合物bb-4的合成取化合物bb-3(1.73g)和碳酸铯(1.7g)加入n,n-二甲基甲酰胺(14ml)中,室温搅拌15分钟后缓慢加入xa-7(1.1g),室温反应约30分钟后停止反应。加入水(30ml),用乙酸乙酯(50mlx3)和饱和食盐水(100mlx3)萃取,无水硫酸钠干燥。减压蒸除溶剂,加入乙酸乙酯(15ml)固体析出,直接抽滤烘干溶剂至恒重得淡黄色固体化合物bb-4(1.2g)。化合物bb-5的合成取d-丝氨酸乙酯盐酸盐(163mg)溶于二氯甲烷与甲醇的混合溶剂(2:1,3ml),加入n,n-二异丙基乙胺(dipea,119mg)室温搅拌20分钟,加入化合物bb-4(150mg)和氰基硼氢化钠(45mg),室温反应12小时后停止反应。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=1:1)纯化,得黄色油状化合物bb-5(85mg)化合物82的合成取化合物bb-5(85mg)溶于甲醇与四氢呋喃的混合溶剂(2:1,3ml),加入一水合氢氧化锂(10mg),室温反应12小时后,减压蒸除溶剂,加入水(5ml),用1n盐酸调节ph=5~6,抽滤,得黄色固体化合物82(44mg):1hnmr(300mhz,dmso-d6)δ8.13(d,j=26.3hz,2h),7.52(dt,j=59.8,20.6hz,6h),7.11(s,1h),6.95(d,j=21.9hz,3h),5.37(d,j=29.0hz,4h),4.06(d,j=18.5hz,4h),3.79–3.52(m,5h),3.29–3.20(m,1h),2.03–1.79(m,2h).ms(esi):m/z710.1[m-h]-.实施例831-(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((2-溴-3'-(3-羟基丙氧基)-[1,1'-联苯]-3-(基)甲氧基)-5-氯苄基)氮杂环丁烷-3-羧酸(化合物83)参照实施例82的方法,将实施例82中的d-丝氨酸乙酯盐酸盐替换成3-甲酸甲酯氮杂环丁烷盐酸盐,制得化合物83:1hnmr(300mhz,dmso-d6)δ8.09(d,j=7.7hz,2h),7.69(d,j=9.4hz,1h),7.61(d,j=7.6hz,1h),7.45(t,j=7.6hz,1h),7.41–7.26(m,3h),7.04(s,1h),6.98(d,j=8.2hz,1h),6.91(d,j=9.0hz,2h),5.37(s,2h),5.27(s,2h),4.08(t,j=6.4hz,2h),3.55(d,j=4.8hz,4h),3.38(d,j=6.9hz,3h),3.22(t,j=6.3hz,2h),3.13(d,j=6.9hz,1h),1.87(t,j=6.3hz,2h).ms(esi):m/z708.1[m-h]-.实施例84(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-5-氯-4-((2-溴-3'-(3-(4-羟基哌啶-1-基))丙氧基)-[1,1'-联苯]-3-基)甲氧基)苄基)-d-丝氨酸乙酯(化合物84)合成路线:化合物ba-5的合成取化合物bb-4(869mg)和4-二甲氨基吡啶(4-dmap,34mg)溶于无水二氯甲烷(10ml)中,冰浴下加入三乙胺(353mg)并缓慢加入对甲基本磺酰氯(432mg),室温搅拌10小时后停止反应。用二氯甲烷(10mlx3)萃取,无水硫酸钠干燥,减压蒸干溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=6:1)纯化,得淡黄色固体化合物bb-5(887mg)。化合物bb-6的合成取化合物bb-5(290mg),4-羟基哌啶(57mg)溶于n,n-二甲基甲酰胺(5ml)中,加入无水碳酸钾(102mg),50℃油浴加热反应8小时,加水(8ml)停止反应。用乙酸乙酯(8mlx3)和饱和食盐水(5mlx3)萃取,无水硫酸钠干燥,减压蒸干溶剂,残余物经硅胶柱层析(洗脱剂:二氯甲烷/甲醇=100:1)纯化,得淡黄色固体化合物bb-6(240mg)。化合物84的合成取d-丝氨酸乙酯盐酸盐(156mg)溶于二氯甲烷与甲醇的混合溶剂(2:1,3ml),加入n,n-二异丙基乙胺(dipea,115mg)室温搅拌20分钟,加入化合物bb-6(150mg)和三乙酰氧基硼氢化钠(stab,194mg),室温反应12小时后停止反应。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:二氯甲烷/甲醇=20:1)纯化,得黄色油状化合物84(131mg):1hnmr(300mhz,methanol-d4)δ7.96(s,1h),7.89(d,j=9.3hz,1h),7.57(dd,j=12.9,8.4hz,2h),7.38–7.29(m,3h),7.22(d,j=7.3hz,1h),6.99–6.87(m,3h),6.79(s,1h),5.25(s,4h),4.14–4.02(m,4h),3.82(d,j=3.5hz,3h),3.72(t,j=4.9hz,2h),3.38(t,j=5.2hz,1h),3.14(t,j=5.8hz,2h),2.95(t,j=7.9hz,2h),2.74(s,2h),2.12(dd,j=9.9,5.5hz,2h),1.92(s,4h),1.72(t,j=6.8hz,2h),1.20(t,j=7.1hz,3h).ms(esi):m/z823.3[m+h]+.实施例85(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-5-氯-4-((2-溴-3'-(3-(4-羟基哌啶-1-基))丙氧基)-[1,1'-联苯]-3-基)甲氧基)苄基)-d-丝氨酸(化合物85)取化合物84(80mg)溶于甲醇与四氢呋喃的混合溶剂(2:1,3ml),加入一水合氢氧化锂(10mg),室温反应12小时后,减压蒸除溶剂,加入水(5ml),用1n盐酸调节ph=5~6,抽滤,得黄色固体化合物85(25mg):1hnmr(300mhz,methanol-d4)δ8.15(s,1h),8.07(d,j=9.3hz,1h),7.72(d,j=9.3hz,1h),7.61(d,j=7.2hz,1h),7.53(s,1h),7.49–7.41(m,1h),7.40–7.31(m,2h),7.08(s,1h),6.97(d,j=8.2hz,1h),6.91(d,j=9.4hz,2h),5.39(s,2h),5.30(s,2h),4.04(d,j=5.8hz,2h),4.00(d,j=5.2hz,2h),3.73–3.66(m,2h),3.66–3.59(m,2h),2.76–2.66(m,2h),2.41(t,j=7.0hz,2h),2.03(t,j=9.9hz,2h),1.93–1.80(m,2h),1.75–1.64(m,2h),1.45–1.28(m,2h).ms(esi):m/z795.3[m+h]+.实施例86(r)-1-(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((2-溴-[1,1'-联苯]-3-基)甲氧基)-5-氟苄基)哌啶-2-羧酸(化合物86)合成路线:化合物h-3的合成取二氯甲基甲醚(2.8ml)溶于无水二氯甲烷(20ml)中,滴加四氯化钛(5.1ml),冰浴下加4-氟间苯二酚(2g),自然升温至室温反应过夜。冰浴下以10ml1nhcl与20ml水淬灭,二氯甲烷萃取(20mlx3),无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=5:1)纯化,得黄色固体化合物h-3(0.48g)。化合物ba-1的合成取化合物h-3(143mg)溶于无水乙腈(10ml)中,加入无水碳酸氢钠(103mg),并缓慢加入b-4(200mg),70℃油浴加热。反应约20小时后停止加热,减压蒸除溶剂,加入乙酸乙酯(10ml)稀释,用水(10mlx3)和饱和食盐水(10mlx2)洗涤,无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=10:1)纯化,得白色固体化合物ba-1(236mg)。化合物ba-2的合成取化合物ba-1(200mg)和碳酸铯(325mg)加入n,n-二甲基甲酰胺(6ml)中,室温搅拌15分钟后缓慢加入xa-7(127mg),室温反应约1小时后停止反应。加入水(30ml),用乙酸乙酯(20mlx3)萃取,无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=2:1)纯化,得白色固体化合物ba-2(170mg)。化合物86的合成参照实施例1的方法,将实施例1中的i-7替换成ba-2,经水解直接制得化合物86(6mg):1hnmr(300mhz,meod-d4)δ7.93(d,j=10.1hz,1h),7.60(d,j=9.3hz,1h),7.54–7.29(m,4h),7.26(d,j=6.3hz,1h),6.96(d,j=6.9hz,1h),5.33(s,2h),4.41(dd,j=36.7,13.2hz,1h),3.50(s,1h),2.95(s,1h),2.20(s,1h),1.79(s,2h),1.29(s,1h).hrms(esi):m/zcalcdforc33h30brfn3o5[m+h]+646.13374or648.13269found648.13257.实施例87(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((2-氯-[1,1'-联苯]-3-基)甲氧基)-5-氟苄基)-d-丝氨酸乙酯盐酸盐(化合物87)合成路线:化合物aa-1的合成取3-溴-2-氯甲苯(7.1g)、溴代丁二酰亚胺(6.7g)和过氧化二苯甲酰(418mg)溶解于50ml四氯化碳中,90℃油浴加热。反应8小时后停止加热,减压蒸出溶剂,用乙酸乙酯30ml稀释后减压抽滤,母液经硅胶柱层析(洗脱剂:石油醚)纯化,得无色油状化合物aa-1(8.9g)。化合物ab-1的合成取化合物aa-1(288mg)溶于无水乙腈(10ml)中,加入无水碳酸氢钠(170mg),缓慢加入s-1(190mg),70℃油浴加热。反应约6小时后停止加热,冷却至室温后减压抽滤,用水5ml洗涤,干燥至恒重,得黄色固体化合物ab-1(300mg)。化合物ab-2的合成取化合物ab-1(150mg)、苯硼酸(61mg)、双三苯基膦二氯化钯(15mg)和无水碳酸钠(89mg),加入到100ml三颈瓶中,氩气保护。室温下加入甲苯(10ml)、甲醇(3ml)和水(1ml)。80℃油浴加热。反应约5小时后停止加热,减压蒸除溶剂,用乙酸乙酯25ml稀释,硅藻土抽滤,母液用乙酸乙酯(45mlx3)萃取,饱和食盐水(30mlx2)洗涤,无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=10:1)纯化,得米色固体化合物ab-2(142mg)。化合物ab-3的合成取化合物ab-2(142mg)和碳酸铯(195mg)加入n,n-二甲基甲酰胺(4ml)中,室温搅拌15分钟后缓慢加入xa-7(93mg),室温反应约30分钟后停止反应。加入水(10ml),析出白色固体,减压抽滤,干燥至恒重,得白色固体化合物ba-2(150mg)。化合物87的合成参照实施例1的方法,将实施例1中的i-6替换成ab-3,不经水解直接制得化合物87的游离碱,再将化合物87的游离碱加入盐酸乙醇溶液中,搅拌过夜,抽滤,得白色固体化合物87(120mg):1hnmr(300mhz,dmso-d6)δ9.45(d,2h),8.17(s,1h),8.11(d,j=9.3hz,1h),7.70(d,j=9.3hz,1h),7.62(d,j=7.0hz,1h),7.58–7.34(m,4h),7.19(d,j=7.3hz,1h),5.63(s,1h),5.37(d,j=2.9hz,4h),4.28(s,2h),4.19–4.04(m,3h),3.92(dd,j=29.1,11.9hz,2h),1.16(t,j=7.1hz,3h).ms(esi):m/z606.2[m+h]+.实施例88(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((2-氯-[1,1'-联苯]-3-基)甲氧基)-5-氟苄基)-d-丝氨酸(化合物88)取化合物87(100mg)溶于甲醇与四氢呋喃的混合溶剂(2:1,3ml),加入一水合氢氧化锂(20mg),室温反应12小时后,减压蒸除溶剂,加入水(2ml),用冰醋酸调节ph=4~5,抽滤,得白色固体化合物88(89mg):1hnmr(300mhz,dmso-d6)δ8.14(s,1h),8.07(d,j=9.3hz,1h),7.71(d,j=9.3hz,1h),7.61(d,j=7.2hz,1h),7.54–7.34(m,8h),7.15(d,j=7.2hz,1h),5.35(s,2h),5.33(s,2h),4.01(s,2h),3.79–3.59(m,2h),3.21(t,j=5.3hz,1h).ms(esi):m/z576.2[m-h]-.实施例89n-(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-5-氯-4-((2-氯-4'-氟-[1,1'-联苯]-3-基)甲氧基)苄基)-d-丝氨酸乙酯(化合物89)合成路线:化合物u-1的合成取化合物3-硝基-2-氯甲苯(25g)溶于150ml乙醇中,依次加入铁粉(24g),浓盐酸(12ml),加热至60℃。反应3小时后停止加热,冷却至室温,经硅藻土抽滤,滤液用饱和碳酸钠溶液调节ph=10,乙酸乙酯萃取(100mlx3),无水硫酸钠干燥。减压蒸除溶剂,得棕黄色油状化合物u-1(20g)。化合物u-2的合成取化合物u-1(14g)溶于水(20ml)中,冰盐浴下缓慢加入盐酸(25ml),搅拌5分钟,缓慢滴加亚硝酸钠(8g)的水溶液,20分钟加毕,冰盐浴下搅拌30分钟后,向反应液中缓慢滴加碘化钾(24g)的水溶液,室温下反应8小时后停止反应。加入硫代硫酸钠的饱和水溶液(100ml),用乙酸乙酯(100mlx3)萃取,饱和食盐水(100mlx3)洗涤,无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚)纯化,得淡黄色油状化合物u-2(17.6g)。化合物u-3的合成取对氟苯硼酸(660mg)、四(三苯基膦)钯(200mg)和无水碳酸钠(839mg)置于三颈瓶中,氩气保护。将化合物u-2(1g)溶于甲苯(8ml)中,加入反应瓶,加入甲苯(22ml)和甲醇(3ml),80℃油浴加热。反应约4小时后停止加热,减压蒸除溶剂,用乙酸乙酯(15ml)稀释,硅藻土抽滤,母液经硅胶柱层析(洗脱剂:石油醚)纯化,得淡黄色油状化合物u-3(816mg)。化合物u-4的合成取化合物u-3(816mg),溶于四氯化碳(15ml)中,加入n-溴代丁二酰亚胺(nbs,691mg),分批加入过氧化苯甲酰(bpo,45mg),90℃油浴加热。反应约6小时后停止加热,减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚)纯化,得无色油状化合物u-4(430mg)。化合物u-5的合成取化合物h-1(370mg)溶于无水乙腈(15ml)中,加入无水碳酸氢钠(241mg),缓慢加入u-4(430mg),70℃油浴加热。反应约6小时后停止反应,冷却至室温后减压抽滤,用水5ml洗涤,干燥至恒重,得黄色固体化合物u-5(395mg)。化合物u-6的合成取化合物u-5(200mg)和碳酸铯(333mg)加入n,n-二甲基甲酰胺(4ml)中,室温搅拌15分钟后缓慢加入xa-7(130mg),室温反应约30分钟后停止反应。加入水(20ml),用乙酸乙酯(15mlx3)萃取,无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=2:1)纯化,得白色固体化合物u-6(240mg)。化合物89的合成取d-丝氨酸乙酯盐酸盐(104mg)溶于二氯甲烷与甲醇的混合溶剂(3:1,4ml),加入n,n-二异丙基乙胺(dipea,79mg)室温搅拌20分钟,加入化合物u-6(80mg)和三乙酰氧基硼氢化钠(stab,129mg),室温反应12小时后停止反应。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=1:1)纯化,得白色固体化合物89(75mg):1hnmr(300mhz,meod-d4)δ8.05(s,1h),7.95(d,j=9.3hz,1h),7.69–7.56(m,3h),7.49–7.39(m,2h),7.38–7.27(m,2h),7.20(t,j=8.7hz,2h),7.00(s,1h),5.40(s,4h),4.53–4.32(m,2h),4.22(dd,j=13.1,6.1hz,2h),4.13(d,j=3.7hz,1h),4.04(s,2h),1.27(t,j=7.1hz,3h).ms(esi):m/z640.2[m+h]+.实施例90(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-5-氯-4-((2-氯-4'-氟-[1,1'-联苯]-3-基)甲氧基)苄基)-d-丝氨酸(化合物90)取化合物89(65mg)溶于甲醇与四氢呋喃的混合溶剂(2:1,3ml),加入一水合氢氧化锂(20mg),室温反应12小时后,减压蒸除溶剂,加入水(3ml),用冰醋酸调节ph=5~6,抽滤,得白色固体化合物90(30mg):1hnmr(300mhz,dmso-d6)δ8.14(s,1h),8.07(d,j=9.2hz,1h),7.72(d,j=9.3hz,1h),7.64(d,j=6.7hz,1h),7.53(s,1h),7.51–7.37(m,4h),7.32(t,j=8.8hz,2h),7.11(s,1h),5.40(s,2h),5.33(s,2h),3.98(s,2h),3.73–3.58(m,2h),3.16(s,1h).ms(esi):m/z610.1[m-h]-.实施例91(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-5-氯-4-((2-氯-3'-氟-[1,1'-联苯]-3-基)甲氧基)苄基)-d-丝氨酸乙酯盐酸盐(化合物91)合成路线:化合物u-1的合成取化合物3-硝基-2-氯甲苯(25g)溶于150ml乙醇中,依次加入铁粉(24g),浓盐酸(12ml),加毕,加热至60℃。反应3小时后停止加热,冷却至室温,经硅藻土抽滤,滤液用饱和碳酸钠溶液调节ph=10,乙酸乙酯萃取(100mlx3),无水硫酸钠干燥。减压蒸除溶剂,得棕黄色化合物u-1(20g)。化合物u-2的合成取化合物u-1(14g)溶于水(20ml)中,冰盐浴下缓慢加入盐酸(25ml),搅拌5分钟,缓慢滴加亚硝酸钠(8g)的水溶液,20分钟加毕,冰盐浴下搅拌30分钟后,向反应液中缓慢滴加碘化钾(24g)的水溶液,室温下反应8小时后停止反应。加入硫代硫酸钠的饱和水溶液(100ml),用乙酸乙酯(100mlx3)萃取,饱和食盐水(100mlx3)洗涤,无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚)纯化,得淡黄色油状化合物u-2(17.6g)。化合物ua-1的合成取间氟苯硼酸(660mg)、四(三苯基膦)钯(200mg)和无水碳酸钠(839mg)置于三颈瓶中,氩气保护。将化合物u-2(1g)溶于甲苯(8ml)中,加入反应瓶,加入甲苯(22ml)和甲醇(3ml),80℃油浴加热。反应约4小时后停止加热,减压蒸除溶剂,用乙酸乙酯(15ml)稀释,硅藻土抽滤,母液经硅胶柱层析(洗脱剂:石油醚)纯化,得无色油状化合物ua-1(900mg)。化合物ua-2的合成取化合物ua-1(900mg),溶于四氯化碳(15ml)中,加入n-溴代丁二酰亚胺(nbs,838mg),分批加入过氧化苯甲酰(bpo,54mg),90℃油浴加热。反应约6小时后停止加热,减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚)纯化,得无色油状化合物ua-2(532mg)。化合物ua-3的合成取化合物h-1(458mg)溶于无水乙腈(15ml)中,加入无水碳酸氢钠(298mg),缓慢加入ua-2(532mg),70℃油浴加热。反应约6小时后停止反应,冷却至室温后减压抽滤,用水5ml洗涤,干燥至恒重,得黄色固体化合物ua-3(686mg)。化合物ua-4的合成取化合物ua-3(200mg)和碳酸铯(333mg)加入n,n-二甲基甲酰胺(4ml)中,室温搅拌15分钟后缓慢加入xa-7(130mg),室温反应约30分钟后停止反应。加入水(20ml),用乙酸乙酯(15mlx3)萃取,无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=2:1)纯化,得白色固体化合物ua-4(145mg)。化合物91的合成取d-丝氨酸乙酯盐酸盐(104mg)溶于二氯甲烷与甲醇的混合溶剂(3:1,4ml),加入n,n-二异丙基乙胺(dipea,79mg)室温搅拌20分钟,加入化合物ua-4(80mg)和三乙酰氧基硼氢化钠(stab,129mg),室温反应12小时后停止反应。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=1:1)纯化,得黄色固体化合物91(65mg):1hnmr(300mhz,meod-d4)δ8.05(s,1h),7.95(d,j=9.4hz,1h),7.69–7.56(m,3h),7.54–7.43(m,1h),7.36(d,j=7.4hz,2h),7.26–7.12(m,3h),7.01(s,1h),5.40(s,4h),4.41(d,j=3.7hz,2h),4.29–4.19(m,2h),4.14(t,1h),4.04(s,2h),1.27(t,j=7.1hz,3h).ms(esi):m/z640.2[m+h]+.实施例92(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-5-氯-4-((2-氯-3'-氟-[1,1'-联苯]-3-基)甲氧基)苄基)-d-丝氨酸(化合物92)取化合物91(55mg)溶于甲醇与四氢呋喃的混合溶剂(2:1,3ml),加入一水合氢氧化锂(17mg),室温反应12小时后,减压蒸除溶剂,加入水(3ml),用冰醋酸调节ph=5~6,抽滤,得白色固体化合物92(23mg):1hnmr(300mhz,dmso-d6)δ8.14(s,1h),8.06(d,j=9.3hz,1h),7.78–7.61(m,2h),7.59–7.48(m,2h),7.47–7.36(m,2h),7.33–7.21(m,3h),7.11(s,1h),5.39(s,2h),5.33(s,2h),4.02(s,2h),3.79–3.59(m,2h),3.25(t,j=5.3hz,1h).ms(esi):m/z640.2[m+k]+.实施例93(s)-3-((2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-5-氯-4-((2-氯-4'-氟-[1,1'-联苯基]-3-基)甲氧基)苄基)氨基)丙烷-1,2-二醇(化合物93)合成路线:化合物u-1的合成取化合物3-硝基-2-氯甲苯(25g)溶于150ml乙醇中,依次加入铁粉(24g),浓盐酸(12ml),加热至60℃。反应3小时后停止加热,冷却至室温,经硅藻土抽滤,滤液用饱和碳酸钠溶液调节ph=10,乙酸乙酯萃取(100mlx3),无水硫酸钠干燥。减压蒸除溶剂,得棕黄色液体20g。化合物u-2的合成取化合物u-1(14g)溶于水(20ml)中,冰盐浴下缓慢加入盐酸(25ml),搅拌5分钟,缓慢滴加亚硝酸钠(8g)的水溶液,20分钟加毕,冰盐浴下搅拌30分钟后,向反应液中缓慢滴加碘化钾(24g)的水溶液,室温下反应8小时后停止反应。加入硫代硫酸钠的饱和水溶液(100ml),用乙酸乙酯(100mlx3)萃取,饱和食盐水(100mlx3)洗涤,无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚)纯化,得淡黄色油状化合物u-2(17.6g)。化合物u-3的合成取对氟苯硼酸(660mg)、四(三苯基膦)钯(200mg)和无水碳酸钠(839mg)置于三颈瓶中,氩气保护。将化合物u-2(1g)溶于甲苯(8ml)中,加入反应瓶,加入甲苯(15ml)和甲醇(3ml),80℃油浴加热。反应约4小时后停止加热,减压蒸除溶剂,用乙酸乙酯(15ml)稀释,硅藻土抽滤,母液经硅胶柱层析(洗脱剂:石油醚)纯化,得淡黄色油状化合物u-3(816mg)。化合物u-4的合成取化合物u-3(816mg),溶于四氯化碳(15ml)中,加入n-溴代丁二酰亚胺(nbs,691mg),分批加入过氧化苯甲酰(bpo,45mg),90℃油浴加热。反应约6小时后停止加热,减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚)纯化,得无色油状化合物u-4(430mg)。化合物u-5的合成取化合物h-1(370mg)溶于无水乙腈(15ml)中,加入无水碳酸氢钠(241mg),缓慢加入u-4(430mg),70℃油浴加热。反应约6小时后停止反应,冷却至室温后减压抽滤,用水5ml洗涤,干燥至恒重,得黄色固体化合物u-5(395mg)。化合物u-6的合成取化合物u-5(200mg)和碳酸铯(333mg)加入n,n-二甲基甲酰胺(4ml)中,室温搅拌15分钟后缓慢加入xa-7(130mg),室温反应约30分钟后停止反应。加入水(20ml),用乙酸乙酯(15mlx3)萃取,无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=2:1)纯化,得白色固体化合物u-6(240mg)。化合物93的合成取(r)-3-氨基-1,2-丙二醇(21mg)溶于二氯甲烷与甲醇的混合溶剂(3:1,4ml),加入化合物u-6(30mg)和三乙酰氧基硼氢化钠(stab,48mg),室温反应12小时后停止反应。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=1:1)纯化,得黄色固体化合物93(22mg):1hnmr(300mhz,meod-d4)δ7.98–7.87(m,2h),7.61(d,j=9.2hz,2h),7.49–7.38(m,3h),7.37–7.26(m,2h),7.19(t,j=8.7hz,2h),6.89(s,1h),5.33(s,2h),5.31(s,2h),3.97–3.73(m,3h),3.51(dd,j=3.3hz,2h),2.84–2.73(m,1h),2.70–2.59(m,1h).ms(esi):m/z598.2[m+h]+.实施例94(s)-3-((2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-5-氯-4-((2-氯-3'-氟-[1,1'-联苯基]-3-基)甲氧基)苄基)氨基)丙烷-1,2-二醇(化合物94)合成路线:化合物u-1的合成取化合物3-硝基-2-氯甲苯(25g)溶于150ml乙醇中,依次加入铁粉(24g),浓盐酸(12ml),加毕,加热至60℃。反应3小时后停止加热,冷却至室温,经硅藻土抽滤,滤液用饱和碳酸钠溶液调节ph=10,乙酸乙酯萃取(100mlx3),无水硫酸钠干燥。减压蒸除溶剂,得棕黄色油状化合物u-1(20g)。化合物u-2的合成取化合物u-1(14g)溶于水(20ml)中,冰盐浴下缓慢加入盐酸(25ml),搅拌5分钟,缓慢滴加亚硝酸钠(8g)的水溶液,20分钟加毕,冰盐浴下搅拌30分钟后,向反应液中缓慢滴加碘化钾(24g)的水溶液,室温下反应8小时后停止反应。加入硫代硫酸钠的饱和水溶液(100ml),用乙酸乙酯(100mlx3)萃取,饱和食盐水(100mlx3)洗涤,无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚)纯化,得淡黄色油状化合物u-2(17.6g)。化合物ua-1的合成取间氟苯硼酸(660mg)、四(三苯基膦)钯(200mg)和无水碳酸钠(839mg)置于三颈瓶中,氩气保护。将化合物u-2(1g)溶于甲苯(8ml)中,加入反应瓶,加入甲苯(15ml),甲醇(3ml),80℃油浴加热。反应约4小时后停止加热,减压蒸除溶剂,用乙酸乙酯(15ml)稀释,硅藻土抽滤,母液经硅胶柱层析(洗脱剂:石油醚)纯化,得无色油状化合物ua-1(900mg)。化合物ua-2的合成取化合物ua-1(900mg),溶于四氯化碳(15ml)中,加入n-溴代丁二酰亚胺(nbs,838mg),分批加入过氧化苯甲酰(bpo,54mg),90℃油浴加热。反应约6小时后停止加热,减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚)纯化,得无色油状化合物ua-2(532mg)。化合物ua-3的合成取化合物h-1(458mg)溶于无水乙腈(15ml)中,加入无水碳酸氢钠(298mg),缓慢加入ua-2(532mg),70℃油浴加热。反应约6小时后停止反应,冷却至室温后减压抽滤,用水5ml洗涤,干燥至恒重,得黄色固体化合物ua-3(686mg)。化合物ua-4的合成取化合物ua-3(200mg)和碳酸铯(333mg)加入n,n-二甲基甲酰胺(4ml)中,室温搅拌15分钟后缓慢加入xa-7(130mg),室温反应约30分钟后停止反应。加入水(20ml),用乙酸乙酯(15mlx3)萃取,无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=2:1)纯化,得白色固体化合物ua-4(145mg)。化合物94的合成取(r)-3-氨基-1,2-丙二醇(21mg)溶于二氯甲烷与甲醇的混合溶剂(3:1,4ml),加入化合物ua-4(30mg)和三乙酰氧基硼氢化钠(stab,48mg),室温反应12小时后停止反应。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=1:1)纯化,得黄色固体化合物94(26mg):1hnmr(300mhz,meod)δ7.96(s,1h),7.92(d,j=9.5hz,1h),7.71–7.57(m,2h),7.48(q,j=6.9hz,1h),7.43–7.28(m,3h),7.27–7.10(m,3h),6.89(s,1h),5.34(s,2h),5.31(s,2h),4.03–3.70(m,3h),3.50(d,j=3.9hz,2h),2.83–2.72(m,1h),2.70–2.57(m,1h).ms(esi):m/z598.2[m+h]+.实施例95(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-5-氯-4-((2-氯-3'-羟基-[1,1'-联苯]-3-基)甲氧基)苄基)-d-丝氨酸(化合物95)合成路线:化合物sa-1的合成取3-溴苯酚(5g)、3-溴-1-丙醇(15ml)和无水碳酸钾(8g)加入100ml茄形瓶中,加入1,4-二氧六环(30ml),室温反应4小时后停止反应。加入水(100ml),用乙酸乙酯(40mlx3)萃取,无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=100:1)纯化,得无色油状化合物sa-1(9.84g)。化合物sb-1的合成取氧化铬(14.6g),冰浴条件下加至丙酮(80ml)中,硫酸(15ml)以水(30ml)稀释后加入反应瓶中,然后加入sa-1(8.4g),自然升温至室温。反应约8小时后经tlc监测反应完全,以异丙醇(10ml)淬灭,由黑色变为绿色。减压抽滤后滤液经乙酸乙酯(100mlx2)萃取,有机相经无水硫酸钠干燥后减压蒸出溶剂,得黄色固体sb-1(8.9g)。化合物sb-2的合成取化合物sb-1(5g)溶于乙醇(15ml)中,冰浴条件下滴加氯化亚砜(1.8ml),5小时后停止反应,减压蒸出溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=20:1)纯化,得无色油状化合物sb-2(4.8g)。化合物sb-3的合成取化合物sb-2(3.1g)、联硼酸频那醇酯(3.5g)、1,1'-双(二苯膦基)二茂铁二氯化钯(ii)二氯甲烷复合物(230mg)和醋酸钾(2.2g)溶于1,4-二氧六环(50ml),加毕,氩气保护,换气三次。缓慢升温至85℃反应8h后,经tlc监测反应完全。冷却至室温,硅藻土抽滤,母液经乙酸乙酯(50mlx3)萃取,饱和食盐水(50mlx3)洗涤,有机层经无水硫酸钠干燥后,减压蒸干溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=20:1),得到黄色油状化合物sb-3(3.9g)。化合物sb-4的合成:取化合物sb-3(262mg)、中间体aa-2(600mg)、1,1'-双(二苯膦基)二茂铁二氯化钯(ii)二氯甲烷复合物(65mg)以及醋酸钾(314mg)溶于甲苯/乙醇/水为3/1/0.5的混合溶液(20ml),氩气保护,换气三次。缓慢升温至85℃反应5h后,经tlc监测反应完全。冷却至室温,硅藻土抽滤,母液中加入水(20ml),经乙酸乙酯(15mlx3)萃取,饱和食盐水(20mlx3)洗涤,有机层经无水硫酸钠干燥后,减压蒸干溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=5:1)得到浅黄色固体化合物sb-4(476mg)。化合物sb-5的合成:取化合物sb-4(476mg)和碳酸铯(475mg)溶于n,n-二甲基甲酰胺dmf(8ml)中,室温搅拌10min,加入中间体xa-7(227mg),室温搅拌15min,白色固体析出。加水(5ml)停止反应,固体析出。抽滤,滤饼经水洗涤,烘干至恒重得到白色固体化合物sb-5(502mg)。化合物sb-6的合成取化合物d-丝氨酸乙酯盐酸盐(103mg)于二氯甲烷/甲醇为2/1(6ml)的混合溶液中,滴加n,n-二异丙基乙胺dipea(79mg)室温搅拌20min后,加入化合物sb-5(90mg)以及三乙酰氧基硼氢化钠stab(128mg),室温反应过夜。监测反应完全,直接经硅胶柱层析(洗脱剂;二氯甲烷/甲醇=30:1)得到无色透明油状化合物sb-6(49mg)。化合物95的合成取化合物sb-6(49mg)于甲醇(4ml)和水(5滴)的混合溶液中,冰浴下加入一水合氢氧化锂(14mg)加毕,室温反应过夜。监测反应完全,减压蒸干溶剂,加水(2ml),以1nhcl调节ph=4~5白色固体析出,抽滤,滤饼经二氯甲烷/无水乙醚打浆得到白色固体化合物95(33mg):1hnmr(300mhz,dmso-d6)δ9.62(s,1h),8.16(s,1h),8.08(d,j=9.3hz,1h),7.72(d,j=9.2hz,1h),7.62(d,j=7.5hz,1h),7.55(s,1h),7.47–7.31(m,2h),7.27(t,j=7.8hz,1h),7.12(s,1h),6.80(s,3h),5.40(s,2h),5.34(s,2h),4.02(s,2h),3.78–3.60(m,2h),3.22(t,1h).ms(esi):m/z608.1[m-h]-.实施例96(s)-3-((2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-5-氯-4-((2-氯-4'-(甲基磺酰基)-[1,1'-联苯基]-3-基)甲氧基)苄基)氨基)丙烷-1,2-二醇(化合物96)合成路线:化合物aa1-1的合成取化合物aa-2(300mg)、对甲磺酰基苯硼酸(192mg)、四三苯基膦钯(46mg)和无水碳酸钠(170mg)加入到100ml三颈瓶中,氩气保护,换气三次。室温下加入甲苯(15ml)和甲醇(3ml)。80℃油浴加热。反应约5小时后停止加热,减压蒸除溶剂,用乙酸乙酯25ml稀释,硅藻土抽滤,母液以饱和食盐水(10mlx2)洗涤,无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=4:1)纯化,得黄色固体化合物aa1-1(170mg)。化合物aa1-2的合成取化合物aa1-1(170mg)和碳酸铯(184mg)加入n,n-二甲基甲酰胺(5ml)中,室温搅拌15分钟后缓慢加入xa-7(88mg),室温反应约30分钟后停止反应。加入水(5ml),固体析出,经减压抽滤后干燥得白色固体化合物aa1-2(187mg)。化合物96的合成取(r)-3-氨基-1,2-丙二醇(19mg)溶于二氯甲烷与甲醇的混合溶剂(3:1,4ml),加入化合物aa1-2(30mg)和三乙酰氧基硼氢化钠(stab,43mg),室温反应12小时后停止反应。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:二氯甲烷/甲醇=15:1)纯化,得黄色固体化合物96(16mg):1hnmr(300mhz,dmso-d6)δ8.14–8.00(m,4h),7.76–7.64(m,4h),7.55–7.40(m,3h),7.10(s,1h),5.38(s,2h),5.34(s,2h),4.62(s,1h),3.76(s,2h),3.58(s,1h),2.62(dd,1h).ms(esi):m/z658.2[m+h]+.实施例97乙基(2-(苯并[c][1,2,5]恶二唑-5-基甲氧基)-5-氯-4-((2-氯-4'-(甲基氨基甲酰基)-[1,1'-联苯]-3-基)甲氧基)苄基)-d-丝氨酸(化合物97)合成路线:化合物aa2-1的合成取化合物aa-2(300mg)、4-(甲基氨基甲酰基)苯硼酸(158mg)、四三苯基膦钯(46mg)和无水碳酸钠(170mg)加入到100ml三颈瓶中,氩气保护,换气三次。室温下加入甲苯(15ml)和甲醇(3ml)。80℃油浴加热。反应约5小时后停止加热,减压蒸除溶剂,用乙酸乙酯25ml稀释,硅藻土抽滤,母液以饱和食盐水(10mlx2)洗涤,无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=4:1)纯化,得白色固体化合物aa2-1(140mg)。化合物aa2-2的合成取化合物aa2-1(140mg)和碳酸铯(153mg)加入n,n-二甲基甲酰胺(5ml)中,室温搅拌15分钟后缓慢加入xa-7(74mg),室温反应约30分钟后停止反应。加入水(5ml),固体析出,经减压抽滤后干燥至恒重,得白色固体化合物aa2-2(175mg)。化合物aa-3的合成取d-丝氨酸乙酯盐酸盐(106mg)溶于二氯甲烷与甲醇的混合溶剂(3:1,4ml),滴加n,n-二异丙基乙胺dipea(79mg)室温搅拌20min后,加入化合物aa2-2(80mg)和三乙酰氧基硼氢化钠(stab,133mg),室温反应12小时后停止反应。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:二氯甲烷/甲醇=20:1)纯化,得化合物97的游离碱(60mg),再将化合物97的游离碱加入盐酸乙醇溶液中,搅拌过夜,抽滤,得黄色固体化合物aa-3(61mg)。化合物97的合成以化合物96的游离碱为关键中间体,参照实施例1的方法进行水解制得化合物97:(21mg):1hnmr(300mhz,dmso-d6)δ8.51(d,j=4.4hz,1h),8.15(s,1h),8.07(d,j=9.4hz,1h),7.93(d,j=8.2hz,2h),7.69(m,j=15.5,7.5hz,2h),7.59–7.36(m,5h),7.12(s,1h),5.40(s,2h),5.35(s,2h),4.01(s,2h),3.68(m,j=17.0,11.2,5.3hz,2h),3.21(t,j=5.4hz,1h),2.82(d,j=4.4hz,3h).ms(esi):m/z649.2[m-h]-.实施例98(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-5-氯-4-((2-氯-4'-(甲基磺酰基)-[1,1'-联苯]-3-基)甲氧基)苄基)-d-丝氨酸(化合物98)合成路线:化合物aa1-1的合成取化合物aa-2(300mg)、对甲磺酰基苯硼酸(192mg)、四三苯基膦钯(46mg)和无水碳酸钠(170mg)加入到100ml三颈瓶中,氩气保护,换气三次。室温下加入甲苯(15ml)和甲醇(3ml)。80℃油浴加热。反应约5小时后停止加热,减压蒸除溶剂,用乙酸乙酯25ml稀释,硅藻土抽滤,母液以饱和食盐水(10mlx2)洗涤,无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=4:1)纯化,得黄色固体化合物aa1-1(170mg)。化合物aa1-2的合成取化合物aa1-1(170mg)和碳酸铯(184mg)加入n,n-二甲基甲酰胺(5ml)中,室温搅拌15分钟后缓慢加入xa-7(88mg),室温反应约30分钟后停止反应。加入水(5ml),固体析出,经减压抽滤后干燥得白色固体化合物aa1-2(187mg)。化合物aa1-3的合成取d-丝氨酸乙酯盐酸盐(52mg)溶于二氯甲烷与甲醇的混合溶剂(3:1,4ml),滴加n,n-二异丙基乙胺(dipea,40mg)室温搅拌20min后,加入化合物aa1-2(80mg)和三乙酰氧基硼氢化钠(stab,65mg),室温反应12小时后停止反应。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:二氯甲烷/甲醇=20:1)纯化,得黄色固体化合物aa1-3(63mg)。化合物98的合成取化合物aa1-3(60mg)于甲醇(3ml)和水(5滴)的混合溶液中,冰浴下加入一水合氢氧化锂(20mg)加毕,室温反应过夜。经监测反应完全,减压蒸干溶剂,加水(1ml),以1nhcl调节ph=5~6白色固体析出,抽滤,滤饼经二氯甲烷/无水乙醚打浆得到白色固体化合物98(35mg):1hnmr(300mhz,dmso-d6)δ8.15(s,1h),8.06(t,j=8.0hz,3h),7.72(d,j=8.5hz,4h),7.54(s,1h),7.52–7.39(m,2h),7.12(s,1h),5.41(s,2h),5.36(s,2h),4.01(s,2h),3.67(m,j=11.0,5.3hz,2h),3.18(t,j=5.3hz,1h).ms(esi):m/z670.1[m-h]-.实施例99(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-5-氯-4-((2-氯-2',3'-二氟-[1,1'-联苯]-3-基)甲氧基)苄基)-d-丝氨酸(化合物99)合成路线:化合物ub-1的合成取化合物u-2(505mg)、2,3-二氟苯硼酸(347mg)、四(三苯基膦)钯(116mg)和无水碳酸钠(424mg)置于三颈瓶中,加入甲苯(22ml)和甲醇(3ml),氩气保护。80℃油浴加热。反应约4小时后停止加热,减压蒸除溶剂,用乙酸乙酯25ml稀释,硅藻土抽滤,母液以饱和食盐水(10mlx2)洗涤,无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚)纯化,得ub-1(252mg)。化合物ub-2的合成取化合物ub-1(252mg),溶于四氯化碳(10ml)中,加入n-溴代丁二酰亚胺(nbs,196mg),分批加入偶氮二异丁腈(aibn,18mg),90℃油浴加热。反应约6小时后停止加热,减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚)纯化,得无色油状化合物ub-2(180mg)。化合物ub-3的合成取化合物h-1(107mg)溶于无水乙腈(10ml)中,加入无水碳酸氢钠(95mg),缓慢加入ub-2(180mg),80℃油浴加热。反应约6小时后停止反应,冷却至室温减压抽滤,用水5ml洗涤,干燥至恒重,得白色固体化合物ub-3(178mg)。化合物ub-4的合成取化合物ub-3(178mg)和碳酸铯(283mg)加入n,n-二甲基甲酰胺(4ml)中,室温搅拌15分钟后缓慢加入xa-7(101mg),室温反应约30分钟后停止反应。加入水(5ml),固体析出,经减压抽滤后干燥至恒重,得白色固体化合物ub-4(204mg)。化合物ub-5的合成取d-丝氨酸乙酯盐酸盐(94mg)溶于二氯甲烷与甲醇的混合溶剂(3:1,4ml),加入n,n-二异丙基乙胺(dipea,72mg)室温搅拌20分钟,加入化合物ub-4(100mg)和三乙酰氧基硼氢化钠(stab,195mg),室温反应12小时后停止反应。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=1:1)纯化,得化合物ub-5的游离碱,加入盐酸乙醇,搅拌1h,减压抽滤,得白色固体化合物ub-5(69mg)。化合物99的合成取化合物ub-5(50mg)于甲醇(4ml)和水(5滴)的混合溶液中,冰浴下加入一水合氢氧化锂(12mg)加毕,室温反应过夜。经tlc监测反应完全,减压蒸干溶剂,加水(1ml),以1nhcl调节ph=5~6白色固体析出,抽滤,滤饼经二氯甲烷/无水乙醚打浆得到白色固体化合物99(45mg):1hnmr(300mhz,dmso-d6)δ8.22(s,1h),8.11(d,j=9.0hz,1h),7.87(d,j=9.1hz,1h),7.72(d,j=6.9hz,1h),7.61–7.50(m,2h),7.49–7.42(m,2h),7.40–7.31(m,1h),7.27–7.19(m,1h),7.15(s,1h),5.47(s,2h),5.35(s,2h),3.99(s,2h),3.79–3.59(m,2h),3.20(t,1h).ms(esi):m/z644.1[m-h]-.实施例100(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-5-氯-4-((2-氯-3'-(2-(二乙氨基)-2-氧乙基)-[1,1'-联苯基]-3-基)甲氧基)苄基)-d-丝氨酸(化合物100)合成路线:化合物v-1的合成取化合物2-(3-溴苯基)乙酸甲酯(2g)、联硼酸频那醇酯(2.6g)、1,1'-双(二苯膦基)二茂铁二氯化钯(ii)二氯甲烷复合物(200mg)和醋酸钾(1.7g)溶于1,4-二氧六环(20ml),氩气保护,换气三次。缓慢升温至85℃反应8h后,经tlc监测反应完全。冷却至室温,硅藻土抽滤,母液减压浓缩后经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=15:1)得到无色油状化合物v-1(2.4g)。化合物v-2的合成取化合物v-1(2.5g)、关键中间体a-2(1.7g)、1,1'-双(二苯膦基)二茂铁二氯化钯(ii)二氯甲烷复合物(31mg)以及醋酸钾(1.5g)溶于1,4-二氧六环(20ml),加毕,氩气保护,换气三次。缓慢升温至85℃反应5h后,经tlc监测反应完全。冷却至室温,硅藻土抽滤,母液减压浓缩后经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=5:1)得到无色油状化合物v-2(603mg)。化合物v-3的合成取化合物v-2(603mg)溶于四氢呋喃(10ml)中,冰浴下分批加入化合物三苯基膦(817mg)以及四溴化碳(1.03g),室温反应过夜。反应完全后直接经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=8:1)得到无色油状化合物v-3(483mg)。化合物v-4的合成取中间体化合物h-1(281mg)溶于乙腈(15ml)中,加入碳酸氢钠(172mg),室温搅拌30min后,加入化合物v-3(483mg),加毕,缓慢升温至80℃反应5h后,白色固体化合物析出。冷却至室温,直减压抽滤,滤饼经水洗涤,烘干至恒重得到黄色固体化合物v-4(401mg)。化合物v-5的合成取化合物v-4(401mg)于甲醇(10ml)中,水(0.5ml)的混合溶液中,加入一水合氢氧化锂(189mg),室温反应过夜。经tlc(石油醚:乙酸乙酯=2:1)监测反应完全,减压蒸干溶剂,加水(5ml),以1nhcl调节ph=3~4白色固体析出,抽滤,滤饼经乙酸乙酯/无水乙醚打浆得到白色固体化合物v-5(395mg)。化合物v-6的合成取化合物v-5(195mg)溶于乙腈/四氢呋喃为(1:1,8ml)的混合溶液中,加入hatu(190mg)和n,n-二异丙基乙胺dipea(146mg)。加毕室温搅拌30min后加入盐酸二乙胺(55mg),加毕室温反应过夜。经tlc监测反应完全,减压蒸除溶剂,以乙酸乙酯(20ml)稀释,饱和氯化钠溶液洗涤(10mlx2),有机相以无水硫酸钠干燥后,减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=3:1)得到白色固体化合物v-6(160mg)。化合物v-7的合成取化合物v-6(160mg)溶于n,n-二甲基甲酰胺(5ml)中,室温搅拌10min,加入中间体xa-7(77mg),室温搅拌15min,白色固体析出。加水(5ml)停止反应,固体析出。抽滤,滤饼经水洗涤,烘干至恒重得到白色固体化合物v-7(200mg)。化合物v-8的合成取化合物d-丝氨酸乙酯盐酸盐(115mg)于二氯甲烷/甲醇(2:1,4ml)的混合溶液中,滴加n,n-二异丙基乙胺(88mg)室温搅拌20min后,加入化合物v-7(100mg)以及三乙酰氧基硼氢化钠(143mg),加毕,室温反应过夜。反应完全后直接经硅胶柱层析(洗脱剂;二氯甲烷/甲醇=30:1)得到无色透明油状化合物v-8(45mg)。化合物100的合成取化合物v-8(45mg)于甲醇(4ml)和水(5滴)的混合溶液中,加入一水合氢氧化锂(27mg),室温反应过夜。监测反应完全,减压蒸干溶剂,加水(1ml),以1nhcl调节ph=5~6白色固体析出,抽滤,滤饼经二氯甲烷/无水乙醚打浆得到浅棕色固体化合物100(33mg):1hnmr(300mhz,dmso-d6)δ8.16(s,1h),8.08(d,j=9.4hz,1h),7.72(d,j=9.3hz,1h),7.64(d,j=7.2hz,1h),7.55(s,1h),7.43(t,j=7.6hz,2h),7.38–7.26(m,4h),7.12(s,1h),5.41(s,2h),5.35(s,2h),4.02(s,2h),3.75(s,2h),3.72–3.57(m,2h),3.43–3.17(m,6h),1.13–0.94(m,6h).ms(esi)m/z:705.2[m-h]-.实施例101(r)-1-(3-(3'-((5-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((((1-羧基-2-羟乙基)氨基)甲基)-2-氯苯氧基)甲基)-2'-氯代-[1,1'-联苯]-3-基)丙酰基)-4-羟基哌啶-4-羧酸(化合物101)合成路线:化合物w-1的合成取化合物3-(3-溴苯基)丙酸(5g),加入乙醇(15ml),冰浴条件下滴加氯化亚砜(3.1g,1.9ml),保持0℃,3小时后停止反应,减压蒸出溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=10:1)纯化,得无色油状化合物w-2(5.5g)。化合物w-2的合成取化合物w-1(5.5g)、联硼酸频那醇酯(6.5g)、1,1'-双(二苯膦基)二茂铁二氯化钯(ii)二氯甲烷复合物(349mg)以及醋酸钾(4.2g)溶于1,4-二氧六环(30ml),加毕,氩气保护,换气三次。缓慢升温至85℃反应8h,冷却至室温,硅藻土抽滤,母液经减压蒸干溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=30:1),得到黄色油状化合物w-2(6.5g)。化合物w-3的合成取化合物w-2(6.5g)、关键中间体a-2(3.7g)、1,1'-双(二苯膦基)二茂铁二氯化钯(ii)二氯甲烷复合物(300mg)以及碳酸钠(3.7g)溶于甲苯/甲醇的混合溶液(4:1,25ml),氩气保护。缓慢升温至85℃反应5h,经tlc(石油醚:乙酸乙酯=5:1)监测反应完全。冷却至室温,硅藻土抽滤,滤液减压蒸干溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=5:1)得到无色油状化合物w-3(3.8g)。化合物w-4的合成取化合物w-3(3.8g)溶于四氢呋喃(40ml)中,冰浴下分批加入化合物三苯基膦(6.25g)以及四溴化碳(7.9g)。经监测反应完全后,减压抽滤,滤液经减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=8:1)得到无色油状化合物w-4(2.9g)。化合物w-5的合成取中间体化合物h-1(1.4g)溶于乙腈(30ml)中,加入碳酸氢钠(1.3g),室温搅拌30min后,加入化合物w-4(2.9g),缓慢升温至80℃反应5h,白色固体化合物析出。反应完全后冷却至室温,减压抽滤,滤饼经水洗涤,烘干至恒重得到白色固体化合物w-5(2.6g)。化合物w-6的合成取化合物w-5(2.6g)于甲醇(20ml),水(1ml)的混合溶液中,加入一水合氢氧化锂(690mg),室温反应过夜。减压蒸干溶剂,加水(10ml),以1nhcl调节ph=3~4白色固体析出,抽滤,滤饼经乙酸乙酯/无水乙醚(1:2)打浆抽滤,得到白色固体化合物w-6(1.8g)。化合物w-7的合成取化合物w-6(120mg)溶于乙腈/四氢呋喃(1:1,4ml)的混合溶液中,加入hatu(83mg)和n,n-二异丙基乙胺(87mg)。加毕室温搅拌30min后加入xx-5(62mg),室温反应过夜。加水(15ml),乙酸乙酯萃取(10mlx3),有机相以1nhcl洗涤(10mlx3),有机相经无水硫酸钠干燥后减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=1:1)得到白色固体化合物w-7(99mg)。化合物w-8的合成取化合物w-7(99mg)溶于n,n-二甲基甲酰胺(3ml)中,室温搅拌10min,加入中间体xa-7(38mg),室温搅拌15min,白色固体析出。加水(5ml)停止反应,固体析出。抽滤,滤饼经水洗涤,烘干至恒重得到白色固体化合物w-8(103mg)。化合物w-9的合成取化合物d-丝氨酸乙酯盐酸盐(72mg)于二氯甲烷/甲醇(1ml:4ml)的混合溶液中,滴加n,n-二异丙基乙胺(55mg)室温搅拌20min后,加入化合物w-8(103mg)以及三乙酰氧基硼氢化钠(stab,119mg),室温反应过夜。经tlc(二氯甲烷/甲醇=10:1)监测反应完全,直接经硅胶柱层析(洗脱剂;二氯甲烷/甲醇=30:1)得到无色透明油状化合物w-9(66mg)。化合物101的合成取化合物w-9(66mg)于甲醇(3ml)和水(5滴)的混合溶液中,加入一水合氢氧化锂(16mg)。经监测反应完全,减压蒸干溶剂,加水(2ml),以1nhcl调节ph=5~6,白色固体析出,抽滤,滤饼经二氯甲烷/无水乙醚打浆得到白色固体化合物101(50mg):1hnmr(300mhz,dmso-d6)δ8.16(s,1h),8.07(d,j=9.2hz,1h),7.72(d,j=9.3hz,1h),7.63(d,j=6.9hz,1h),7.55(s,1h),7.48–7.21(m,6h),7.12(s,1h),5.40(s,2h),5.34(s,2h),4.11(d,j=12.8hz,1h),4.03(s,2h),3.78–3.56(m,3h),3.24(s,2h),2.97–2.83(m,3h),2.68(s,2h),1.79–1.60(m,2h),1.52(d,j=13.3hz,2h).ms(esi)m/z:791.3[m-h]-.实施例102(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-5-氯-4-((2-氯-3'-(3-(4-氰基-4-羟基哌啶-1-基)-3-氧丙基)-[1,1'-联苯基]-3-基)甲氧基)苄基)-d-丝氨酸(化合物102)合成路线:化合物w-10的合成取化合物w-6(300mg)溶于乙腈/四氢呋喃(1:1,6ml)的混合溶液中,加入hatu(282mg)和n,n-二异丙基乙胺(218mg)。加毕室温搅拌30min后加入4-羟基哌啶-4-甲腈(120mg),室温反应过夜。加水(15ml),乙酸乙酯萃取(10mlx3),有机相以1nhcl洗涤(10mlx3),有机相经无水硫酸钠干燥后减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=1:1)得到白色固体化合物w-10(261mg)。化合物w-11的合成取化合物w-10(210mg)溶于n,n-二甲基甲酰胺(4ml)中,室温搅拌10min,加入中间体xa-7(88mg),加毕,室温搅拌15min,白色固体析出。加水(20ml)停止反应,乙酸乙酯萃取(10mlx3),合并有机相,以饱和nacl溶液洗涤(10mlx3),无水硫酸钠干燥后减压蒸除溶剂,残余物经硅胶柱层析得到白色固体化合物w-11(64mg)。化合物w-12的合成取化合物d-丝氨酸乙酯盐酸盐(48mg)于二氯甲烷/甲醇(1ml:4ml)的混合溶液中,滴加n,n-二异丙基乙胺(33mg)室温搅拌20min后,加入化合物w-11(64mg)以及三乙酰氧基硼氢化钠(stab,99mg),室温反应过夜。反应液经硅胶柱层析(洗脱剂;二氯甲烷/甲醇=30:1)得到无色透明油状化合物w-12(32mg)。化合物102的合成取化合物w-12(32mg)于甲醇(3ml)和水(5滴)的混合溶液中,加入一水合氢氧化锂(8mg)。监测反应完全,减压蒸干溶剂,加水(2ml),以1nhcl调节ph=5~6,白色固体析出,抽滤,滤饼经二氯甲烷/无水乙醚打浆得到白色固体化合物102(21mg):1hnmr(300mhz,dmso-d6)δ8.09(s,1h),8.01(d,j=9.4hz,1h),7.69(d,j=9.6hz,1h),7.55(d,j=10.6hz,2h),7.42–7.17(m,6h),7.04(s,1h),5.40(s,2h),5.30(s,2h),4.40(d,j=14.2hz,1h),4.20(q,j=26.9,13.4hz,2h),3.90(m,2h),3.76–3.62(m,2h),3.48(m,1h),3.35(m,j=4.2hz,1h),3.27(m,1h),2.92(t,1h),2.86(t,j=7.2hz,2h),2.67(t,j=6.7hz,2h),1.96(m,2h).ms(esi)m/z:772.2[m-h]-.实施例103(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-5-氯-4-((2-氯-3'-(3-(二乙氨基)-3-氧丙氧基)-[1,1'-联苯基]-3-基)甲氧基)苄基)-d-丝氨酸(化合物103)合成路线:化合物sc-1的合成取化合物sb-1(1.9g)溶于乙腈/四氢呋喃为(1:1,20ml)的混合溶液中,加入hatu(3.3g)和n,n-二异丙基乙胺(2.5g)。加毕室温搅拌30min后加入盐酸二乙胺(937mg),室温反应过夜。加水(50ml),乙酸乙酯萃取(30mlx3),有机相以1nhcl洗涤(50mlx3),有机相经无水硫酸钠干燥后减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=2:1)得到白色固体化合物sc-1(2.2g)。化合物sc-2的合成取化合物sc-1(2.2g)、联硼酸频那醇酯(2.3g)、1,1'-双(二苯膦基)二茂铁二氯化钯(ii)二氯甲烷复合物(150mg)和醋酸钾(1.4g)溶于1,4-二氧六环(40ml),氩气保护,换气三次。升温至85℃反应8h,冷却至室温,硅藻土抽滤,滤液减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=5:1),得到白色固体化合物sc-2(2.2g)。化合物sc-3的合成取化合物sc-2(2.1g)、关键中间体a-2(1.1g)、1,1'-双(二苯膦基)二茂铁二氯化钯(ii)二氯甲烷复合物(170mg)以及碳酸钠(1.1g)溶于甲苯/甲醇(4:1,25ml)混合溶液,氩气保护。缓慢升温至85℃反应5h后,经tlc(石油醚:乙酸乙酯=1:1)监测反应完全。冷却至室温,硅藻土抽滤,母液减压蒸干溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=3:1)得到黄色油状化合物sc-3(1.3g)。化合物sc-4的合成取化合物sc-3(1.3g)溶于四氢呋喃(20ml)中,冰浴下分批加入化合物三苯基膦(3.4g)以及四溴化碳(2.7g)。反应结束后,反应液经减压抽滤后浓缩后经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=8:1)得到无色油状化合物sc-4(620mg)。化合物sc-5的合成取中间体化合物h-1(274mg)溶于乙腈(10ml)中,加入碳酸氢钠(235mg),室温搅拌30min后,加入化合物sc-4(600mg),缓慢升温至80℃反应5h后,白色固体化合物析出。经监测反应完全,冷却至室温,减压抽滤,滤饼经水洗涤,烘干至恒重得到白色固体化合物sc-5(359mg)。化合物sc-6的合成取化合物sc-5(110mg)溶于n,n-二甲基甲酰胺(3ml)中,室温搅拌10min,加入中间体xa-7(51mg),室温搅拌15min,白色固体析出。反应完全后加水(5ml)停止反应,固体析出。抽滤,滤饼经水洗涤,烘干至恒重得到白色固体化合物sc-6(120mg)。化合物sc-7的合成取化合物d-丝氨酸乙酯盐酸盐(127mg)于二氯甲烷/甲醇(2:1,3ml)的混合溶液中,滴加n,n-二异丙基乙胺(98mg)室温搅拌20min后,加入化合物sc-6(120mg)以及三乙酰氧基硼氢化钠(stab,159mg),室温反应过夜。反应液减压蒸除溶剂残余物经硅胶柱层析(洗脱剂:二氯甲烷/甲醇=50:1)得到无色透明油状化合物sc-7(92mg)。化合物103的合成取化合物sc-7(92mg)于甲醇(4ml)和水(5滴)的混合溶液中,加入一水合氢氧化锂(21mg)。经tlc(二氯甲烷:甲醇=10:1)监测反应完全,减压蒸干溶剂,加水(2ml)),以1nhcl调节ph=4~5白色固体析出,抽滤,滤饼经丙酮/甲醇打浆得到白色固体化合物103(26mg):1hnmr(300mhz,dmso-d6)δ8.14(s,1h),8.06(d,j=9.3hz,1h),7.71(d,j=9.5hz,1h),7.62(d,j=6.6hz,1h),7.53(s,1h),7.40(dd,j=16.8,7.8hz,3h),7.10(s,1h),6.95(m,3h),5.39(s,2h),5.33(s,2h),4.26(t,j=6.1hz,2h),3.99(s,2h),3.75–3.55(m,2h),3.31(m,j=13.9,6.7hz,4h),3.19(d,j=5.3hz,1h),2.78(t,j=6.1hz,2h),1.12(t,j=7.0hz,3h),1.01(t,j=7.0hz,3h).ms(esi)m/z:735.3[m-h]-.实施例104(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-5-氯-4-((2-氟-[1,1'-联苯]-3-基)甲氧基)苄基)-l-亮氨酰-d-亮氨酸(化合物104)合成路线:化合物x-1的合成取化合物n-boc-l-异亮氨酸(1g)溶于四氢呋喃(15ml)中,加入hatu(1.8g)和n,n-二异丙基乙胺(1.4g)。加毕室温搅拌30min后加入l-异亮氨酸甲酯盐酸盐(864mg),加毕室温反应过夜。经tlc监测反应完全。反应液经硅胶柱层析纯化(洗脱剂:石油醚/乙酸乙酯=2:1)得到黄色油状化合物x-1(1.3g)。化合物x-2的合成取化合物x-1(1.3g)于反应瓶中,加入hcl乙醇溶液(5ml),室温搅拌过夜。经tlc检测反应完全,减压蒸去溶剂,将所得固体干燥至恒重,得浅棕色固体化合物x-2(1.0g)。化合物ia-3的合成取化合物x-2(127mg)于二氯甲烷/甲醇(2:1,3ml)的混合溶液中,滴加n,n-二异丙基乙胺(53mg)室温搅拌20min后,加入化合物ia-2(100mg)以及三乙酰氧基硼氢化钠(stab,173mg),室温反应过夜。经tlc监测反应完全,反应液经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=3:1)得到无色透明油状化合物ia-3(108mg)。化合物104的合成取化合物ia-3(103mg)于甲醇(4ml),水(5滴)的混合溶液中,加入一水合氢氧化锂(30mg)加毕,室温反应过夜。反应完全后减压蒸干溶剂,加水(1ml),以1nhcl调节ph=5~6,白色固体析出,抽滤,滤饼经二氯甲烷/无水乙醚打浆得到白色固体化合物104(55mg):1hnmr(300mhz,dmso-d6)δ8.07(d,j=10.7hz,3h),7.63(d,j=9.2hz,1h),7.60–7.36(m,8h),7.26(t,j=7.5hz,1h),7.10(s,1h),5.33(d,j=4.3hz,4h),4.29(d,j=5.9hz,1h),3.64(dd,j=33.8,13.8hz,2h),3.08(t,1h),1.91(s,1h),1.78–1.64(m,1h),1.61–1.43(m,3h),1.32(d,j=7.4hz,2h),1.23(s,1h),0.81(m,j=18.4,6.1hz,12h).ms(esi)m/z:715.3[m-h]-.实施例105(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-5-氯-4-((2-氯-[1,1'-联苯]-3-基)甲氧基)苄基)-o-(叔丁基)-d-丝基-d-丝氨酸(化合物105)合成路线:化合物y-1的合成取化合物n-boc-o-叔丁基-d-丝氨酸酸(1g)溶于四氢呋喃(20ml)中,加入hatu(1.4g)和n,n-二异丙基乙胺(1.2g)。室温搅拌30min后加入d-丝氨酸乙酯盐酸盐(649mg),室温反应过夜。反应完全后反应液经硅胶柱层析(洗脱剂:二氯甲烷/甲醇=100:1)得到黄色油状化合物y-1(1.1g)。化合物y-2的合成取化合物y-1(1.1g)于反应瓶中,加入hcl乙醇溶液(5ml),室温搅拌过夜。经检测反应完全,减压蒸去溶剂,后将所得固体干燥至恒重,得黄色固体化合物y-2(880mg)。化合物ii-2的合成取化合物ii-1(100mg)于二氯甲烷/甲醇(2:1,3ml)的混合溶液中,滴加n,n-二异丙基乙胺(51mg)室温搅拌20min后,加入化合物ii-1(100mg)以及三乙酰氧基硼氢化钠(stab,167mg),室温反应过夜。反应完成后经硅胶柱层析(洗脱剂:二氯甲烷)得到白色固体化合物ii-2(44mg)。化合物105的合成取化合物ii-2(44mg)于甲醇(4ml),水(5滴)的混合溶液中,加入一水合氢氧化锂(11mg)室温反应过夜。反应结束,减压蒸干溶剂,加水(1ml),以1nhcl调节ph=5~6,固体析出,抽滤,滤饼经二氯甲烷/无水乙醚打浆得到白色固体化合物105(21mg):1hnmr(300mhz,dmso-d6)δ8.18–7.97(m,3h),7.77–7.59(m,2h),7.55–7.29(m,8h),7.09(s,1h),5.38(s,2h),5.33(s,2h),4.21(s,1h),3.79(s,2h),3.73(s,1h),3.54(dd,j=10.4,4.1hz,1h),3.44(d,j=3.9hz,1h),3.38–3.25(m,3h),3.16(s,1h),1.04(d,j=2.8hz,9h).ms(esi):m/z735.1[m-h]-.实施例106(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-5-氯-4-((2-氯-[1,1'-联苯]-3-基)甲氧基)苄基)-l-亮氨酰-d-亮氨酸(化合物106)合成路线:化合物ii-3的合成取化合物x-2(122mg)于二氯甲烷/甲醇(2:1,3ml)的混合溶液中,滴加n,n-二异丙基乙胺(51mg)室温搅拌20min后,加入化合物ii-2(100mg)以及三乙酰氧基硼氢化钠(stab,168mg),室温反应过夜。经tlc监测反应完全,反应液经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=3:1)得到无色透明油状化合物ii-3(138mg)。化合物106的合成取化合物ii-3(138mg)于甲醇(4ml),水(5滴)的混合溶液中,加入一水合氢氧化锂(38mg)室温反应过夜。减压蒸干溶剂,加水(1ml),以1nhcl调节ph=5~6,固体析出,抽滤,滤饼经二氯甲烷/无水乙醚打浆得到白色固体化合物106(33mg):1hnmr(300mhz,dmso-d6)δ8.06(t,3h),7.65(d,j=10.3hz,2h),7.55–7.31(m,8h),7.07(s,1h),5.35(s,2h),5.32(s,2h),4.27(m,j=7.0hz,1h),3.65(dd,j=33.8,13.9hz,2h),3.07(t,1h),1.76–1.63(m,1h),1.64–1.43(m,3h),1.32(m,j=8.2hz,2h),1.24(d,1h),0.97–0.56(m,12h).ms(esi):m/z731.3[m-h]-.实施例107(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((3'-(3-(双(2-羟乙基)氨基)丙氧基)-2-溴-[1,1'-联苯基]-3-基)甲氧基)-5-氯苄基)-d-丝氨酸(化合物107)参考实施例84的合成方法,以化合物ba-5为底物,以二乙醇胺代替4-羟基哌啶进行亲核取代反应,后与d-丝氨酸乙酯盐酸盐经三乙酰氧基硼氢化钠还原胺化,碱水解得到化合物106(23mg):1hnmr(300mhz,dmso-d6)δ8.14(s,1h),8.07(d,j=9.2hz,1h),7.72(d,j=9.5hz,1h),7.61(d,j=7.4hz,1h),7.49(d,j=6.4hz,1h),7.44(d,j=7.6hz,1h),7.43–7.29(m,2h),7.07(s,1h),7.03–6.93(m,1h),6.91(d,j=8.5hz,2h),5.38(s,2h),5.29(s,2h),4.05(t,j=6.3hz,2h),3.96–3.88(m,2h),3.58(d,j=6.0hz,3h),3.42(t,j=6.3hz,6h),3.03(t,j=5.9hz,2h),2.62(t,j=6.9hz,2h),2.50(s,6h),1.88–1.79(m,2h).ms(esi)m/z:801.2[m+h]+.实施例108(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((2-氯-3'-(羧基甲氧基)-[1,1'-联苯]-3-基)甲氧基)-5-氯苄基)-d-丝氨酸(化合物108)合成路线:化合物z-1的合成取化合物3-溴苯酚(10g),联硼酸频那醇酯(16g),1,1'-双(二苯膦基)二茂铁二氯化钯(ii)二氯甲烷复合物(272mg),醋酸钾(11.3g)溶于1,4-二氧六环中(50ml),氩气保护,换气三次。缓慢升温至85℃反应8h,冷却至室温,硅藻土抽滤,滤液减压蒸干溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=10:1),得到黄色油状化合物z-1(14g)。化合物z-2的合成取化合物z-1(5g),溴乙酸乙酯(5.67g)以及碳酸钾(6.3g)溶于n,n-二甲基甲酰胺(15ml)中,升温至66℃反应8h,冷却至室温。加水(75ml)停止反应,经乙酸乙酯(50mlx3)萃取,有机相合并后以饱和食盐水(50mlx3)洗涤,经无水硫酸钠干燥后,减压蒸干溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=15:1)得到白色固体化合物z-2(3.3g)。化合物z-3的合成取化合物z-2(3.2g)、关键中间体a-2(1.5g)、1,1'-双(二苯膦基)二茂铁二氯化钯(ii)二氯甲烷复合物(284mg)以及醋酸钾(1.36g)溶于甲苯/乙醇/水(3:1:0.5,18ml)的混合溶液,氩气保护。升温至85℃反应5h,冷却至室温,硅藻土抽滤,滤液经减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=5:1)得到棕色油状化合物z-3(1.7g)。化合物z-4的合成取化合物z-3(1.7g)溶于四氢呋喃(20ml)中,冰浴下分批加入化合物三苯基膦(3.5g)以及四溴化碳(4.5g),室温反应过夜。经监测反应完全,反应液抽滤,滤液浓缩后经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=8:1)纯化得到无色油状化合物z-4(1.6g)。化合物z-5的合成取中间体化合物h-1(828mg)溶于乙腈(20ml)中,加入碳酸氢钠(880mg),室温搅拌30min后,加入化合物z-4(1.6g),升温至80℃反应5h,白色固体化合物析出。冷却至室温,减压抽滤,滤饼经水洗涤,烘干至恒重得到白色固体化合物z-5(818mg)。化合物z-6的合成取化合物z-5(300mg)溶于n,n-二甲基甲酰胺(5ml)中,室温搅拌10min,加入中间体xa-7(175mg),室温搅拌15min,白色固体析出。加水(5ml)停止反应,固体析出。抽滤,滤饼经水洗涤,烘干至恒重得到黄色固体化合物z-6(405mg)。化合物z-7的合成取化合物d-丝氨酸乙酯盐酸盐(210mg)于二氯甲烷/甲醇(2:1,6ml)的混合溶液中,滴加n,n-二异丙基乙胺(154mg),室温搅拌20min后,加入化合物z-6(180mg)以及三乙酰氧基硼氢化钠(262mg),室温反应过夜。反应液浓缩后经硅胶柱层析(洗脱剂:二氯甲烷/甲醇=30:1)纯化得到无色透明油状化合物z-7(158mg)。化合物108的合成取化合物z-7(158mg)于甲醇(4ml),水(5滴)的混合溶液中,加入一水合氢氧化锂(23mg),室温反应过夜。经tlc监测反应完全,减压蒸干溶剂,加水(1ml),以1nhcl调节ph=4~5白色固体析出,抽滤,滤饼经二氯甲烷/无水乙醚打浆得到白色固体化合物108(111mg):1hnmr(300mhz,dmso-d6)δ8.15(s,1h),8.06(d,j=9.3hz,1h),7.71(d,j=9.3hz,1h),7.63(dd,j=7.3,1.9hz,1h),7.57(s,1h),7.47–7.33(m,3h),7.12(s,1h),7.04–6.92(m,3h),5.40(s,2h),5.34(s,2h),4.71(s,2h),4.08(s,2h),3.78(dd,j=11.3,4.3hz,1h),3.69(dd,j=11.4,5.9hz,1h),3.36–3.28(m,1h).ms(esi)m/z:670.2[m+h]+.实施例109(r)-4-((3'-((5-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-(((1-羧基-2-羟乙基)氨基)甲基)-2-氯苯氧基)甲基)-2'-氯-[1,1'-联苯]-3-基)氧基)丁酸(化合物109)合成路线:化合物z-1的合成取化合物3-溴苯酚(10g)、联硼酸频那醇酯(16g)、1,1'-双(二苯膦基)二茂铁二氯化钯(ii)二氯甲烷复合物(272mg)和醋酸钾(11.3g)溶于1,4-二氧六环中(50ml),氩气保护,换气三次。缓慢升温至85℃反应8h冷却至室温,硅藻土抽滤,滤液减压蒸干溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=10:1),得到黄色油状化合物z-1(14g)。化合物za-1的合成取化合物z-1(5g)、溴丁酸乙酯(6.6g)以及碳酸钾(6.3g)溶于n,n-二甲基甲酰胺(15ml)中,升温至66℃反应8h,冷却至室温。加水(75ml)停止反应,经乙酸乙酯(50mlx3)萃取,有机相合并后以饱和食盐水(50mlx3)洗涤,经无水硫酸钠干燥后,减压蒸干溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=15:1)得到白色固体化合物za-1(6.15g)。化合物za-2的合成取化合物za-1(5g)、关键中间体a-2(2.2g)、1,1'-双(二苯膦基)二茂铁二氯化钯(ii)二氯甲烷复合物(408mg)以及醋酸钾(1.96g)溶于甲苯/乙醇/水(3:1:0.5,36ml)的混合溶液,氩气保护。升温至85℃反应5h,冷却至室温,硅藻土抽滤,滤液经减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=5:1)得到棕色油状化合物za-2(3.2g)。化合物za-3的合成:取化合物za-2(3.2g)溶于四氢呋喃(30ml)中,冰浴下分批加入化合物三苯基膦(6.0g)以及四溴化碳(7.6g),室温反应过夜。经监测反应完全,反应液抽滤,滤液浓缩后经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=8:1)纯化得到无色油状化合物za-3(3.6g)。化合物za-4的合成:取中间体化合物h-1(1.5g)溶于乙腈(30ml)中,加入碳酸氢钠(1.5g),室温搅拌30min后,加入化合物za-3(3.6g),升温至80℃反应5h,白色固体化合物析出。冷却至室温,减压抽滤,滤饼经水洗涤,烘干至恒重得到白色固体化合物za-4(1.8g)。化合物za-5的合成:取化合物za-4(300mg)溶于n,n-二甲基甲酰胺(5ml)中,室温搅拌10min,加入中间体xa-7(164mg),室温搅拌15min,白色固体析出。加水(5ml)停止反应,固体析出。抽滤,滤饼经水洗涤,烘干至恒重得到黄色固体化合物za-5(368mg)。化合物za-6的合成:取化合物d-丝氨酸乙酯盐酸盐(180mg)于二氯甲烷/甲醇(2:1,6ml)的混合溶液中,滴加n,n-二异丙基乙胺(132mg),室温搅拌20min后,加入化合物za-5(160mg)以及三乙酰氧基硼氢化钠(224mg),室温反应过夜。反应液浓缩后经硅胶柱层析(洗脱剂:二氯甲烷/甲醇=30:1)纯化得到无色透明油状化合物za-6(143mg)。化合物109的合成取化合物za-6(143mg)于甲醇(4ml)和水(5滴)的混合溶液中,加入一水合氢氧化锂(20mg),室温反应过夜。,减压蒸干溶剂,加水(1ml),以1nhcl调节ph=4~5白色固体析出,抽滤,滤饼经二氯甲烷/无水乙醚打浆得到白色固体化合物109(91mg):1hnmr(300mhz,dmso-d6)δ8.16(s,1h),8.07(d,j=9.3hz,1h),7.72(d,j=9.4hz,1h),7.63(d,j=7.2hz,1h),7.56(s,1h),7.49–7.30(m,3h),7.12(s,1h),7.03–6.90(m,3h),5.40(s,2h),5.34(s,2h),4.11–3.99(m,4h),3.76(dd,j=11.5,4.5hz,1h),3.67(dd,j=11.3,5.2hz,1h),3.33–3.23(m,1h),2.39(t,j=7.3hz,2h),2.03–1.91(m,2h).ms(esi)m/z:696.1[m+h]+.实施例110(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-5-氯-4-((2-氯-3'-(4-(二乙氨基)-4-氧丁氧基)-[1,1'-联苯基]-3-基)甲氧基)苄基)-d-丝氨酸(化合物110)合成路线:化合物za-7的合成取化合物za-4(185mg)于甲醇(5ml)和水(1ml)的混合溶液中,加入一水合氢氧化锂(42mg),室温反应过夜。经tlc(石油醚:乙酸乙酯=2:1)监测反应完全,减压蒸干溶剂,加水(5ml),以1nhcl调节ph=3~4白色固体析出,抽滤,滤饼干燥至恒重,得到白色固体化合物za-7(160mg)。化合物za-8的合成取化合物za-7(160mg)溶于乙腈/四氢呋喃(1:1,4ml)的混合溶液中,加入hatu(142mg)和n,n-二异丙基乙胺(106mg)。加毕室温搅拌30min后加入二乙胺盐酸盐(41mg),室温反应过夜。反应完成后加水(15ml),乙酸乙酯萃取(10mlx3),有机相以1nhcl洗涤(10mlx3),有机相经无水硫酸钠干燥后减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=1:1)得到白色固体化合物za-8(98mg)。化合物za-9的合成取化合物za-8(98mg)和碳酸铯(123mg)溶于n,n-二甲基甲酰胺(3ml)中,室温搅拌10min,加入中间体xa-7(51mg),室温搅拌15min,白色固体析出。加水(5ml)停止反应,固体析出。抽滤,滤饼经水洗涤,烘干至恒重得到白色固体化合物za-9(102mg)。化合物za-10的合成取化合物d-丝氨酸乙酯盐酸盐(108mg)于二氯甲烷/甲醇(1ml:4ml)的混合溶液中,滴加n,n-二异丙基乙胺(80mg)室温搅拌20min后,加入化合物za-9(102mg)以及三乙酰氧基硼氢化钠(stab,135mg),室温反应过夜。反应液减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:二氯甲烷/甲醇=30:1)纯化得到无色透明油状化合物za-10(95mg)。化合物110的合成:取化合物za-10(95mg)于甲醇(4ml)和水(5滴)的混合溶液中,加入一水合氢氧化锂(13mg),室温反应过夜。减压蒸干溶剂,加水(1ml),以1nhcl调节ph=4~5白色固体析出,抽滤,滤饼经二氯甲烷/无水乙醚打浆得到白色固体化合物110(46mg):1hnmr(300mhz,dmso-d6)δ8.05(d,j=7.3hz,2h),7.64(dd,j=17.2,8.2hz,2h),7.39(dd,j=13.7,6.2hz,4h),7.10–6.88(m,4h),5.35(s,2h),5.29(s,2h),4.04(t,j=6.5hz,2h),3.85(d,j=14.5hz,1h),3.65–3.52(m,4h),3.37–3.20(m,4h),2.48–2.37(m,2h),2.02–1.90(m,2h),1.08(t,j=7.1hz,3h),0.99(t,j=7.0hz,3h).ms(esi)m/z:751.3[m+h]+.实施例111(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-5-氯-4-((2-氟-3'-(3-(4-甲基哌嗪-1-基)丙氧基)-[1,1'-联苯]-3-基)甲氧基)苄基)-d-丝氨酸(化合物111)合成路线:化合物ma-1的合成取化合物m-5(200mg)、无水碳酸钾(119mg)和n-甲基哌嗪(135mg)加入n,n-二甲基甲酰胺(4ml)中,60℃油浴加热。约4小时后停止反应。加入水(20ml),用乙酸乙酯(10mlx3)萃取,无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:二氯甲烷/甲醇=20:1)纯化,得黄色固体化合物ma-1(77mg)。化合物ma-2的合成取d-丝氨酸乙酯盐酸盐(145mg)溶于二氯甲烷与甲醇的混合溶剂(3:1,4ml),加入n,n-二异丙基乙胺(dipea,84mg)室温搅拌20分钟,加入化合物ma-1(77mg)和三乙酰氧基硼氢化钠(stab,144mg),室温反应12小时后停止反应。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:二氯甲烷/甲醇=20:1)纯化,得黄色油状化合物ma-2(60mg)。化合物111的合成取化合物ma-2(60mg)于甲醇(4ml)和水(5滴)的混合溶液中,加入一水合氢氧化锂(12mg),室温反应过夜。反应结束后,以冰醋酸调节反应液ph=5~6,经c18反向柱纯化(水/甲醇=3:7)得白色固体化合物111(17mg):1hnmr(300mhz,dmso-d6)δ8.13(d,j=30.8hz,3h),7.73(s,1h),7.60–7.46(m,2h),7.45–7.34(m,2h),7.28(d,j=7.8hz,2h),7.09(d,j=8.8hz,3h),6.98(d,j=8.2hz,1h),5.35(s,2h),5.32(s,2h),4.85(s,1h),4.17–3.95(m,4h),3.42(d,j=26.2hz,1h),3.23–2.87(m,2h),2.67(d,j=28.1hz,2h),2.44–2.24(m,8h),2.12(s,3h),1.87(d,j=16.5hz,2h).ms(esi)m/z:734.3[m+h]+.实施例112(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((3'-(4-(双(2-羟乙基)氨基)-4-氧丁氧基)-2-氟-[[1,1'-联苯]-3-基)甲氧基)-5-氯苄基)-d-丝氨酸(化合物112)合成路线:化合物a1-1的合成取3-溴-2-氟苯甲酸(25g)溶于四氢呋喃(50ml)中,冰浴下缓慢加入硼烷四氢呋喃溶液(1mol/l,17ml),反应约5h后停止反应,加水(50ml)淬灭,以乙酸乙酯萃取(30mlx3),合并有机相,以1nhcl洗涤(30mlx2),经无水硫酸钠干燥后减压蒸除溶剂,得黄色油状化合物a1-1(22.7g)化合物a1-2的合成取化合物a1-1(5g)、苯硼酸(4g)、四三苯基膦钯(563mg)以及无水碳酸钾(6.7g)溶于甲苯/甲醇(4:1,25ml)混合溶液中,氩气保护。升温至85℃反应5h,冷却至室温,硅藻土抽滤,滤液减压蒸干溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=5:1)得到黄色油状化合物a1-2(4.3g)。化合物a1-3的合成取化合物a1-2(4.3g)溶于n,n-二甲基甲酰胺(15ml)中,加入溴丁酸乙酯(8.7g)和无水碳酸钾(6.5g),升温至66℃反应8h停止反应。加水(75ml),经乙酸乙酯(50mlx3)萃取,合并有机相,饱和食盐水(50mlx3)洗涤,经无水硫酸钠干燥后,减压蒸干溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=5:1)得无色透明油状化合物a1-3(4.2g)。化合物a1-4的合成取化合物a1-3(4.2g)溶于四氢呋喃(30ml)中,冰浴下分别加入三苯基膦(6.6g)以及四溴化碳(8.4g),室温反应过夜。反应液抽滤,滤液减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=8:1)得到淡黄色油状化合物a1-4(6.6g)。化合物a1-5的合成取中间体化合物h-1(2.1g)溶于乙腈(30ml)中,加入碳酸氢钠(2.1g),室温搅拌30min后,加入化合物a1-4-3(6.6g),升温至80℃反应5h,白色固体化合物析出。冷却至室温,减压抽滤,滤饼经水洗涤,烘干至恒重得到白色固体化合物a1-5(4g)。化合物a1-6的合成取化合物a1-5(4g)于甲醇(20ml)和水(2ml)的混合溶液中,加入一水合氢氧化锂(924mg),室温反应过夜。减压蒸干溶剂,加水(10ml),以1nhcl调节ph=3~4白色固体析出,抽滤,滤饼干燥至恒重,得到白色固体化合物a1-6(3.4g)。化合物a1-7的合成取化合物a1-6(500mg)溶于乙腈/四氢呋喃(1:1,6ml)的混合溶液中,加入hatu(498mg)和n,n-二异丙基乙胺(370mg)。加毕室温搅拌30min后加入二乙醇胺(137mg),室温反应过夜。反应完成后加水(20ml),乙酸乙酯萃取(10mlx3),有机相以1nhcl洗涤(10mlx3),有机相经无水硫酸钠干燥后减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=1:1)得到白色固体化合物a1-7(538mg)。化合物a1-8的合成取化合物a1-7(100mg)和碳酸铯(130mg)溶于n,n-二甲基甲酰胺(3ml)中,室温搅拌10min,加入中间体xa-7(46mg),室温搅拌15min,白色固体析出。加水(5ml)停止反应,固体析出。抽滤,滤饼经水洗涤,烘干至恒重得到白色固体化合物a1-8(105mg)。化合物a1-9的合成取化合物d-丝氨酸乙酯盐酸盐(145mg)于二氯甲烷/甲醇(2:1,6ml)的混合溶液中,滴加n,n-二异丙基乙胺(84mg)室温搅拌20min后,加入化合物a1-8(105mg)以及三乙酰氧基硼氢化钠(stab,144mg),室温反应过夜。反应液减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:二氯甲烷/甲醇=30:1)纯化得到无色透明油状化合物a1-9(95mg)。化合物112的合成:取化合物a1-9(95mg)于甲醇(4ml)和水(0.5ml)的混合溶液中,加入一水合氢氧化锂(13mg),室温反应过夜。监测反应完全,减压蒸干溶剂,加水(1ml),以1nhcl调节ph=4~5白色固体析出,抽滤,滤饼经二氯甲烷/甲醇打浆得到白色固体化合物112(19mg):1hnmr(300mhz,dmso-d6)δ8.12–8.01(m,2h),7.68(d,j=9.6hz,1h),7.50(dt,j=15.7,7.2hz,2h),7.43–7.32(m,2h),7.23(t,j=7.5hz,1h),7.06(d,j=7.2hz,2h),7.01(s,1h),6.96(d,j=8.0hz,1h),5.32(d,j=4.0hz,4h),4.00(t,j=6.6hz,2h),3.86–3.59(m,6h),3.56–3.25(m,6h),2.77(t,j=6.5hz,1h),2.10(t,j=7.1hz,2h),1.89(t,j=6.8hz,2h).ms(esi)m/z:678.2[m-h]-.实施例113(e)-3-(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((2-溴-[1,1'-联苯]-3-基)甲氧基)-5-氯苯基)丙烯酸乙酯(化合物113)合成路线:化合物113的合成取化合物j-1(300mg)溶于甲苯(30ml)中,加入乙氧甲酰基亚甲基三苯基膦(420mg),油浴80℃反应2小时后停止反应,减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=10:1)纯化,得白色固体化合物113:1hnmr(300mhz,chloroform-d)δ7.97(d,j=16.1hz,1h),7.87(d,j=9.4hz,1h),7.83(s,1h),7.64(s,1h),7.59(dd,j=7.6,1.8hz,1h),7.55–7.34(m,7h),7.29(d,j=7.5hz,1h),6.58(s,1h),6.44(d,j=16.1hz,1h),5.28(s,2h),5.21(s,2h),4.27(q,j=7.1hz,2h),1.35(t,j=7.1hz,3h).ms(esi):m/z619.1[m+h]+.实施例114(e)-3-(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((2-溴-[1,1'-联苯]-3-基)甲氧基)-5-氯苯基)丙烯酸(化合物114)化合物114的合成取化合物113(50mg)于甲醇(2ml),水(3滴)的混合溶液中,冰浴下加入一水合氢氧化锂(6mg)加毕,室温反应过夜。次日,经tlc(二氯甲烷:甲醇=10:1)冰醋酸1滴,监测反应完全,减压蒸干溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=10:1)纯化,得到白色固体化合物114:1hnmr(300mhz,dmso-d6)δ12.26(s,1h),8.11(d,j=9.4hz,1h),8.08(s,1h),7.88(s,1h),7.80(d,j=16.1hz,1h),7.65(d,j=9.8hz,1h),7.63–7.59(m,1h),7.51–7.31(m,8h),7.13(s,1h),6.52(d,j=16.0hz,1h),5.47(s,2h),5.35(s,2h).ms(esi):m/z589.3[m-h]-.实施例115(e)-3-(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((2-溴-[1,1'-联苯]-3-基)甲氧基)-5-氯苯基)丙烯酸甲酯(化合物115)取化合物113(52mg)于甲醇(2ml),水(3滴)的混合溶液中,冰浴下加入一水合氢氧化锂(5mg)加毕,室温反应过夜。次日,经tlc(二氯甲烷:甲醇=10:1)冰醋酸1滴,监测反应完全,减压蒸干溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=20:1)纯化,得到白色固体化合物115:1hnmr(300mhz,chloroform-d)δ7.97(d,j=16.1hz,1h),7.88(dd,j=9.3,1.1hz,1h),7.83(t,j=1.3hz,1h),7.63(s,1h),7.59(dd,j=7.6,1.8hz,1h),7.52–7.34(m,7h),7.30(d,j=7.3hz,1h),6.58(s,1h),6.44(d,j=16.1hz,1h),5.28(s,2h),5.21(d,j=1.3hz,2h),3.82(s,3h).ms(esi):m/z603.4[m-h]-.实施例1161-(3-((3'-((5-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-2-氯-4-(羟甲基)苯氧基)甲基)-2'-溴-[[1,1'-联苯]-3-基)氧基)丙基)哌啶-4-醇(化合物116)合成路线:化合物116的合成取化合物bb-6(30mg)溶于四氢呋喃(2ml)中,冰浴下缓慢加入硼氢化钠(4mg),薄层层析检测反应完全后停止反应,加入1n盐酸淬灭反应,经乙酸乙酯萃取,饱和食盐水洗涤,有机层经无水硫酸钠干燥后,减压蒸干溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=5/1)得到化合物116:1hnmr(300mhz,methanol-d4)δ7.85(d,j=9.3hz,2h),7.52(d,j=1.5hz,1h),7.51–7.47(m,1h),7.40(s,1h),7.35–7.24(m,2h),7.19(dd,j=7.6,1.8hz,1h),6.92(dd,j=7.9,2.4hz,1h),6.89–6.83(m,2h),6.73(s,1h),5.19(s,4h),4.64(s,2h),4.03(t,j=6.1hz,2h),3.63(dt,j=9.4,4.8hz,1h),3.31(p,j=1.6hz,2h),2.85(dd,j=11.2,5.5hz,2h),2.57(dd,j=9.3,6.2hz,2h),2.23(t,j=11.1hz,2h),1.97(p,j=6.2hz,2h),1.86(dd,j=13.2,4.6hz,2h).ms(esi):m/z710.2[m+h]+.实施例117(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((2-溴-3'-甲氧基-[1,1'-联苯]-3-基)甲氧基)-5-氯苄基)-d-丝氨酸(化合物117)合成路线:化合物bba-2的合成取间甲氧基苯硼酸(97mg)、四(三苯基膦)钯(30mg)和无水碳酸钠(113mg)置于三颈瓶中,氩气保护。将化合物bb-2(250mg)溶于甲苯(9ml)中,加入反应瓶,加入乙醇(3ml)和水(1.5ml),80℃油浴加热。反应约4小时后停止加热,减压蒸除溶剂,乙酸乙酯(15ml)稀释,硅藻土抽滤,母液经硅胶柱层析(洗脱剂:石油醚:乙酸乙酯=8:1)纯化,得白色固体化合物bba-2(157mg)。化合物bba-3的合成取化合物bba-2(151mg)和碳酸铯(217mg)加入n,n-二甲基甲酰胺(6ml)中,室温搅拌15分钟后缓慢滴加xa-7(86mg),室温搅拌约30分钟后停止反应。加入水(20ml),用乙酸乙酯(20mlx3)萃取,合并有机相,用饱和食盐水(30mlx3)洗涤,再合并有机相,无水硫酸钠干燥。减压蒸除溶剂,加入乙腈(6ml)固体析出,直接抽滤烘干溶剂至恒重得白色固体化合物bba-3(249mg)粗品,用于下一步。化合物bba-4的合成取d-丝氨酸乙酯盐酸盐(280mg)溶于二氯甲烷与甲醇的混合溶剂(2:1,3ml)中,加入n,n-二异丙基乙胺(dipea,214mg)室温搅拌20分钟,加入化合物bba-3(240mg)和氰基硼氢化钠(521mg),室温反应12小时后停止反应。减压蒸除溶剂,残余物经柱层析(洗脱剂:石油醚/乙酸乙酯=1:1)纯化,得白色固体化合物bba-4(60mg)。化合物117的合成取化合物bba-4(60mg)溶于甲醇(5ml),加入一水合氢氧化锂(18mg),室温反应12小时后,减压蒸除溶剂,加入水(5ml),用冰醋酸调节ph=5~6,抽滤,得白色固体化合物117(22mg):1hnmr(300mhz,dmso-d6):δ8.16(s,1h),8.07(d,j=9.3hz,1h),7.72(d,j=9.3hz,1h),7.61(d,j=7.6hz,1h),7.55(s,1h),7.41(dt,j=29.8,7.9hz,3h),7.09(s,1h),7.04–6.86(m,2h),5.40(s,2h),5.30(s,2h),4.03(s,2h),3.80(s,3h),3.80–3.59(m,2h),3.25(t,1h).ms(esi):m/z668.1[m+h]+.实施例118(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((2-溴-3'-氯代-[1,1'-联苯]-3-基)甲氧基)-5-氯苄基)-d-丝氨酸(化合物118)合成路线:化合物bbc-2的合成取间氯基苯硼酸(127mg)、四(三苯基膦)钯(39mg)和无水碳酸钠(142mg)置于三颈瓶中,氩气保护。将化合物b-2(200mg)溶于甲苯(9ml)中,加入反应瓶,加入甲苯(6ml)、乙醇(5ml)和水(2.5ml),80℃油浴加热。反应约4小时后停止加热,减压蒸除溶剂,用乙酸乙酯(20ml)稀释,硅藻土抽滤,母液经硅胶柱层析(洗脱剂:石油醚)纯化,得无色油状化合物bbc-2(184mg)。化合物bbc-3的合成取化合物bbc-2(184mg)溶于四氯化碳(10ml)中,加入n-溴代丁二酰亚胺(nbs,122mg),分批加入过氧化苯甲酰(bpo,8mg),90℃油浴加热。反应约6小时后停止加热,减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚)纯化,得无色油状化合物bbc-3(136mg)。化合物bbc-4的合成取实施例4中化合物h-1(98mg)溶于无水乙腈(6ml)中,加入无水碳酸氢钠(64mg),缓慢加入bbc-3(136mg),70℃油浴加热。反应约12小时后停止加热,减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=5:1)纯化,得白色固体化合物bbc-4(142mg)。化合物bbc-5的合成取化合物bbc-4(142mg)和碳酸铯(205mg)加入n,n-二甲基甲酰胺(6ml)中,室温搅拌15分钟后缓慢加入xa-7(80mg),室温反应约30分钟后停止反应。加入水(20ml),乙酸乙酯(20mlx3)萃取,合并有机相用饱和食盐水(30mlx3)洗涤,再合并有机相,无水硫酸钠干燥。减压蒸除溶剂,加入石油醚与乙酸乙酯(5:1,6ml)的混合溶剂(5:1,6ml),固体析出,直接抽滤烘干溶剂至恒重得白色固体化合物bbc-5(175mg)。化合物bbc-6的合成取d-丝氨酸乙酯盐酸盐(203mg)溶于二氯甲烷与甲醇的混合溶剂(2:1,3ml)中,加入n,n-二异丙基乙胺(dipea,155mg),室温搅拌20分钟,加入化合物bbc-5(175mg)和氰基硼氢化钠(381mg),室温反应12小时后停止反应。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=1:1)纯化,得白色固体化合物bbc-6(72mg)。化合物118的合成取化合物bbc-6(72mg)溶于甲醇(5ml),加入一水合氢氧化锂(22mg),室温反应12小时后,减压蒸除溶剂,加入水(5ml),用冰醋酸调节ph=5~6,抽滤,得白色固体化合物118(40mg):1hnmr(300mhz,dmso-d6)δ8.13(s,1h),8.05(d,j=9.4hz,1h),7.72(d,j=9.3hz,1h),7.63(d,j=7.7hz,1h),7.53–7.46(m,3h),7.44(s,1h),7.39–7.32(m,2h),7.06(s,1h),5.38(s,2h),5.28(s,2h),3.91(s,2h),3.58(m,j=5.9hz,2h),3.04(t,j=5.9hz,1h).ms(esi):m/z672.0[m-h]-.实施例119(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((2-溴-3'-甲基-[1,1'-联苯]-3-基)甲氧基)-5-氯苄基)-d-丝氨酸(化合物119)合成路线:化合物bbd-2的合成取间甲基苯硼酸(87mg)、四(三苯基膦)钯(31mg)和无水碳酸钠(112mg)置于三颈瓶中,氩气保护。将化合物bb-2(250mg)溶于甲苯(9ml)中,加入反应瓶,加入乙醇(3ml)和水(1.5ml),80℃油浴加热。反应约4小时后停止加热,减压蒸除溶剂,用乙酸乙酯(15ml)稀释,硅藻土抽滤,母液经柱层析(洗脱剂:石油醚:乙酸乙酯=8:1)纯化,得白色固体化合物bbd-2(148mg)。化合物bbd-3的合成取化合物bbd-2(68mg)和碳酸铯(104mg)加入n,n-二甲基甲酰胺(6ml)中,室温搅拌15分钟后缓慢加入xa-7(40mg),室温反应约30分钟后停止反应。加入水(20ml),乙酸乙酯(20mlx3)萃取,合并有机相用饱和食盐水(30mlx3)洗涤,再合并有机相,无水硫酸钠干燥。减压蒸除溶剂,加入石油醚与乙酸乙酯的混合溶剂(5:1,6ml),固体析出,直接抽滤烘干溶剂至恒重得白色固体化合物bbd-3(72mg)。化合物bbd-4的合成取d-丝氨酸乙酯盐酸盐(87mg)溶于二氯甲烷与甲醇的混合溶剂(2:1,3ml),加入n,n-二异丙基乙胺(dipea,65mg)室温搅拌20分钟,加入化合物bbd-3(72mg)和氰基硼氢化钠(165mg),室温反应12小时后停止反应。减压蒸除溶剂,加入盐酸乙酸乙酯(5ml)析出白色固体,直接抽滤烘干溶剂至恒重得白色固体化合物bbd-4(33mg)。化合物119的合成取化合物bbd-4(33mg)溶于甲醇(5ml),加入一水合氢氧化锂(19mg),室温反应12小时后,减压蒸除溶剂,加入水(5ml),用冰醋酸调节ph=5~6,抽滤,得白色固体化合物119(24mg):1hnmr(300mhz,dmso-d6)δ8.15(s,1h),8.07(d,j=9.3hz,1h),7.72(d,j=9.2hz,1h),7.61(d,j=7.5hz,1h),7.53(s,1h),7.45(t,j=7.6hz,1h),7.34(q,j=8.2,7.8hz,2h),7.23(d,j=7.8hz,1h),7.16(d,j=7.4hz,2h),7.08(s,1h),5.39(s,2h),5.29(s,2h),3.99(s,2h),3.73–3.57(m,2h),3.18(t,j=5.7hz,1h),2.37(s,3h).ms(esi):m/z650.1[m-h]-.实施例120(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((2-溴-3'-甲基-[1,1'-联苯]-3-基)甲氧基)-5-氯苄基)-d-丝氨酸(化合物120)合成路线:化合物bbe-2的合成取2,3-二氟苯硼酸(126mg)、四(三苯基膦)钯(31mg)和无水碳酸钠(112mg)置于三颈瓶中,氩气保护。将化合物bb-2(250mg)溶于甲苯(9ml)中,加入反应瓶,加入乙醇(3ml)和水(1.5ml),80℃油浴加热。反应约4小时后停止加热,减压蒸除溶剂,用乙酸乙酯(15ml)稀释,硅藻土抽滤,母液经硅胶柱层析(洗脱剂:石油醚:乙酸乙酯=8:1)纯化,得白色固体化合物bbe-2(120mg)。化合物bbe-3的合成取化合物bbe-2(116mg)和碳酸铯(167mg)加入n,n-二甲基甲酰胺(6ml)中,室温搅拌15分钟后缓慢滴加xa-7(65mg),室温反应约30分钟后停止反应。加入水(20ml),乙酸乙酯(20mlx3)萃取,合并有机相用饱和食盐水(30mlx3)洗涤,再合并有机相,无水硫酸钠干燥。减压蒸除溶剂,加入乙腈(5ml),固体析出,直接抽滤烘干溶剂至恒重得白色固体化合物bbe-3(122mg)。化合物bbe-4的合成取d-丝氨酸乙酯盐酸盐(141mg)溶于二氯甲烷与甲醇的混合溶剂(2:1,3ml),加入n,n-二异丙基乙胺(dipea,108mg)室温搅拌20分钟,加入化合物bbe-3(122mg)和氰基硼氢化钠(265mg),室温反应12小时后停止反应。减压蒸除溶剂,加入盐酸乙酸乙酯(5ml)析出白色固体,直接抽滤烘干溶剂至恒重得白色固体化合物bbe-4(89mg)。化合物120的合成取化合物bbe-4(89mg)溶于甲醇(5ml),加入一水合氢氧化锂(40mg),室温反应12小时后,减压蒸除溶剂,加入水(5ml),用冰醋酸调节ph=5~6,抽滤,得白色固体化合物120(50mg):1hnmr(300mhz,dmso-d6)δ8.16(s,1h),8.08(d,j=9.3hz,1h),7.76–7.68(m,2h),7.59–7.49(m,3h),7.41(dd,j=7.6,1.7hz,1h),7.34(d,j=5.2hz,1h),7.24–7.15(m,1h),7.11(s,1h),5.40(s,2h),5.32(s,2h),4.01(s,2h),3.75–3.61(m,2h),3.21(t,j=5.4hz,1h).ms(esi):m/z674.2[m+h]+.实施例121(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((2-溴-3'-氟-[1,1'-联苯]-3-基)甲氧基)-5-氯苄基)-d-丝氨酸(化合物121)合成路线:化合物bbf-2的合成取3-氟苯硼酸(108mg)、四(三苯基膦)钯(37mg)和无水碳酸钠(136mg)置于三颈瓶中,氩气保护。将化合物bb-2(300mg)溶于甲苯(9ml)中,加入反应瓶,加入乙醇(3ml)和水(1.5ml),80℃油浴加热。反应约4小时后停止加热,减压蒸除溶剂,用乙酸乙酯(15ml)稀释,硅藻土抽滤,母液经硅胶柱层析(洗脱剂:石油醚:乙酸乙酯=8:1)纯化,得白色固体化合物bbf-2(153mg)。化合物bbf-3的合成取化合物bbf-2(153mg)和碳酸铯(228mg)加入n,n-二甲基甲酰胺(6ml)中,室温搅拌15分钟后缓慢加入xa-7(90mg),室温反应约30分钟后停止反应。加入水(20ml),用乙酸乙酯(20mlx3)萃取,合并有机相用饱和食盐水(30mlx3)洗涤,再合并有机相,无水硫酸钠干燥。减压蒸除溶剂,加入乙腈(5ml),固体析出,直接抽滤烘干溶剂至恒重得白色固体化合物bbf-3(134mg)。化合物bbf-4的合成取d-丝氨酸乙酯盐酸盐(160mg)溶于二氯甲烷与甲醇的混合溶剂(2:1,3ml),加入n,n-二异丙基乙胺(dipea,121mg)室温搅拌20分钟,加入化合物bbf-3(134mg)和氰基硼氢化钠(199mg),室温反应12小时后停止反应。减压蒸除溶剂,加入盐酸乙酸乙酯(5ml)析出白色固体,直接抽滤烘干溶剂至恒重得白色固体化合物bbf-4(99mg)。化合物121的合成取化合物bbf-4(90mg)溶于甲醇(5ml),加入一水合氢氧化锂(26mg),室温反应12小时后,减压蒸除溶剂,加入水(5ml),用冰醋酸调节ph=5~6,抽滤,得白色固体化合物121(50mg):1hnmr(300mhz,dmso-d6)δ8.16(s,1h),8.08(d,j=9.4hz,1h),7.72(d,j=9.4hz,1h),7.64(d,j=7.6hz,1h),7.57–7.43(m,3h),7.37(d,j=7.4hz,1h),7.31–7.18(m,2h),7.10(s,1h),5.40(s,2h),5.31(s,2h),4.02(s,2h),3.76–3.58(m,2h),3.25(t,j=5.1hz,1h).ms(esi):m/z654.1[m-h]-.实施例122(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((3'-(3-(双(2-羟乙基)氨基)丙氧基)-2-氯-[1,1'-联苯基]-3-基)甲氧基)-5-氯苄基)-d-丝氨酸(化合物122)合成路线:化合物aa5-1的合成取化合物aa-5(200mg)、三乙胺(96mg)和4-羟基哌啶(100mg)加入n,n-二甲基甲酰胺(4ml)中,60℃油浴加热。约4小时后停止反应。加入水(30ml),用乙酸乙酯(10mlx3)萃取,无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:二氯甲烷/甲醇=20:1)纯化,得黄色固体化合物aa5-1(154mg)。化合物aa5-2的合成取d-丝氨酸乙酯盐酸盐(120mg)溶于二氯甲烷与甲醇的混合溶剂(2:1,6ml),加入n,n-二异丙基乙胺(dipea,92mg)室温搅拌20分钟,加入化合物aa5-1(154mg)和三乙酰氧基硼氢化钠(stab,150mg),室温反应12小时后停止反应。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:二氯甲烷/甲醇=20:1)纯化,得黄色油状化合物aa5-2(111mg)。化合物122的合成取化合物aa5-2(111mg)溶于甲醇(3ml)中,加水(0.5ml),加入一水合氢氧化锂(24mg),室温反应12小时后,减压蒸除溶剂,加入水(2ml),用1n盐酸调节ph=5~6,抽滤,所得固体经c18反向硅胶柱层析纯化(水/甲醇=3:7)得黄色固体化合物122(25mg):1hnmr(300mhz,dmso-d6)δ8.10(s,1h),8.05(d,j=9.3hz,1h),7.70(d,j=9.2hz,1h),7.62(d,j=6.0hz,1h),7.46(s,1h),7.43–7.33(m,3h),7.06(s,1h),6.97(t,3h),5.36(s,2h),5.31(s,2h),4.32(s,2h),4.05(t,j=6.0hz,2h),3.84(q,j=14.2hz,2h),3.55(d,j=5.4hz,2h),3.42(t,j=6.1hz,4h),2.96(t,1h),2.61(t,j=6.7hz,2h),1.98–1.64(m,2h).ms(esi):m/z755.3[m+h]+.实施例123(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-5-氯-4-((2-氯-3'-(3-(二乙氨基)丙氧基)-[1,1'-联苯基]-3-基)甲氧基)苄基)-d-丝氨酸(化合物123)合成路线:化合物aa5-3的合成取化合物aa-5(300mg)、三乙胺(289mg)和二乙胺盐酸盐(156mg)加入n,n-二甲基甲酰胺(4ml)中,60℃油浴加热。约4小时后停止反应。加入水(30ml),用乙酸乙酯(10mlx3)萃取,无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:二氯甲烷/甲醇=20:1)纯化,得黄色固体化合物aa5-3(109mg)。化合物aa5-4的合成取d-丝氨酸乙酯盐酸盐(89mg)溶于二氯甲烷与甲醇的混合溶剂(2:1,6ml),加入n,n-二异丙基乙胺(dipea,68mg)室温搅拌20分钟,加入化合物aa5-3(109mg)和三乙酰氧基硼氢化钠(stab,111mg),室温反应12小时后停止反应。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:二氯甲烷/甲醇=20:1)纯化,得黄色油状化合物aa5-4(100mg)。化合物123的合成取化合物aa5-4(100mg)溶于甲醇(3ml)中,加水(5滴),加入一水合氢氧化锂(17mg),室温反应12小时后,减压蒸除溶剂,加入水(2ml),用1n盐酸调节ph=5~6,抽滤,所得固体经c18反向硅胶柱层析纯化(水/甲醇=3:7)得黄色固体化合物123(53mg):1hnmr(300mhz,dmso-d6)δ8.15(s,1h),8.07(d,j=9.3hz,1h),7.72(d,j=9.5hz,1h),7.63(d,j=6.1hz,1h),7.53(s,1h),7.46–7.30(m,3h),7.11(s,1h),7.03–6.83(m,3h),5.40(s,2h),5.34(s,2h),3.76–3.59(m,2h),3.45(q,j=6.7hz,4h),3.18(t,1h),2.57(t,2h),1.92–1.71(m,2h),0.96(t,j=7.1hz,6h).hrms(esi):m/zcalcdforc37h41cl2n4o7+[m+h]+723.23468found723.23422.实施例124.(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((3'-(4-(双(2-羟乙基)氨基)丁氧基)-2-氯-[1,1'-联苯基]-3-基)甲氧基)-5-氯苄基)-d-丝氨酸(化合物124)合成路线:化合物a2-1的合成取2-氯-3-硝基苯甲酸(25g),加入乙醇(50ml),冰浴条件下滴加氯化亚砜(9.9ml),5小时后停止反应,减压蒸出溶剂,得黄色油状化合物a2-1(25.7g),不经进一步纯化直接用于下一步。化合物a2-2的合成取化合物a2-1(25.7g)溶于50ml乙醇中,依次加入铁粉(20.8g)和浓盐酸(10ml),加热至60℃。反应3小时后停止加热,冷却至室温,经硅藻土抽滤,滤液用饱和碳酸钠溶液调节ph=10,乙酸乙酯萃取(100mlx3),无水硫酸钠干燥。减压蒸除溶剂,得棕黄色液体(24.2g)。化合物a2-3的合成取化合物a2-2(24.2g)溶于水(20ml)中,冰盐浴下缓慢加入盐酸(25ml),搅拌5分钟,缓慢滴加亚硝酸钠(9.9g)的水溶液,冰盐浴下搅拌30分钟后,向反应液中缓慢滴加碘化钾(39.8g)的水溶液,室温下反应8小时后停止反应。加入硫代硫酸钠的饱和水溶液(100ml),用乙酸乙酯(100mlx3)萃取,饱和食盐水(100mlx3)洗涤,无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚)纯化,得淡黄色油状化合物a2-3(30.4g)。化合物a2-4的合成取化合物a2-3(5g)、化合物za-1(6.4g)、1,1'-双(二苯膦基)二茂铁二氯化钯(ii)二氯甲烷复合物(400mg)和无水碳酸钠(3.4g)置于三颈瓶中,氩气保护。加入甲苯(30ml)和甲醇(6ml),80℃油浴加热。反应约4小时后停止加热,减压蒸除溶剂,用乙酸乙酯(15ml)稀释,硅藻土抽滤,母液经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=10:1)纯化,得淡黄色油状化合物a2-4(2.3g)。化合物a2-5的合成取化合物a2-4(2.3g)溶于四氢呋喃(20ml)中,冰浴条件下加入氢化铝锂(330mg),反应约2h,依次加入水(0.1ml)、10%氢氧化钠溶液(0.1ml)和水(0.3ml)淬灭反应,然后以无水硫酸镁干燥,减压抽滤,滤液减压蒸除溶剂,得无色油状化合物a2-5(2.1g)。化合物a2-6的合成取化合物a2-5(2.1g)溶于四氢呋喃(30ml)中,冰浴下分别加入三苯基膦(5.6g)以及四溴化碳(7.0g),室温反应过夜。经tlc(石油醚/乙酸乙酯=15:1)监测反应完全。反应液抽滤,滤液减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=20:1)得到无色油状化合物a2-6(1.6g)。化合物a2-7的合成取化合物h-1(718mg)溶于无水乙腈(15ml)中,加入无水碳酸氢钠(638mg),缓慢加入a2-6(1.6g),80℃油浴加热。反应约6小时后停止反应,冷却至室温后减压抽滤,用水(5ml)洗涤,干燥至恒重,得黄色固体化合物a2-7(1.0g)。化合物a2-8的合成取化合物a2-7(1.0g)和碳酸铯(929mg)加入n,n-二甲基甲酰胺(8ml)中,室温搅拌15分钟后缓慢加入xa-7(455mg),室温反应约30分钟后停止反应。加入水(10ml),固体析出,抽滤,所得固体以甲醇(6ml)打浆,经减压抽滤干燥后得浅棕色固体化合物a2-8(986mg)。化合物a2-9的合成取化合物a2-8(300mg)和二乙醇胺(245mg)加入n,n-二甲基甲酰胺(4ml)中,60℃油浴加热。约3小时后停止反应。加入水(30ml),用乙酸乙酯(10mlx3)萃取,无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:二氯甲烷/甲醇=20:1)纯化,得黄色油状化合物a2-9(195mg)。化合物a2-10的合成取d-丝氨酸乙酯盐酸盐(148mg)溶于二氯甲烷与甲醇的混合溶剂(2:1,6ml),加入n,n-二异丙基乙胺(dipea,114mg)室温搅拌20分钟,加入化合物a2-9(195mg)和三乙酰氧基硼氢化钠(stab,185mg),室温反应12小时后停止反应。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:二氯甲烷/甲醇=20:1)纯化,得无色油状化合物a2-10(142mg)。化合物124的合成取化合物a2-10(142mg)溶于甲醇(5ml)中,加水(0.5ml),加入一水合氢氧化锂(30mg),室温反应12小时后,减压蒸除溶剂,加入水(2ml),用1n盐酸调节ph=5~6,抽滤,所得固体经c18反向硅胶柱层析(水/甲醇=3:7)得黄色固体化合物124(42mg):1hnmr(300mhz,dmso-d6)δ9.67(s,1h),8.17(s,1h),8.09(d,j=9.3hz,1h),7.79–7.67(m,2h),7.64(dd,j=5.2hz,1h),7.49–7.30(m,3h),7.16(s,1h),7.06–6.83(m,3h),5.43(s,2h),5.37(s,1h),5.31(s,2h),4.27(s,2h),4.06(t,j=5.7hz,2h),4.01–3.83(m,3h),3.84–3.67(m,4h),3.31–3.11(m,6h),1.99–1.55(m,4h).ms(esi):m/z767.3[m-h]-.实施例125(2-(苯并[c][1,2,5]噻二唑-5-基甲氧基)-4-((2-甲基-[[1,1'-联苯]-3-基)甲氧基)苄基)-d-丝氨酸乙酯盐酸盐(化合物125)合成路线:化合物k1-1的合成取2,4-二羟基苯甲醛(2.7g)溶于无水乙腈(30ml)中,加入无水碳酸氢钠(3.3g),缓慢加入k-2(5.7g),80℃油浴加热。反应约6小时后停止反应,冷却至室温后减压抽滤,用水(30ml)洗涤,干燥至恒重,得白色固体化合物k1-1(5.3g)。化合物k1-2的合成取化合物k1-1(210mg)、碳酸铯(410mg)加入n,n-二甲基甲酰胺(4ml)中,室温搅拌15分钟后缓慢加入xc-3(158mg),室温反应约30分钟后停止反应。加入水(5ml),固体析出,抽滤,干燥至恒重,得白色固体化合物k1-2(297mg)。化合物125的合成取d-丝氨酸乙酯盐酸盐(316mg)溶于二氯甲烷与甲醇的混合溶剂(2:1,6ml),滴加n,,n-二异丙基乙胺(242mg)室温搅拌20min后,加入化合物k1-2(290mg)和三乙酰氧基硼氢化钠(stab,394mg),室温反应12小时后停止反应。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:二氯甲烷/甲醇=50:1)纯化,得化合物120的游离碱,再将化合物120的游离碱加入盐酸乙醇溶液中,搅拌过夜,抽滤,得白色固体化合物120(231mg):1hnmr(300mhz,dmso-d6)δ9.41(d,j=37.9hz,2h),8.21(s,1h),8.11(d,j=9.0hz,1h),7.83(d,j=9.0hz,1h),7.52–7.33(m,5h),7.28(d,j=7.3hz,2h),7.25–7.12(m,2h),6.90(s,1h),6.75(d,j=8.4hz,1h),5.62(s,1h),5.40(s,2h),5.16(s,2h),4.24(s,2h),4.14–3.94(m,3h),3.87(q,j=19.3hz,2h),2.15(s,3h),1.10(t,j=7.1hz,3h).hrms(esi):m/zcalcdforc33h34n3o5s+[m+h]+584.22137found584.22077.实施例126(2-(苯并[c][1,2,5]噻二唑-5-基甲氧基)-4-((2-甲基-[[1,1'-联苯]-3-基)甲氧基)苄基)-d-丝氨酸(化合物126)以化合物125的游离碱为关键中间体,参照实施例1的方法进行水解制得化合物126(144mg):1hnmr(300mhz,dmso-d6)δ8.23(s,1h),8.11(d,j=9.1hz,1h),7.86(d,j=9.1hz,1h),7.54–7.35(m,5h),7.34–7.27(m,2h),7.26–7.14(m,2h),6.91(s,1h),6.76(d,j=8.3hz,1h),5.42(s,2h),5.17(s,2h),4.23(d,j=3.4hz,2h),3.94–3.78(m,2h),3.75(d,1h),2.17(s,3h).hrms(esi):m/zcalcdforc31h30n3o5s+[m+h]+556.19007found556.19012.实施例127(2-(苯并[c][1,2,5]噁二唑-4-基甲氧基)-4-((2-甲基-[1,1'-联苯]-3-基)甲氧基)苄基)-d-丝氨酸乙酯盐酸盐(化合物127)合成路线:化合物k1-3的合成取化合物k1-1(210mg)、碳酸铯(410mg)加入n,n-二甲基甲酰胺(4ml)中,室温搅拌15分钟后缓慢加入xb-4(158mg),室温反应约30分钟后停止反应。加入水(5ml),固体析出,抽滤,干燥至恒重,得白色固体化合物k1-3(295mg)。化合物127的合成取d-丝氨酸乙酯盐酸盐(329mg)溶于二氯甲烷与甲醇的混合溶剂(2:1,6ml),滴加n,n-二异丙基乙胺(251mg)室温搅拌20min后,加入化合物k1-3(291mg)和三乙酰氧基硼氢化钠(stab,410mg),室温反应12小时后停止反应。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:二氯甲烷/甲醇=50:1)纯化,得化合物127的游离碱,再将化合物127的游离碱加入盐酸乙醇溶液中,搅拌过夜,抽滤,得白色固体化合物127(224mg):1hnmr(300mhz,dmso-d6)δ9.31(d,j=53.4hz,2h),8.06(d,j=9.0hz,1h),7.76(d,j=6.2hz,1h),7.67(d,j=6.8hz,1h),7.53–7.14(m,8h),7.01(s,1h),6.79(d,j=8.2hz,1h),5.58(s,2h),5.20(s,2h),4.22(s,2h),4.09–3.72(m,5h),2.20(s,3h),1.13(t,j=7.0hz,3h).ms(esi):m/z568.8[m+h]+.实施例128(2-(苯并[c][1,2,5]噁二唑-4-基甲氧基)-4-((2-甲基-[1,1'-联苯]-3-基)甲氧基)苄基)-d-丝氨酸(化合物128)以化合物127的游离碱为关键中间体,参照实施例1的方法进行水解制得化合物128(144mg):1hnmr(300mhz,dmso-d6)δ8.01(d,j=9.1hz,1h),7.79(d,j=6.1hz,1h),7.62(t,1h),7.51–7.42(m,3h),7.42–7.34(m,2h),7.31(d,j=7.8hz,2h),7.27–7.15(m,2h),6.96(s,1h),6.73(d,j=8.1hz,1h),5.58(s,2h),5.17(s,2h),4.08(s,2h),3.83–3.51(m,3h),3.20(t,1h),2.18(s,3h).hrms(esi):m/zcalcdforc31h30n3o6+[m+h]+540.21291found540.21309.实施例129(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((3'-(2-(双(2-(羟乙基)氨基)乙氧基)-2-氯-[1,1'-联苯基]-3-基)甲氧基)-5-氯苄基)-d-丝氨酸(化合物129)合成路线:化合物sd-1的合成取3-溴苯酚(5g)、2-溴乙醇(4.8g)和无水碳酸钾(9.6g)加入100ml茄形瓶中,加入n,n-二甲基甲酰胺(15ml),室温反应4小时后停止反应。加入水(100ml),用乙酸乙酯(40mlx3)萃取,无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=5:1)纯化,得无色油状化合物sd-1(4.4g)。化合物sd-2的合成取化合物sd-1(4.4g)、联硼酸频那醇酯(6.2g)、1,1'-双(二苯膦基)二茂铁二氯化钯(ii)二氯甲烷复合物(828mg)和醋酸钾(3.9g)加入到25ml三颈瓶中,氩气保护。室温下加入1,4-二氧六环(30ml),80℃油浴加热。反应约5小时后停止加热,减压蒸除溶剂,用乙酸乙酯25ml稀释,硅藻土抽滤,母液中加入水(50ml),用乙酸乙酯(10mlx3)萃取,无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=5:1)纯化,得无色油状化合物sd-2(4.7g)。化合物uc-1的合成取化合物u-2(10g)溶于四氯化碳(50ml),加入n-溴代琥珀酰亚胺(nbs,7.4g)和偶氮二异丁腈(aibn,325mg),加热回流反应,8小时后停止反应,减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚)纯化,淡黄色油状化合物uc-1(8.0g)。化合物uc-2的合成取中间体化合物h-1(4.5g)溶于乙腈(30ml)中,加入碳酸氢钠(4g),室温搅拌30min后,加入化合物uc-1(8g),升温至80℃反应5h,白色固体化合物析出。冷却至室温,减压抽滤,滤饼经水洗涤,烘干至恒重得到白色固体化合物uc-2(9.2g)。化合物uc-3的合成取化合物uc-2(3.7g)、化合物sd-2(3.5g)、1,1'-双(二苯膦基)二茂铁二氯化钯(ii)二氯甲烷复合物(359mg)和无水碳酸钾(2.4g)置于三颈瓶中,氩气保护。加入甲苯(30ml)和甲醇(6ml),80℃油浴加热。反应约4小时后停止加热,减压蒸除溶剂,用乙酸乙酯(15ml)稀释,硅藻土抽滤,母液经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=2:1)纯化,得米白色固体化合物uc-3(2.8g)。化合物uc-4的合成取化合物uc-3(2.8g)和碳酸铯(3.2g)加入n,n-二甲基甲酰胺(20ml)中,室温搅拌15分钟后缓慢加入xa-7(1.5g),室温反应约30分钟后停止反应。加入水(15ml),固体析出,抽滤,所得固体以二氯甲烷/无水乙醚(2ml:10ml)打浆,得白色固体化合物uc-4(2g)。化合物uc-5的合成取化合物uc-4(615mg)、吡啶(172mg)和三乙胺(220mg)溶于无水二氯甲烷(10ml)中,冰浴下缓慢加入甲基磺酰氯(188mg),室温搅拌10小时后停止反应。加入无水乙醚(5ml)稀释,减压抽滤,所得固体用甲醇(5ml)打浆,得淡黄色固体化合物uc-5(587mg)。化合物uc-6的合成取化合物uc-5(250mg)和二乙醇胺(435mg)溶于n,n-二甲基甲酰胺(5ml)中,50℃油浴加热反应3小时,加水(25ml)停止反应。用乙酸乙酯(8mlx3)萃取,饱和食盐水(5mlx3)洗涤,无水硫酸钠干燥,减压蒸干溶剂,残余物经硅胶柱层析(洗脱剂:二氯甲烷/甲醇=20:1)纯化,得白色固体化合物uc-6(217mg)。化合物uc-7的合成取d-丝氨酸乙酯盐酸盐(172mg)溶于二氯甲烷与甲醇的混合溶剂(2:1,6ml),加入n,n-二异丙基乙胺(dipea,132mg)室温搅拌20分钟,加入化合物uc-6(217mg)和三乙酰氧基硼氢化钠(stab,215mg),室温反应12小时后停止反应。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:二氯甲烷/甲醇=20:1)纯化,得无色油状化合物uc-7(97mg)。化合物129的合成取化合物uc-7(97mg)溶于甲醇(5ml)中,加水(0.5ml),加入一水合氢氧化锂(21mg),室温反应12小时后,减压蒸除溶剂,加入水(2ml),用1n盐酸调节ph=5~6,抽滤,所得固体经c18反向硅胶柱层析纯化(水/甲醇=3:7)得黄色固体化合物129(52mg):1hnmr(300mhz,dmso-d6)δ8.15(s,1h),8.07(d,j=9.3hz,1h),7.72(d,j=9.4hz,1h),7.64(d,j=7.1hz,1h),7.54(s,1h),7.47–7.30(m,3h),7.11(s,1h),7.04–6.86(m,3h),5.40(s,2h),5.34(s,2h),4.07(t,j=6.0hz,2h),4.00(s,2h),3.79–3.58(m,2h),3.45(t,j=6.2hz,4h),3.20(t,j=4.8hz,1h),2.92(t,j=5.9hz,2h),2.65(t,j=6.2hz,4h).ms(esi):m/z739.2[m-h]-.实施例130(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-5-氯-4-((2-氯-3'-(2-羟基乙氧基)-[1,1'-联苯]-3-基)甲氧基)苄基)-d-丝氨酸(化合物130)合成路线:化合物uc-8的合成取d-丝氨酸乙酯盐酸盐(135mg)溶于二氯甲烷与甲醇的混合溶剂(2:1,6ml),加入n,n-二异丙基乙胺(dipea,103mg)室温搅拌20分钟,加入化合物uc-4(150mg)和三乙酰氧基硼氢化钠(stab,169mg),室温反应12小时后停止反应。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:二氯甲烷/甲醇=80:1)纯化,得无色油状化合物uc-8(153mg)。化合物130的合成取化合物uc-8(153mg)溶于甲醇(5ml)中,加水(0.5ml),加入一水合氢氧化锂(38mg),室温反应12小时后,减压蒸除溶剂,加入水(2ml),用1n盐酸调节ph=5~6,抽滤,所得固体经丙酮(2ml)打浆,干燥至恒重,得黄色固体化合物130(45mg):1hnmr(300mhz,dmso-d6)δ8.16(s,1h),8.08(d,j=9.3hz,1h),7.72(d,j=9.4hz,1h),7.68–7.60(m,2h),7.48–7.32(m,33h),7.14(s,1h),7.04–6.84(m,3h),5.41(s,2h),5.36(s,2h),4.16(s,2h),4.04(t,j=4.9hz,2h),3.89–3.76(m,2h),3.73(t,j=4.8hz,2h),3.60(t,1h).ms(esi):m/z652.1[m-h]-.实施例131(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-5-氯-4-((2-氯-3'-(4-(4-羟基哌啶-1-基)丁氧基)-[[1,1'-联苯]-3-基)甲氧基)苄基)-d-丝氨酸(化合物131)合成路线:化合物a3-1的合成取化合物a2-8(300mg)和4-羟基哌啶(471mg)溶于n,,n-二甲基甲酰胺(5ml)中,50℃油浴加热反应3小时,加水(25ml)停止反应。用乙酸乙酯(10mlx3)萃取,饱和食盐水(10mlx3)洗涤,无水硫酸钠干燥,减压蒸干溶剂,残余物经硅胶柱层析(洗脱剂:二氯甲烷/甲醇=20:1)纯化,得白色固体化合物a3-1(231mg)。化合物a3-2的合成取d-丝氨酸乙酯盐酸盐(177mg)溶于二氯甲烷与甲醇的混合溶剂(2:1,6ml),加入n,n-二异丙基乙胺(dipea,135mg)室温搅拌20分钟,加入化合物a3-1(231mg)和三乙酰氧基硼氢化钠(stab,220mg),室温反应12小时后停止反应。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:二氯甲烷/甲醇=20:1)纯化,得无色油状化合物a3-2(200mg)。化合物131的合成取化合物a3-2(200mg)溶于甲醇(5ml)中,加水(0.5ml),加入一水合氢氧化锂(43mg),室温反应12小时后,减压蒸除溶剂,加入水(2ml),用1n盐酸调节ph=5~6,抽滤,所得固体经c18反向硅胶柱层析(水/甲醇=3:7)得黄色固体化合物131(39mg):1hnmr(300mhz,dmso-d6)δ8.14(s,1h),8.07(d,j=9.3hz,1h),7.71(d,j=9.4hz,1h),7.63(d,j=7.2hz,1h),7.52(s,1h),7.45–7.33(m,3h),7.10(s,1h),7.04–6.89(m,3h),5.39(s,2h),5.33(s,2h),4.02(t,j=6.2hz,2h),3.96(s,2h),3.69–3.55(m,2h),3.44–3.38(m,1h),3.14(t,j=5.8hz,1h),2.69(t,j=11.6hz,2h),2.31(t,j=7.2hz,2h),1.99(t,j=10.0hz,2h),1.79–1.64(m,4h),1.62–1.49(m,2h),1.43–1.28(m,2h).ms(esi):m/z763.2[m-h]-.实施例132(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-5-氯-4-((2-氯3'-(2-(4-羟基哌啶-1-基)乙氧基)-[[1,1'-联苯]-3-基)甲氧基)苄基)-d-丝氨酸(化合物132)合成路线:化合物ud-1的合成取化合物uc-5(200mg),4-羟基哌啶(316mg)溶于n,n-二甲基甲酰胺(5ml)中,50℃油浴加热反应3小时,加水(25ml)停止反应。用乙酸乙酯(10mlx3)萃取,饱和食盐水(10mlx3)洗涤,无水硫酸钠干燥,减压蒸干溶剂,残余物经硅胶柱层析(洗脱剂:二氯甲烷/甲醇=20:1)纯化,得黄色固体化合物ud-1(115mg)。化合物ud-2的合成取d-丝氨酸乙酯盐酸盐(91mg)溶于二氯甲烷与甲醇的混合溶剂(2:1,6ml),加入n,n-二异丙基乙胺(dipea,69mg)室温搅拌20分钟,加入化合物ud-1(115mg)和三乙酰氧基硼氢化钠(stab,112mg),室温反应12小时后停止反应。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:二氯甲烷/甲醇=20:1)纯化,得黄色色油状化合物ud-2(98mg)化合物132的合成取化合物ud-2(98mg)溶于甲醇(5ml)中,加水(0.5ml),加入一水合氢氧化锂(22mg),室温反应12小时后,减压蒸除溶剂,加入水(2ml),用1n盐酸调节ph=5~6,抽滤,所得固体经c18反向硅胶柱层析纯化(水/甲醇=3:7)得黄色固体化合物132(28mg):ms(esi):m/z735.2[m-h]-.实施例133(3-((3'-((5-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((((r)-1-羧基-2-羟乙基)氨基)甲基)-2-氯苯氧基)甲基)-2'-氯-[1,1'-联苯]-3-基)氧基)丙基)-d-丝氨酸(化合物133)合成路线:化合物se-1的合成取化合物sa-1(5g)溶于二氯甲烷(30ml)中,冰浴下缓慢加入戴斯马丁试剂(11g),反应过夜。反应液减压抽滤,滤液依次用饱和硫代硫酸钠溶液(20mlx2)和饱和碳酸氢钠溶液(20mlx2)洗涤,无水硫酸钠干燥后蒸干溶剂,残余物经硅胶柱层析(石油醚/乙酸乙酯=8:1)纯化,得黄色油状化合物se-1(4.2g)。化合物se-2的合成取化合物se-1(4.2g),加入原甲酸三乙酯(3.2g),搅拌10分钟后加入对甲苯磺酸一水合物(32mg),室温反应过夜。反应完成后加入无水碳酸钾(50mg),搅拌1h,加水(50ml),用乙酸乙酯(30mlx3)萃取,饱和食盐水(50mlx3)洗涤,无水硫酸钠干燥,减压蒸干溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=20:1)纯化,得无色油状化合物se-2(4.1g)。化合物se-3的合成取化合物se-2(4.1g)、联硼酸频那醇酯(4.2g)、1,1'-双(二苯膦基)二茂铁二氯化钯(ii)二氯甲烷复合物(283mg)和醋酸钾(2.7g)加入到250ml三颈瓶中,氩气保护。室温下加入1,4-二氧六环(50ml),80℃油浴加热。反应约5小时,减压蒸除溶剂,用乙酸乙酯(25ml)稀释,硅藻土抽滤,母液中加入水(50ml),用乙酸乙酯(10mlx3)萃取,无水硫酸钠干燥。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=15:1)纯化,得无色油状化合物se-3(5.6g)。化合物se-4的合成取化合物se-3(2.5g)、化合物uc-2(2.1g)、1,1'-双(二苯膦基)二茂铁二氯化钯(ii)二氯甲烷复合物(215mg)和无水碳酸钾(1.1g)置于三颈瓶中,氩气保护。加入甲苯(30ml)和甲醇(6ml),80℃油浴加热。反应约4小时后停止加热,减压蒸除溶剂,用乙酸乙酯(15ml)稀释,硅藻土抽滤,母液经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=5:1)纯化,得黄色固体化合物se-4(933mg)。化合物se-5的合成取化合物se-4(933mg)和碳酸铯(880mg)加入n,n-二甲基甲酰胺(5ml)中,室温搅拌15分钟后缓慢加入xa-7(418mg),室温反应约30分钟后停止反应。加入水(5ml),固体析出,抽滤,所得固体以二氯甲烷/无水乙醚(1ml:5ml)打浆,得白色固体化合物se-5(854mg)。化合物se-6的合成取化合物se-5(200mg)溶于丙酮(3ml)中,加入对甲苯磺酸一水合物(35mg),反应约3h,加入饱和碳酸钠溶液(3ml),搅拌10分钟后减压蒸除丙酮,残余物直接抽滤,经干燥后得白色固体化合物se-6(169mg)。化合物se-7的合成取d-丝氨酸乙酯盐酸盐(497mg)溶于二氯甲烷与甲醇的混合溶剂(2:1,6ml),加入n,n-二异丙基乙胺(dipea,375mg)室温搅拌20分钟,加入化合物se-6(169mg)和三乙酰氧基硼氢化钠(stab,247mg),室温反应12小时后停止反应。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:二氯甲烷/甲醇=20:1)纯化,得无色油状化合物se-7(50mg)。化合物133的合成取化合物se-7(50mg)溶于甲醇(5ml)中,加水(0.5ml),加入一水合氢氧化锂(10mg),室温反应12小时后,减压蒸除溶剂,加入水(2ml),用1n盐酸调节ph=5~6,抽滤,所得固体经二氯甲烷/甲醇(2ml:0.2ml)打浆纯化得黄色固体化合物133(30mg):1hnmr(300mhz,dmso-d6)δ8.15(s,1h),8.07(d,j=9.4hz,1h),7.71(d,j=9.1hz,1h),7.63(d,j=7.1hz,1h),7.54(s,1h),7.39(t,j=8.2hz,3h),7.11(s,1h),7.05–6.90(m,3h),5.40(s,2h),5.34(s,2h),4.10(t,2h),4.02(s,2h),3.85–3.49(m,4h),3.27(t,1h),3.21(t,1h),3.13–2.98(m,2h),2.20–1.91(m,2h).ms(esi):m/z753.2[m-h]-.实施例134(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-5-氯-4-((2-氯-3'-(3-((2-羟乙基)氨基)丙氧基)-[[1,1'-联苯]-3-基)甲氧基)苄基)-d-丝氨酸(化合物134)合成路线:化合物aa5-5的合成取化合物aa-5(300mg)和乙醇胺(236mg)加入n,n-二甲基甲酰胺(4ml)中,60℃油浴加热。约4小时后停止反应,加入水(4ml),固体析出,减压抽滤,干燥至恒重,得黄色固体化合物aa5-5(121mg)。化合物aa5-6的合成取d-丝氨酸乙酯盐酸盐(165mg)溶于二氯甲烷与甲醇的混合溶剂(2:1,6ml),加入n,n-二异丙基乙胺(dipea,127mg)室温搅拌20分钟,加入化合物aa5-5(121mg)和三乙酰氧基硼氢化钠(stab,207mg),室温反应12小时后停止反应。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:二氯甲烷/甲醇=20:1)纯化,得黄色油状化合物aa5-6(106mg)化合物134的合成取化合物aa5-6(106mg)溶于甲醇(3ml)中,加水(0.5ml),加入一水合氢氧化锂(24mg),室温反应12小时后,减压蒸除溶剂,加入水(2ml),用1n盐酸调节ph=5~6,抽滤,所得固体经c18反向硅胶柱层析(水/甲醇=3:7)得白色固体化合物134(33mg):1hnmr(300mhz,dmso-d6)δ8.12(s,1h),8.06(d,j=9.5hz,1h),7.71(d,j=9.3hz,1h),7.64(d,j=6.9hz,1h),7.47(s,1h),7.45–7.31(m,3h),7.07(s,1h),7.04–6.91(m,3h),5.37(s,2h),5.32(s,2h),4.07(t,j=6.1hz,2h),3.87(s,2h),3.60–3.54(m,2h),3.49(t,2h),2.98(t,1h),2.76(t,j=6.9hz,2h),2.67(t,j=5.6hz,2h),1.97–1.71(m,2h).ms(esi):m/z709.1[m-h]-.实施例135(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-5-氯-4-((2-氟-3'-(2-羟基乙氧基)-[1,1'-联苯]-3-基)甲氧基)苄基)-d-丝氨酸(化合物135)合成路线:化合物mb-1的合成取化合物m-3(1g)、化合物sd-2(878mg)、1,1'-双(二苯膦基)二茂铁二氯化钯(ii)二氯甲烷复合物(114mg)和无水碳酸钠(594mg)置于三颈瓶中,氩气保护。加入甲苯(15ml)和甲醇(5ml),80℃油浴加热。反应约4小时后停止加热,减压蒸除溶剂,用乙酸乙酯(15ml)稀释,硅藻土抽滤,母液经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=2:1)纯化,得米白色固体化合物mb-1(611mg)。化合物mb-2的合成取化合物mb-1(150mg)和碳酸铯(176mg)加入n,n-二甲基甲酰胺(5ml)中,室温搅拌15分钟后缓慢加入xa-7(84mg),室温反应约30分钟后停止反应。加入水(5ml),固体析出,抽滤,所得固体干燥至恒重,得白色固体化合物mb-2(183mg)。化合物mb-3的合成取d-丝氨酸乙酯盐酸盐(227mg)溶于二氯甲烷与甲醇的混合溶剂(2:1,6ml),加入n,n-二异丙基乙胺(dipea,173mg)室温搅拌20分钟,加入化合物mb-2(183mg)和三乙酰氧基硼氢化钠(stab,282mg),室温反应12小时后停止反应。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:二氯甲烷/甲醇=50:1)纯化,得黄色油状化合物mb-3(148mg)。化合物135的合成取化合物mb-3(148mg)溶于甲醇(3ml)中,加水(0.5ml),加入一水合氢氧化锂(37mg),室温反应12小时后,减压蒸除溶剂,加入水(2ml),用1n盐酸调节ph=5~6,抽滤,所得固体经c18反向硅胶柱层析(水/甲醇=3:7)得白色固体化合物135(73mg):1hnmr(300mhz,dmso-d6)δ8.13(s,1h),8.07(d,j=9.3hz,1h),7.70(d,j=9.3hz,1h),7.61–7.47(m,3h),7.40(t,j=7.9hz,1h),7.27(t,j=7.7hz,1h),7.18–7.06(m,3h),7.01(d,j=8.3hz,1h),5.39(s,2h),5.35(s,2h),4.05(t,j=5.0hz,2h),3.98(s,2h),3.74(t,j=4.9hz,2h),3.70–3.56(m,2h),3.17(t,1h).ms(esi):m/z636.3[m-h]-.实施例136(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-5-氯-4-((2-氯-3'-(3-羟基丙氧基)-[1,1'-联苯]-3-基)甲氧基)苄基)-d-丝氨酸(化合物136)合成路线:化合物ue-1的合成取化合物uc-2(1g)、化合物sa-2(1.6g)、1,1'-双(二苯膦基)二茂铁二氯化钯(ii)二氯甲烷复合物(204mg)和无水碳酸钠(1g)置于三颈瓶中,氩气保护。加入甲苯(20ml)和甲醇(5ml),80℃油浴加热。反应约4小时后停止加热,减压蒸除溶剂,用乙酸乙酯(15ml)稀释,硅藻土抽滤,母液经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=2:1)纯化,得米白色固体化合物ue-1(1.3g)。化合物ue-2的合成取化合物ue-1(180mg)和碳酸铯(144mg)加入n,n-二甲基甲酰胺(5ml)中,室温搅拌15分钟后缓慢加入xa-7(69mg),室温反应约30分钟后停止反应。加入水(5ml),固体析出,抽滤,所得固体干燥至恒重,得白色固体化合物ue-2(219mg)。化合物ue-3的合成取d-丝氨酸乙酯盐酸盐(264mg)溶于二氯甲烷与甲醇的混合溶剂(2:1,6ml),加入n,n-二异丙基乙胺(dipea,202mg),室温搅拌20分钟,加入化合物ue-2(183mg)和三乙酰氧基硼氢化钠(stab,329mg),室温反应12小时后停止反应。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:二氯甲烷/甲醇=50:1)纯化,得黄色油状化合物ue-3(119mg)化合物136的合成取化合物ue-3(119mg)溶于甲醇(3ml)中,加水(0.5ml),加入一水合氢氧化锂(29mg),室温反应12小时后,减压蒸除溶剂,加入水(2ml),用1n盐酸调节ph=5~6,抽滤,所得固体经c18反向硅胶柱层析(水/甲醇=3:7)得白色固体化合物136(39mg):1hnmr(300mhz,dmso-d6)δ8.16(s,1h),8.08(d,j=9.3hz,1h),7.72(dd,j=9.3hz,1h),7.64(dd,1h),7.55(s,1h),7.48–7.31(m,3h),7.12(s,1h),7.05–6.89(m,3h),5.41(s,2h),5.35(s,2h),4.09(t,j=6.4hz,2h),4.03(s,2h),3.80–3.61(m,2h),3.57(t,j=6.2hz,2h),3.21(t,1h),1.96–1.73(m,2h).ms(esi):m/z666.1[m-h]-.实施例137(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-5-氯-4-((2-氯-[1,1'-联苯]-3-基)甲氧基)苄基)-2-(哌啶-1-基)乙-1-胺二盐酸盐(化合物137)合成路线:化合物137的合成取2-乙胺哌啶(46mg)溶于二氯甲烷与甲醇的混合溶剂(3:1,4ml),滴加冰醋酸(30ul),加入化合物ii-2(53mg)和三乙酰氧基硼氢化钠(stab,75mg),室温反应12小时后停止反应。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:二氯甲烷/甲醇=80:1)纯化,得化合物137的游离碱,再将化合物137的游离碱加入盐酸乙醇溶液中,搅拌过夜,抽滤,得白色固体化合物137(59mg):1hnmr(300mhz,dmso-d6)δ10.69(s,1h),9.58(s,2h),8.22(s,1h),8.11(d,j=9.3hz,1h),7.78(d,j=9.3hz,1h),7.73(s,1h),7.64(dd,j=7.3,1.9hz,1h),7.54–7.46(m,3h),7.45–7.34(m,4h),7.18(s,1h),5.48(s,2h),5.38(s,2h),4.25(s,2h),3.54–3.42(m,6h),2.91(t,2h),1.87–1.73(m,4h),1.71–1.64(m,1h),1.50–1.33(m,1h).ms(esi):m/z617.2[m+h]+.实施例138(r)-5-((((2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-5-氯-4-((2-氯-[1,1'-联苯]-3-基)甲氧基)苯基)氨基)甲基吡咯烷-2-酮盐酸盐(化合物138)合成路线:化合物a4-1的合成取l-焦谷氨酸甲酯(5g)溶于四氢呋喃/甲醇(10ml:10ml)中,冰浴条件下缓慢加入硼氢化钠(2.6g),反应8h后,加水淬灭至无气泡冒出,加入无水硫酸镁干燥,减压抽滤,滤液浓缩后经硅胶柱层析(二氯甲烷/甲醇=20:1)纯化,得无色油状化合物a4-1(3.8g)。化合物a4-2的合成取化合物a4-1(3.8g)和三乙胺(3.3g)溶于二氯甲烷(20ml)中,冰浴下缓慢加入甲基磺酰氯(2.9g),反应过夜。加水(1ml)淬灭,减压抽滤,滤液加入无水硫酸镁干燥,后经减压蒸干溶剂,残余物经硅胶柱层析(二氯甲烷/甲醇=30:1)纯化,得黄色固体化合物a4-2(6.2g)。化合物a4-3的合成取化合物a4-2(2.7g)溶于n,n二甲基甲酰胺(20ml)中,加入叠氮化钠(4.5g),升温至85℃反应2h。冷却至室温,加水(120ml),乙酸乙酯萃取(100mlx6)合并有机相,饱和氯化钠洗涤(100mlx2),有机相经无水硫酸钠干燥后减压浓缩,残余物经硅胶柱层析(二氯甲烷/甲醇=50:1)纯化,得淡黄色油状化合物a4-3(3.9g)化合物a4-4的合成取化合物a4-3(3.9g)和钯碳(390mg)溶于四氢呋喃(30ml)中,氢气氛围下反应过夜。反应液经减压抽滤,滤液直接浓缩得黄色油状化合物(3.8g),不经进一步纯化直接用于下一步。化合物a4-5的合成取化合物a4-4(3.8g)溶于乙酸乙酯(5ml)中,加入盐酸乙醇溶液(10ml),搅拌1h后,减压蒸除溶剂,加入无水乙醚(15ml)打浆,抽滤后干燥得黄色固体化合物a4-5(2.9g)。化合物138的合成取化合物a4-5(36mg)溶于二氯甲烷与甲醇的混合溶剂(3:1,4ml),滴加n,n-二异丙基乙胺(31mg),加入化合物ii-2(60mg)和三乙酰氧基硼氢化钠(stab,51mg),室温反应12小时后停止反应。减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:二氯甲烷/甲醇=80:1)纯化,得化合物138的游离碱,再将化合物138的游离碱加入盐酸乙醇溶液中,搅拌过夜,抽滤,得白色固体化合物138(40mg):1hnmr(300mhz,dmso-d6)δ9.14(s,2h),8.18(s,1h),8.11(d,j=9.3hz,1h),7.77(s,2h),7.72(d,j=9.3hz,1h),7.64(dd,1h),7.44(m,j=16.5,7.5,4.3hz,7h),7.18(s,1h),5.45(s,2h),5.39(s,2h),4.21(s,2h),4.01–3.77(m,1h),3.16–2.90(m,2h),2.25–2.05(m,3h),1.83–1.65(m,1h).ms(esi):m/z589.1[m+h]+.实施例1391-(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((2-溴-[1,1'-联苯]-3-基)甲氧基)-5-氯苄基)哌啶-4-醇(化合物139)参照实施例8的方法,将实施例8中的d-丝氨酸乙酯盐酸盐替换为4-羟基哌啶,制得化合物139:1hnmr(300mhz,dmso-d6)δ8.13(s,1h),8.10(d,j=9.3hz,1h),7.72–7.61(m,2h),7.54–7.43(m,4h),7.43–7.32(m,4h),7.10(s,1h),5.39(s,2h),5.30(s,2h),4.59(s,1h),3.48(s,3h),2.77(s,2h),2.29–2.08(m,2h),1.74(s,2h),1.43(s,3h).ms(esi):m/z634.7[m+h]+。实施例1405-((5-((2-溴-[1,1'-联苯]-3-基)甲氧基)-4-氯-2-((4-甲基哌嗪-1-基)甲基)苯氧基)甲基)苯并[c][1,2,5]噁二唑(化合物140)参照实施例8的方法,将实施例8中的d-丝氨酸乙酯盐酸盐替换为n-甲基哌嗪,制得化合物144:1hnmr(300mhz,dmso-d6)δ8.16–8.03(m,2h),7.70–7.58(m,2h),7.52–7.41(m,4h),7.40–7.32(m,4h),7.09(s,1h),5.39(s,2h),5.29(s,2h),3.49(s,2h),2.42(s,8h),2.19(s,3h).ms(esi):m/z633.2[m+h]+。实施例141(r)-3-((2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((2-溴-[1,1'-联苯]-3-基)甲氧基)-5-氯苄基)氨基)丙烷-1,2-二醇(化合物141)参照实施例8的方法,将实施例8中的d-丝氨酸乙酯盐酸盐替换为(r)-3-氨基-1,2-丙二醇,制得化合物145:1hnmr(300mhz,dmso-d6)δ8.15–8.04(m,2h),7.72–7.59(m,2h),7.52–7.42(m,5h),7.41–7.31(m,3h),7.07(s,1h),5.39(s,2h),5.30(s,2h),4.66(s,2h),3.80(s,3h),3.59(s,2h),2.68(dd,j=8.0hz,1h),2.53(s,1h).ms(esi):m/z624.3[m+h]+。实施例1424-((2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((2-溴-[1,1'-联苯]-3-基)甲氧基)-5-氯苄基)氨基)环己-1-醇(化合物142)参照实施例8的方法,将实施例8中的d-丝氨酸乙酯盐酸盐替换为对氨基环己醇,制得化合物142:1hnmr(300mhz,dmso-d6)δ8.09(s,2h),7.66(d,j=9.3hz,3h),7.46(s,6h),7.39(s,3h),7.05(s,1h),5.37(s,2h),5.29(s,2h),3.71(s,4h),2.33(s,1h),2.02(s,2h),1.90(s,1h),1.87–1.80(m,3h),1.78–1.70(m,2h).ms(esi):m/z648.1[m+h]+。实施例143(s)-3-((2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((2-溴-[1,1'-联苯]-3-基)甲氧基)-5-氯苄基)氨基)丙烷-1,2-二醇(化合物143)参照实施例8的方法,将实施例8中的d-丝氨酸乙酯盐酸盐替换为(s)-3-氨基-1,2-丙二醇,制得化合物143:1hnmr(300mhz,dmso-d6)δ8.09(d,j=8.9hz,2h),7.68(d,j=9.5hz,1h),7.63(d,j=7.6hz,1h),7.44(d,j=13.5hz,5h),7.41–7.29(m,3h),7.08(s,1h),5.39(s,2h),5.30(s,2h),3.84(s,3h),3.62(s,4h),2.72(d,j=11.7hz,2h),2.54(s,1h).ms(esi):m/z624.3[m+h]+。实施例144(2-((2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((2-溴-[1,1'-联苯]-3-基]甲氧基)-5-氯苄基)氨基)乙基)乙酰胺(化合物144)参照实施例8的方法,将实施例8中的d-丝氨酸乙酯盐酸盐替换为n-(2-氨基乙基)乙酰胺,制得化合物144的游离碱。再将化合物144的游离碱加入盐酸乙醇溶液中,搅拌2h,抽滤,得化合物144:1hnmr(300mhz,dmso-d6)δ9.00(s,2h),8.18(s,2h),8.11(d,j=9.4hz,1h),7.74(d,j=9.4hz,1h),7.67(s,1h),7.62(d,j=7.5hz,1h),7.46(dd,j=11.8,6.9hz,4h),7.36(t,j=7.7hz,3h),5.46(s,2h),5.34(s,2h),4.20(s,2h),3.36(s,2h),3.01(s,2h),1.81(s,3h).ms(esi):m/z635.1[m+h]+。实施例145(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((2-溴-[1,1'-联苯]-3-基)甲氧基)-5-氯苄基)甘氨酸(化合物145)参照实施例9的方法,将实施例9中的d-丝氨酸乙酯盐酸盐替换为甘氨酸甲酯盐酸盐,得淡黄色固体化合物149(60mg):1hnmr(300mhz,dmso-d6)δ8.16(s,1h),8.09(d,j=9.6hz,1h),7.71(d,j=9.6hz,1h),7.62(d,j=7.1hz,1h),7.53(s,1h),7.46(dd,j=12.1,6.2hz,4h),7.41–7.31(m,3h),7.10(s,1h),5.40(s,2h),5.31(s,2h),3.99(s,2h),3.15(s,3h).ms(esi):m/z606.5[m-h]-。实施例1462-((2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((2-溴-[1,1'-联苯]-3-基)甲氧基)-5-氯苄基)氨基)乙烷-1-磺酰胺(化合物146)参照实施例8的方法,将实施例8中的d-丝氨酸乙酯盐酸盐替换为2-氨基乙烷-1-磺酰胺,制得化合物146:ms(esi):m/z657.1[m+h]+。实施例147(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-5-氯-4-((2-氟-3'-(4-吗啉代丁氧基)-[1,1'-联苯]-3-基)甲氧基)苄基)-d-丝氨酸乙酯(化合物147);(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-5-氯-4-((2-氟-3'-(4-吗啉代丁氧基)-[1,1'-联苯]-3-基)甲氧基)苄基)-d-丝氨酸(化合物148)合成路线:化合物za-2的合成室温下,将化合物za-1(300mg,0.470mmol)、碳酸钾(78mg,0.564mmol)、n,n-二甲基甲酰胺(6ml)和吗啉(84mg,0.940mmol)加入100ml单口瓶中,60℃加热回流过夜。反应结束后,加入50ml的水,15ml的乙酸乙酯萃取三次,合并有机相后,饱和食盐水洗一次。无水硫酸钠干燥,减压蒸除溶剂,柱层析(洗脱剂:二氯甲烷:甲醇=40:1),得化合物za-2(黄色固体,280mg)。化合物147的合成室温下,将化合物12(150mg,0.232mmol)、d-丝氨酸乙酯盐酸盐(120mg,0.697mmol)、三乙胺(75mg,0.697mmol)和n,n-二甲基甲酰胺(4ml,含5%醋酸)加入50ml单口瓶中,室温搅拌一小时后,加入三乙酰氧基硼氢化钠(150mg,0.697mmol),反应约12小时后完成。加入20ml饱和碳酸氢钠溶液,搅拌10分钟后,用15ml二氯甲烷萃取三次,合并有机相,用饱和食盐水洗一次,无水硫酸钠干燥,减压蒸除溶剂,得化合物147(138mg):ms(esi):m/z763.87[m+h]+.化合物148的合成室温下,将化合物147(150mg,0.197mmol)、一水合氢氧化锂(90mg,1.97mmol)和四氢呋喃的水溶液(3ml,四氢呋喃:水=4:1)加入25ml的单口瓶,室温搅拌过夜。反应结束后,用甲酸调节ph至7-8,减压蒸除溶剂,反相制备柱纯化(0.5%甲酸水和乙腈,水:乙腈=9:1-3:2),得化合物148(39mg):1hnmr(400mhz,dmso-d6)δ1.53-1.62(m,2h),1.70-1.79(m,2h),2.20-2.40(m,6h),3.21-3.25(m,1h),3.54(s,1h),3.63-3.70(m,2h),3.72-3.79(m,2h),4.02(s,2h),4.07(s,2h),5.35(s,2h),5.40(s,2h),6.99(d,j=4.0hz,1h),7.05-7.15(m,3h),7.26(t,j=4.0hz,1h),7.40(t,j=8.0hz,1h),7.47-7.58(m,3h),7.70(d,j=4.0hz,1h),8.04-8.10(m,1h),8.15(s,1h).ms(esi):m/z733.76[m-h]-.实施例148(r)-1-(4-((3'-((5-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((((1-羧基甲酯-2-羟乙基)氨基)甲基)-2-氯苯氧基)甲基)-2'-氟-[1,1'-联苯]-3-基)氧基)丁基)-4-羟基哌啶-4-羧酸甲酯(化合物149);(r)-1-(4-((3'-((5-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((((1-羧基-2-羟乙基)氨基)甲基)-2-氯苯氧基)甲基)-2'-氟-[1,1'-联苯]-3-基)氧基)丁基)-4-羟基哌啶-4-羧酸(化合物150)合成路线:化合物zb-2的合成将化合物zb-1(10.0g)溶于无水n-甲基吡咯烷酮(110ml)中,然后逐滴加入三甲基氰硅烷(10.5g),室温搅拌过夜。反应结束后,减压蒸除溶剂,将水(50ml)加入其中,然后用乙酸乙酯(80ml×3)萃取,饱和食盐水(50ml)洗涤,无水硫酸钠干燥。减压蒸除溶剂,得化合物zb-2(黄色固体,13.5g)。化合物zb-3的合成将化合物zb-2(5.28g)溶于浓盐酸(50ml)中,室温搅拌1小时,再升温至60℃搅拌1小时,停止反应。减压蒸除浓盐酸,得到化合物zb-3的盐酸盐粗品(4.3g),将其直接用于下一步反应。化合物zb-4的合成将化合物zb-3(3.22g)溶于甲醇(30ml),加入浓硫酸(0.30ml),室温反应4小时。反应结束后加入水(30ml)稀释,然后调节反应液ph至弱碱性,用乙酸乙酯(50ml×3)萃取,再用饱和食盐水(30ml)洗涤,无水硫酸钠干燥,减压蒸除溶剂,得到化合物zb-4的粗品(2.50g)。化合物zb-5的合成将化合物zb-4(2.1g)溶于甲醇(70ml)加入高压釜中,然后再加入5%pd/c(200mg)和氢氧化钯碳(200mg),通入氢气并置换气体三次,在2.0mpa,45℃下反应8小时,结束反应后,硅藻土过滤,减压蒸除溶剂,得到白色固体化合物zb-5(1.1g)。化合物zb-6的合成将化合物zb-5(330mg)、化合物za-1(250mg)、无水碳酸钾(572mg)和无水乙腈(8ml)充分混合溶解,然后再加入碘化钾(50mg),氮气保护并置换气体三次,60℃反应过夜。反应结束后,减压蒸除溶剂,加入饱和碳酸氢钠溶液(5ml),然后用二氯甲烷(10ml×3)萃取,饱和食盐水(5ml)洗涤,无水硫酸钠干燥,减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:二氯甲烷)纯化,得黄色固体化合物zb-6(220mg)。化合物149的合成将化合物zb-6(182mg)和d-丝氨酸乙酯盐酸盐(313mg)溶于n,n-二甲基乙酰胺(3ml,含5%醋酸)中,然后加入4a分子筛,再加入三乙胺(154mg),氮气保护并置换气体三次,室温搅拌半小时后加入醋酸硼氢化钠(323mg),再次置换气体,反应过夜。当反应结束后,过滤除去4a分子筛,减压蒸除溶剂,得到化合物149(700mg):ms(esi):m/z835.14[m+h]+.化合物150的合成将化合物149(700mg)溶于含水量为20%的四氢呋喃溶液(10ml)中,然后加入一水合氢氧化锂(200mg),室温搅拌一小时。反应结束后,用甲酸调节ph至7-8,减压蒸除溶剂,残余物经反相柱层析(洗脱剂:0.5%甲酸水和乙腈,水:乙腈=9:1-3:2)纯化,得黄色固体化合物150(30mg):1hnmr(400mhz,dmso-d6)δ1.26-1.27(m,2h),1.30-1.31(m,2h),1.39-1.51(m,2h),1.70(s,1h),1.93-2.03(m,2h),2.16-2.19(m,1h),2.28-2.35(m,1h),2.60-2.68(m,2h),2.88-2.90(m,2h),4.05(s,4h),4.25-4.28(m,1h),5.25-5.36(m,4h),6.68(s,1h),6.99-7.01(m,1h),7.05-7.11(m,2h),7.23-7.28(m,1h),7.38-7.43(m,1h),7.46-7.60(m,2h),7.70-7.72(m,1h),8.05-8.13(m,1h),8.26(s,2h).ms(esi):m/z791.32[m-h]-.实施例149(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-5-氯-4-((2-氟-3'-(3-吗啉丙氧基)-[1,1'-联苯]-3-基)甲氧基)苄基)-d-丝氨酸乙酯(化合物151);(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-5-氯-4-((2-氟-3'-(3-吗啉丙氧基)-[1,1'-联苯]-3-基)甲氧基)苄基)-d-丝氨酸(化合物152)合成路线:化合物zc-1的合成将化合物m-5(125.2mg,0.2mmol)、碳酸钾(55.6mg,0.4mmol)、吗啉(26.1mg,0.3mmol)和n,n-二甲基乙酰胺(2ml)加入到10ml反应瓶中,将反应瓶放入60℃油浴锅中反应过夜,反应结束后,体系冷却至室温,加入水(20ml),并用乙酸乙酯(30ml×3)萃取,合并有机相,有机相用饱和食盐水洗一次,硫酸钠干燥,过滤,减压脱溶,硅胶柱层析(洗脱剂:石油醚:二氯甲烷=4:1),得到化合物zc-1(黄色固体,101mg)。化合物151的合成将化合物d-丝氨酸乙酯盐酸盐(67.4mg,0.4mmol)、二异丙基乙胺(51.6mg,0.4mmol)和二氯甲烷(2ml)加入到反应瓶中并搅拌10分钟,然后加入化合物zc-1(101mg,0.16mmol)和一滴乙酸,室温搅拌30分钟,然后分批加入三乙酰基硼氢化钠(84.8mg,0.4mmol),室温搅拌2小时,反应结束后,加入水(10ml),并用二氯甲烷(20ml×3)萃取,合并有机相,有机相用饱和食盐水洗一次,硫酸钠干燥,过滤,减压脱溶,硅胶柱层析(洗脱剂:二氯甲烷:甲醇=100:1),得到化合物151(黄色固体,81mg)。化合物152的合成将化合物151(81mg,0.08mmol)、一水氢氧化锂(26.7mg,0.64mmol)、四氢呋喃(4ml)和水(1ml)加入到反应瓶中,室温搅拌2小时,反应结束后,用1n盐酸调节体系ph至5-6,减压脱溶,n,n-二甲基甲酰胺(2ml)溶解,用反相柱分离(洗脱剂:0.2%甲酸水:乙腈=72:28),得到化合物152(白色固体,45mg):1hnmr(400mhz,dmso-d6)δ8.20(s,1h),8.14(s,1h),8.08(d,j=9.2hz,1h),7.70(d,j=9.2hz,1h),7.60-7.47(m,3h),7.42-7.38(m,1h),7.26(t,j=7.6hz,1h),7.16-7.07(m,3h),6.99(dd,j=8.0,2.2hz,1h),5.39(s,2h),5.35(s,2h),4.08-4.05(m,2h),4.02(s,2h),3.77-3.61(m,3h),3.60-3.51(m,4h),3.21-3.18(m,1h),2.44-2.42(m,2h),2.36-2.34(m,3h),1.95-1.84(m,2h).ms(esi):m/z721.75[m+h]+.实施例150(3-((3'-((5-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((((r)-1-羧基甲酯-2-羟乙基)氨基)甲基)-2-氯苯氧基)甲基)-2'-氟-[1,1'-联苯]-3-基)氧基)丙基)-d-丝氨酸乙酯(化合物153);(3-((3'-((5-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((((r)-1-羧基-2-羟乙基)氨基)甲基)-2-氯苯氧基)甲基)-2'-氟-[1,1'-联苯]-3-基)氧基)丙基)-d-丝氨酸(化合物154)合成路线:化合物m-1的合成将化合物2-氟-3甲基苯胺(12.5g,100mmol)加入到250ml反应瓶中,体系降温至0-5℃,滴加37%盐酸(29.6g,300mmol)并控制内温在下5℃以下,将化合物亚硝酸钠(7.59g,110mmol)溶于h2o(50ml)中,并缓慢滴加到上述体系中,控制内温5℃以下,用淀粉碘化钾试纸检测,试纸迅速变蓝停止滴加,反应继续搅拌1小时。将碘化钾(24.9g,150mmol)溶于h2o(50ml)中,并缓慢滴加到上述体系中,控制内温5℃以下,滴加完毕后将反应体系缓慢升至室温搅拌过夜,反应结束后,加入亚硫酸钠淬灭体系,并用乙酸乙酯(100ml×3)萃取,合并有机相,有机相用过饱和食盐水洗一次,无水硫酸钠干燥,过滤,减压脱溶,硅胶柱层析(洗脱剂:石油醚:乙酸乙酯=100:1),得到化合物m-1(无色液体,13.11g)。化合物m-2的合成将化合物m-1(2.36g,10mmol)、n-溴代丁亚酰亚胺(1.42g,8mmol)、偶氮二异丁腈(52.5mg,0.32mmol)和二氯乙烷(10ml)加入到100ml反应瓶中,反应瓶在70℃油浴锅中回流反应12小时,反应结束后,体系冷却至室温,加入水(50ml),并用二氯乙烷(30ml×3)萃取,合并有机相,有机相用饱和食盐水洗一次,硫酸钠干燥,过滤,减压脱溶,硅胶柱层析(洗脱剂:石油醚:乙酸乙酯=100:1),得到化合物m-2(白色固体,1.9g)。化合物m-3的合成将化合物h-1(2.59g,15mmol)、碳酸氢钠(1.68g,20mmol)和无水乙腈(20ml)加入到250ml反应瓶中,并将化合物m-2(3.15g,10mmol)溶于n,n-二甲基乙酰胺(3ml)中,并滴加到上述体系,滴加完毕后,将反应瓶放入70℃油浴锅中反应8小时,反应结束后,体系冷却至室温,加入水(50ml),并用乙酸乙酯(80ml×3)萃取,合并有机相,有机相用饱和食盐水洗一次,硫酸钠干燥,过滤,减压脱溶,硅胶柱层析(洗脱剂:石油醚:乙酸乙酯=8:1),得到化合物m-3(灰色固体,5g)。化合物zd-1的合成将化合物m-5(125.2mg,0.2mmol)、碳酸钾(83.2mg,0.6mmol)、d-丝氨酸乙酯盐酸盐(67.6mg,0.4mmol)、碘化钾(3.3mg,0.02mmol)和无水乙腈(2ml)加入到10ml反应瓶中,将反应瓶放入60℃油浴锅中反应过夜,反应结束后,体系冷却至室温,加入水(20ml),并用乙酸乙酯(30ml×3)萃取,合并有机相,有机相用饱和食盐水洗一次,硫酸钠干燥,过滤,减压脱溶,硅胶柱层析(洗脱剂:石油醚:二氯甲烷=5:1),得到化合物zd-1(黄色固体,95mg)。化合物153的合成将化合物d-丝氨酸乙酯盐酸盐(67.4mg,0.4mmol)、二异丙基乙胺(51.6mg,0.4mmol)和二氯甲烷(2ml)加入到反应瓶中并搅拌10分钟,然后加入化合物zd-1(95mg,0.14mmol)和一滴乙酸,室温搅拌30分钟,然后分批加入三乙酰基硼氢化钠(84.8mg,0.4mmol),室温搅拌2小时,反应结束后,加入水(10ml),并用二氯甲烷(20ml×3)萃取,合并有机相,有机相用饱和食盐水洗一次,硫酸钠干燥,过滤,减压脱溶,硅胶柱层析(洗脱剂:二氯甲烷:甲醇=100:1),得到化合物153(黄色固体,80mg):ms(esi):m/z795.86[m+h]+.化合物154的合成将化合物153(80mg,0.1mmol)、一水氢氧化锂(33.6mg,0.8mmol)、四氢呋喃(4ml)和水(1ml)加入到反应瓶中,室温搅拌2小时,反应结束后,用1n盐酸调节体系ph至5-6,减压脱溶,n,n-二甲基甲酰胺(2ml)溶解,用反相柱分离(洗脱剂:0.2%甲酸水:乙腈=70:30),得到化合物154(白色固体,50mg):1hnmr(400mhz,dmso)δ8.14(s,1h),8.07(d,j=9.2hz,1h),7.70(d,j=9.2hz,1h),7.58-7.46(m,3h),7.40(t,j=7.8hz,1h),7.26(t,j=7.8hz,1h),7.14-7.08(m,3h),7.00(d,j=8.0hz,1h),5.39(s,2h),5.35(s,2h),4.10(t,j=8hz,2h),4.02(s,2h),3.81-3.77(m,1h),3.71-3.62(m,3h),3.29-3.26(m,1h),3.21-3.18(m,1h),3.08-3.06(m,2h),2.11-2.08(m,2h).ms(esi):m/z739.84[m+h]+.实施例151(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-5-氯-4-((2-氟-3'-(3-((5-氟-2-氧-2,3-二氢嘧啶-4-基)氨基)丙氧基)-[1,1'-联苯]-3-基)甲氧基)苄基)-d-丝氨酸乙酯(化合物155);(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-5-氯-4-((2-氟-3'-(3-((5-氟-2-氧代-2,3-二氢嘧啶-4-基)氨基)丙氧基)-[1,1'-联苯]-3-基)甲氧基)苄基)-d-丝氨酸(化合物156)合成路线:化合物ze-1的合成将化合物m-5(125.2mg,0.2mmol)、碳酸钾(55.6mg,0.4mmol)、5-氟胞嘧啶(38.7mg,0.3mmol)和n,n-二甲基乙酰胺(2ml)加入到10ml反应瓶中,将反应瓶放入60℃油浴锅中反应过夜,反应结束后,体系冷却至室温,加入水(20ml),并用乙酸乙酯(30ml×3)萃取,合并有机相,有机相用饱和食盐水洗一次,硫酸钠干燥,过滤,减压脱溶,硅胶柱层析(洗脱剂:石油醚:二氯甲烷=3:1),得到化合物ze-1(黄色固体,80mg)。化合物155的合成将化合物d-丝氨酸乙酯盐酸盐(67.4mg,0.4mmol)、二异丙基乙胺(51.6mg,0.4mmol)和二氯甲烷(2ml)加入到反应瓶中并搅拌10分钟,然后加入化合物ze-1(80mg,0.12mmol)和一滴乙酸,室温搅拌30分钟,然后分批加入三乙酰基硼氢化钠(84.8mg,0.4mmol),室温搅拌2小时,反应结束后,加入水(10ml),并用二氯甲烷(20ml×3)萃取,合并有机相,有机相用饱和食盐水洗一次,硫酸钠干燥,过滤,减压脱溶,硅胶柱层析(洗脱剂:二氯甲烷:甲醇=120:1),得到化合物155(黄色固体,65mg)。化合物156的合成将化合物155(65mg,0.08mmol)、一水氢氧化锂(26.7mg,0.64mmol)、四氢呋喃(4ml)和水(1ml)加入到反应瓶中,室温搅拌2小时,反应结束后,用1n盐酸调节体系ph至5-6,减压脱溶,n,n-二甲基甲酰胺(2ml)溶解,用反相柱分离(洗脱剂:0.2%甲酸水:乙腈=70:30),得到化合物156(白色固体,40mg):1hnmr(400mhz,dmso-d6)δ8.14(s,1h),8.07(d,j=8.0hz,1h),7.95(d,j=6.6hz,1h),7.70(d,j=8.0hz,1h),7.54-7.49(m,3h),7.40(t,j=8.0hz,1h),7.27(t,j=7.6hz,1h),7.13-7.07(m,3h),6.98(d,j=8.0hz,1h),5.39(s,2h),5.35(s,2h),4.07-4.03(m,2h),3.98(s,2h),3.81-3.78(m,2h),3.65-3.63(m,2h),3.15-3.13(m,1h),2.08-2.05(m,2h).ms(esi):m/z763.90[m+h]+.实施例152(r)-1-(3-((3'-((5-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-(((1-羧基甲酯-2-羟乙基)氨基)甲基)-2-氯苯氧基)甲基)-2'-氟-[1,1'-联苯]-3-基)氧基)丙基)-4-羟基哌啶-4-羧酸甲酯(化合物157);(r)-1-(3-((3'-((5-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-(((1-羧基-2-羟乙基)氨基)甲基)-2-氯苯氧基)甲基)-2'-氟-[1,1'-联苯]-3-基)氧基)丙基)-4-羟基哌啶-4-羧酸(化合物158)合成路线:化合物zf-1的合成将化合物m-5(125.2mg,0.2mmol)、碳酸钾(55.6mg,0.4mmol)、4-羟基哌啶-4羧酸甲酯(47.7mg,0.3mmol)和n,n-二甲基乙酰胺(2ml)加入到10ml反应瓶中,将反应瓶放入60℃油浴锅中反应过夜,反应结束后,体系冷却至室温,加入水(20ml),并用乙酸乙酯(30ml×3)萃取,合并有机相,有机相用饱和食盐水洗一次,硫酸钠干燥,过滤,减压脱溶,硅胶柱层析(洗脱剂:石油醚:二氯甲烷=3:1),得到化合物zf-1(黄色固体,105mg)。化合物157的合成将化合物d-丝氨酸乙酯盐酸盐(67.4mg,0.4mmol)、二异丙基乙胺(51.6mg,0.4mmol)和二氯甲烷(2ml)加入到反应瓶中并搅拌10分钟,然后加入化合物zf-1(105mg,0.15mmol)和一滴乙酸,室温搅拌30分钟,然后分批加入三乙酰基硼氢化钠(84.8mg,0.4mmol),室温搅拌2小时,反应结束后,加入水(10ml),并用二氯甲烷(20ml×3)萃取,合并有机相,有机相用饱和食盐水洗一次,硫酸钠干燥,过滤,减压脱溶,硅胶柱层析(洗脱剂:二氯甲烷:甲醇=120:1),得到化合物157(黄色固体,85mg)。化合物158的合成将化合物157(85mg,0.1mmol)、一水氢氧化锂(33.6mg,0.8mmol)、四氢呋喃(4ml)和水(1ml)加入到反应瓶中,室温搅拌2小时,反应结束后,用1n盐酸调节体系ph至5-6,减压脱溶,n,n-二甲基甲酰胺(2ml)溶解,用反相柱分离(洗脱剂:0.2%甲酸水:乙腈=65:35),得到化合物158(白色固体,60mg):1hnmr(400mhz,dmso-d6)δ8.14(s,1h),8.06(d,j=9.4hz,1h),7.69(d,j=9.4hz,1h),7.57-7.46(m,3h),7.39(t,j=8.0hz,1h),7.24(t,j=7.6hz,1h),7.14(s,1h),7.12-7.03(m,2h),6.98(dd,j=8.0,2.2hz,1h),5.38(s,2h),5.34(s,2h),4.11-4.03(m,4h),3.74(dd,j=11.2,4.3hz,2h),3.66(dd,j=11.2,6.3hz,2h),3.25-3.22(m,1h),2.85(s,2h),2.70(s,2h),1.97-1.95(m,4h),1.48-1.47(m,2h).ms(esi):m/z779.78[m+h]+.实施例153(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-5-氯-4-((2-氟-3'-(4-(4-羟基哌啶-1-基)丁氧基)-[[1,1'-联苯]-3-基)甲氧基)苄基)-d-丝氨酸乙酯(化合物159);(2-(苯并[c][1,2,5]噁二唑-5-甲氧基)-5-氯-4-((2-氟-3'-(4-(4-羟基哌啶-1-基)丁氧基)-[1,1'-联苯]-3-基)甲氧基)苄基)-d-丝氨酸(化合物160)合成路线:化合物zg-1的合成室温下,将3-溴苯酚(20.0g,116mmol)、1.4-二溴丁烷(65.1g,302mmol)、碳酸铯(56.5g,173mmol)、n,n-二甲基甲酰胺(120ml)依次加入250ml单口瓶,40℃加热搅拌反应。反应结束后,抽滤,用乙酸乙酯(80ml)冲洗滤饼,加入500ml的水,分液,水相用乙酸乙酯(50ml)萃取重复4到5次,合并有机相用饱和食盐水(400ml)洗一次,无水硫酸钠干燥,减压蒸除溶剂,柱层析(洗脱剂:石油醚)纯化得浅黄色无色油状透明液体粗品zg-1(50g)。化合物zg-2的合成室温下,将1-溴-3-(4-溴代丁氧基)苯(20.0g,65.0mmol)、双联频哪醇硼酸酯(24.7g,97.4mmol)、醋酸钾(19.1g,195mmol)、1,1′-双(二苯基膦)二茂铁]二氯化钯(ii)络合物(0.760g,1.04mmol)加入500ml三口瓶,氮气置换气体三次,加入1,4-二氧六环(200ml),100℃加热回流反应。反应5小时后,停止加热,移至室温冷却。冷却后,减压蒸除溶剂,硅藻土过滤,向滤液中加入水(300ml),用乙酸乙酯(120ml×3)萃取,无水硫酸钠干燥。减压蒸除溶剂,柱层析(洗脱剂:石油醚)纯化,得无色透明油状液体zg-2(12.5g)。化合物zg-3的合成室温下,将化合物zg-2(2.37g,6.71mmol)、碳酸钾(1.93g,14.0mmol)、4-((3-溴-2-氟苄基)氧基)-5-氯-2-羟基苯甲醛(2.00g,5.59mmol)、1,1′-双(二苯基膦)二茂铁]二氯化钯(ii)二氯甲烷络合物(0.465g,0.559mmol)和18-冠-6-醚(0.147g,0.559mmol)加入100ml单口瓶,氮气置换气体三次,加入甲苯(20ml)、水(3ml)和乙醇(6ml),90℃加热回流。反应2小时后停止加热,移至室温冷却,减压蒸除溶剂,用乙酸乙酯20ml冲洗滤饼。母液中加入40ml的水,分液,加入乙酸乙酯(20ml)萃取3次。最后,用饱和食盐水(60ml)洗一次,无水硫酸钠干燥,减压蒸除溶剂,柱层析(洗脱剂:石油醚:乙酸乙酯=50:1)纯化,得白色固体zg-3(0.67g)。化合物zg-4的合成室温下,将化合物zg-3(200mg,0.400mmol)、碳酸钾(82mg,0.593mmol)和n,n-二甲基甲酰胺(4ml)加入25ml单口瓶,室温搅拌15分钟。然后,加入xa-7(88mg,0.412mmol),约半小时反应结束。向反应液中加入30ml的水,乙酸乙酯(10ml)萃取5次。合并有机相后,用饱和食盐水洗一次,用乙酸乙酯萃取饱和食盐水相2次。合并有机相后,加入无水硫酸钠干燥,减压蒸除溶剂,得产品zg-4(260mg)。化合物zg-5的合成室温下,将化合物zg-4(0.600g,0.940mmol)、碳酸钾(0.155g,1.13mmol)、n,n-二甲基甲酰胺(6ml)、哌啶-4-醇(0.190g,1.88mmol)加入100ml单口瓶,60℃加热回流。反应结束后,加入50ml的水,15ml的乙酸乙酯萃取三次,合并有机相后,饱和食盐水洗一次。无水硫酸钠干燥后,减压蒸除溶剂,柱层析(洗脱剂:二氯甲烷:甲醇=40:1)得黄色固体zg-5(395mg)。化合物159的合成室温下,将化合物zg-5(300mg,0.455mmol),d-丝氨酸乙酯盐酸盐(154mg,0.910mmol),三乙胺(92mg,0.910mmol),n,n-二甲基甲酰胺(4ml,含5%醋酸)加入50ml单口瓶,室温搅拌一小时后,加入三乙酰氧基硼氢化钠(145mg,0.683mmol),反应约12小时后完成。加入20ml饱和碳酸氢钠溶液,搅拌10分钟后,用15ml二氯甲烷萃取三次,合并后,用饱和食盐水洗一次,无水硫酸钠干燥后,减压蒸除溶剂,得化合物159(340mg):ms(esi):m/z778.07[m+h]+.化合物160的合成室温下,将化合物159(340mg,0.438mmol),一水合氢氧化锂(102mg,2.43mmol),四氢呋喃的水溶液(3ml,四氢呋喃:水=4:1)加入25ml的单口瓶,室温搅拌过夜。反应结束后,用甲酸调节ph至7-8,减压蒸除溶剂,反相柱层析(洗脱剂:0.5%甲酸水和乙腈,水:乙腈=9:1-3:2)纯化得化合物160(82mg):1hnmr(400mhz,dmso-d6)δ1.35-1.48(m,2h),1.53-1.65(m,2h),1.67-1.79(m,4h),2.15-2.30(m,2h),2.40-2.50(m,2h),2.75-2.85(m,2h),3.20-3.25(m,1h),3.50(s,1h),3.60-3.67(m,2h),3.67-3.77(m,2h),3.99-4.08(m,4h),5.35(s,2h),5.39(d,j=4.0hz,2h),6.96-7.02(m,1h),7.04-7.07(m,1h),7.07-7.11(m,1h),7.14(s,1h),7.26(t,j=8.0hz,1h),7.39(t,j=8.0hz,1h),7.47-7.59(m,3h),7.69(d,j=8.0hz,1h),8.07(d,j=8.0hz1h),8.14(s,1h),8.24(s,1h).ms(esi):m/z747.67[m-h]-.实施例154(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((3'-(4-(双(2-羟乙基)氨基)丁氧基)-2-氟-[1,1'-联苯基]-3-基)甲氧基)-5-氯苄基)-d-丝氨酸乙酯(化合物161);(2-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((3'-(4-(双(2-羟乙基)氨基)丁氧基)-2-氟-[1,1'-联苯基]-3-基)甲氧基)-5-氯苄基)-d-丝氨酸(化合物162)合成路线:化合物zh-1的合成将化合物za-1(450mg)和乙二醇胺(223mg)溶于无水乙腈(4ml)中,再加入无水碳酸钾(584mg)和碘化钾(35mg),氮气保护并置换气体三次,60℃过夜。反应结束后减压蒸除溶剂,加入饱和碳酸氢钠溶液(5ml),然后用二氯甲烷(20ml×3)萃取,饱和食盐水(5ml)洗涤,无水硫酸钠干燥,减压蒸除溶剂,残余物经柱层析(洗脱剂:二氯甲烷/甲醇=1:0-10:1)纯化,得黄色固体化合物zh-1(365mg)。化合物161的合成将化合物zh-1(300mg)和d-丝氨酸乙酯(180mg)溶于n,n-二甲基乙酰胺(3ml,含5%醋酸),然后加入4a分子筛,氮气保护并置换气体三次,室温搅拌半小时后加入醋酸硼氢化钠(323mg),再次置换气体,反应过夜。当反应结束后,过滤除去4a分子筛,减压蒸除溶剂,得到化合物161(500mg):ms(esi):m/z781.76[m+h]+.化合物162的合成将化合物161(500mg)溶于含水量为20%的四氢呋喃溶液(6ml)中,然后加入一水合氢氧化锂(100mg),室温搅拌一小时。反应结束后,用甲酸调节ph至7-8,减压蒸除溶剂,残余物经反相柱层析(洗脱剂:0.03mol/l碳酸氢铵水溶液和乙腈,水:乙腈=9:1-3:2)纯化,得到淡黄色固体化合物162(10mg):1hnmr(400mhz,dmso-d6)δ1.52-1.60(m,2h),1.69-1.76(m,3h),2.55-2.59(m,5h),3.19-3.22(m,4h),3.62-3.66(m,5h),3.71-3.75(m,2h),4.01-4.04(m,4h),5.31-5.43(m,4h),6.98-7.01(m,1h),7.08(t,j=8hz,2h),7.15(s,1h),7.26(t,j=8hz,1h),7.40(t,j=8hz,1h),7.50-7.58(m,3h),7.70(d,j=12hz,1h),8.07(d,j=8hz,1h),8.15(s,1h).ms(esi):m/z751.96[m-h]-.实施例155(4-((3'-((5-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((((r)-1-羧基-2-羟乙基)氨基)甲基)-2-氯苯氧基)甲基)-2'-氟-[1,1'-联苯]-3-基)氧基)丁基)-d-丝氨酸乙酯(化合物163);(4-((3'-((5-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-((((s)-1-羧基-2-羟乙基)氨基)甲基)-2-氯苯氧基)甲基)-2'-氟-[1,1'-联苯]-3-基)氧基)丁基)-d-丝氨酸(化合物164)合成路线:化合物zi-1的合成室温下,将化合物za-1(100mg,0.157mmol)、碳酸钾(130mg,0.940mmol)、乙腈(4ml)、d-丝氨酸乙酯盐酸盐(80mg,0.470mmol)、碘化钾(25mg,0.0784mmol)加入100ml单口瓶,60℃加热回流过夜。反应结束后,减压蒸除溶剂,加入10ml饱和碳酸氢钠溶液,二氯甲烷10ml萃取3次,合并有机相后,用饱和食盐水洗一次。有机相用无水硫酸钠干燥后,减压蒸除溶剂,柱层析(洗脱剂:二氯甲烷/甲醇=1:0-10:1),得黄色固体zi-1(73mg)。化合物163的合成室温下,将化合物zi-1(240mg,0.347mmol)、d-丝氨酸乙酯盐酸盐(176mg,1.04mmol)、三乙胺(105mg,1.04mmol)、n,n-二甲基甲酰胺(3ml,含5%醋酸)加入50ml单口瓶,室温搅拌一小时后,加入三乙酰氧基硼氢化钠(221mg,1.04mmol),反应约12小时后完成。加入20ml饱和碳酸氢钠溶液,搅拌10分钟后,用15ml二氯甲烷萃取三次,合并后,用饱和食盐水洗一次,无水硫酸钠干燥后,减压蒸除溶剂,得化合物163(232mg):ms(esi):m/z809.88[m+h]+.化合物164的合成室温下,将化合物163(300mg,0.371mmol),一水合氢氧化锂(156mg,3.71mmol),四氢呋喃的水溶液(4ml,四氢呋喃:水=4:1)加入25ml的单口瓶,室温搅拌过夜。反应结束后,用甲酸调节ph至7-8,减压蒸除溶剂,反相柱层析(洗脱剂:0.5%甲酸水和乙腈,水:乙腈=9:1-3:2),得化合物164(28mg):1hnmr(400mhz,dmso-d6)δ1.76(s,4h),2.94(s,2h),3.20-3.29(m,2h),3.66-3.68(m,2h),3.70-3.74(m,2h),3.75-3.80(m,2h),4.00-4.05(m,2h),4.05(s,2h),5.35(s,2h),5.39(s,2h),6.95-7.03(m,1h),7.06(s,1h),7.10(d,j=8.0hz,1h),7.15(s,1h),7.26(t,j=8.0hz,1h),7.39(t,j=8.0hz,1h),7.46-7.61(m,3h),7.71(d,j=8.0hz,2h),8.07(d,j=8.0hz,1h),8.14(s,1h).ms(esi):m/z751.58[m-h]-.实施例156(s)-1-(4-((3'-((5-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-(((((r)-1-羧基乙酯-2-羟乙基)氨基)甲基)-2-氯苯氧基)甲基)-2'-氟-[1,1'-联苯]-3-基)氧基)丁基)哌啶-2-羧酸甲酯(化合物165);(s)-1-(4-((3'-((5-(苯并[c][1,2,5]噁二唑-5-基甲氧基)-4-(((((r)-1-羧基-2-羟乙基)氨基)甲基)-2-氯苯氧基)甲基)-2'-氟-[1,1'-联苯]-3-基)氧基)丁基)哌啶-2-羧酸(化合物166)合成路线:化合物zj-1的合成将化合物za-1(300mg,0.470mmol)、化合物24(253mg,1.77mmol)、无水碳酸钾(390mg,2.82mmol)、碘化钾(30.0mg,0.181mmol)和乙腈(4ml)加入单口瓶中60℃搅拌6小时。反应结束后减压蒸除溶剂,然后加入饱和碳酸氢钠溶液(10ml),再用二氯甲烷萃取(20ml×3),合并有机相,用饱和食盐水(30ml)洗一次,然后用无水硫酸钠干燥,减压蒸除溶剂,粗品经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=2:1)纯化,得化合物zj-1(283mg)。化合物165的合成将化合物zj-1(260mg,0.371mmol),d-丝氨酸乙酯盐酸盐(189mg,1.12mmol)和三乙胺(113mg,1.12mmol)溶于n,n-二甲基甲酰胺(3ml,含5%醋酸),氮气保护并置换气体三次,室温搅拌半小时后加入醋酸硼氢化钠(236mg,1.12mmol),再次置换气体,室温搅拌过夜。当反应结束后,加入饱和碳酸氢钠溶液(3ml)搅拌5分钟,然后用二氯甲烷(6ml×3)萃取,合并有机相,用饱和食盐水(5ml)洗一次,无水硫酸钠干燥,减压蒸除溶剂得化合物165(250mg):ms(esi):m/z819.40[m+h]+.化合物166的合成将化合物165(230mg,0.281mmol)溶于四氢呋喃的水溶液(3ml,四氢呋喃:水=4:1)中,然后加入一水合氢氧化锂(400mg,16.6mmol),室温搅拌一小时。反应结束后,用甲酸调节ph至7-8,减压蒸除溶剂,残余物经制备液相(洗脱剂:2.0%甲酸水/乙腈=9/1-3/2)纯化,得到化合物166(白色固体,8mg):1hnmr(400mhz,dmso-d6)δ1.24(s,2h),1.36-1.38(m,1h),1.61-1.64(m,3h),1.71-1.77(m,3h),1.90-1.93(m,1h),1.97-2.03(m,1h),2.08(s,1h),2.61-2.67(m,2h),2.77-2.84(m,2h),3.95-4.04(m,6h),5.37(d,j=8hz,4h),6.99(d,j=6hz,1h),7.06(s,1h),7.10(d,j=4hz,1h),7.14(s,1h),7.26(t,j=8hz,1h),7.40(t,j=8hz,1h),7.49-7.56(m,3h),7.70(d,j=8hz,1h),8.07(d,j=8hz,1h),8.14(s,1h).ms(esi):m/z775.80[m-h]-.实施例157化合物对pd-1/pd-l1蛋白-蛋白相互作用的抑制活性评价实验目的:使用pd-1/pd-l1bindingassaykit检测试剂盒(cisbio公司),检测式(i)或式(ii)化合物抑制pd-1和pd-l1相互作用的活性。实验原理:通过使用anti-tag1-europium(htrf供体)和anti-tag2-xl665(htrf受体)检测tag1-pd-l1和tag2-pd-1之间的相互作用。当htrf供体和htrf受体由于pd-l1和pd-1结合而紧密接近时,htrf供体的激发触发朝向htrf受体的荧光共振能量转移(fret),htrf受体又在665nm处发射荧光。该特定信号与pd-1/pd-l1相互作用的程度成正比。因此,阻断pd-1/pd-l1相互作用的化合物或抗体将导致htrf信号的减少。实验材料:384孔细胞培养板购自nunc公司;均相时间分辨荧光homogeneoustime-resolvedfluorescence(htrf)试剂盒购自cisbio公司;dmso购自sigma公司。实验仪器:perkinelmerenvision多功能酶标检测仪。受试药物:阳性药(bms-1016)和本发明的化合物。样品用dmso配制成母液使用,使用时用diluentbuffer稀释使用,dmso终浓度不超过0.1%。测定化合物抑制率时使用diluentbuffer稀释为10nm的溶液;测定化合物ic50值时,根据抑制率高低将化合物配置成一系列浓度梯度(10μm-0.05nm)的化合物溶液进行活性测试。实验方案:pd-1/pd-l1蛋白-蛋白相互作用抑制试验采用htrf试剂盒。设立阴性对照组、阳性对照组和给药组,每组3个复孔。在阴性对照组中,向384孔板中依次加入2μldiluentbuffer、4μl经diluentbuffer稀释的tag1-pd-l1和4μl经diluentbuffer稀释的tag2-pd-1;在阳性对照组中,向384孔板中依次加入2μldiluentbuffer、4μl经diluentbuffer稀释的tag1-pd-l1和4μldiluentbuffer;在给药组中,向384孔板中依次加入2μl经diluentbuffer稀释的受试药物[阳性药、式(i)或式(ii)化合物],随后每孔各加入4μl经diluentbuffer稀释的tag1-pd-l1和4μl经diluentbuffer稀释的tag2-pd-1。待各组加完后,将384孔板于37℃下孵育15分钟后,再向每孔加入5μl经detectionbuffer稀释的anti-tag1-eu3+和anti-tag2-xl665。于37℃下孵育1小时至过夜后,用perkinelmerenvision多功能酶标检测仪测定在665nm和620nm处的荧光值,htrfratio=(665nm/620nm)*104。每个化合物检测8-10个浓度梯度,使用graphpad软件计算化合物的ic50。本实验选用bms公司专利wo2015160641中的化合物1016(bms-1016)为阳性药。活性测试结果见表2。表2化合物抑制pd-1与pd-l1相互作用的蛋白水平活性注:“nd”表示未检测。实验结果表明,本发明的化合物具有显著的pd-1/pd-l1蛋白-蛋白相互作用的抑制活性。例如,化合物5(ic50=2.10nm)、化合物9(ic50=2.29nm)、化合物10(ic50=2.52nm)、化合物24(ic50=6.11nm)、化合物25(ic50=7.36nm)、化合物37(ic50=2.58nm)、化合物38(ic50=6.42nm)、化合物39(ic50=8.51nm)、化合物41(ic50=3.86nm)、化合物43(ic50=6.16nm)、化合物45(ic50=8.10nm)、化合物47(ic50=2.58nm)、化合物48(ic50=6.64nm)、化合物49(ic50=4.33nm)、化合物56(ic50=1.97nm)、化合物74(ic50=4.76nm)、化合物78(ic50=8.11nm)、化合物80(ic50=3.5nm)、化合物81(ic50=1.69nm)、化合物85(ic50=2.33nm)和化合物117(ic50=7.8nm)等的活性显著优于阳性对照bms-1016(ic50=34.31nm)。本发明中的其他化合物也都显示了显著的pd-1/pd-l1相互作用的抑制活性。化合物8、36、46、65、67、71、73、75、77、79、84、87、89、91、125、127、147、149、151、153、155、157、159、161、163和165等可作为相应的羧酸化合物的酯类前药而在体内发挥pd-1/pd-l1相互作用的抑制活性。实施例158化合物阻断hpd-l1蛋白抑制人外周血单个核细胞(pbmc)分泌ifn-γ作用的实验人外周血单个核细胞(pbmc)活化后会分泌ifn-γ、il-2和tnf-α等细胞因子,而hpd-l1(人源pd-l1)蛋白与人pbmc上的pd-1蛋白结合会抑制pbmc的活化,降低细胞因子分泌。本实验的目的是检测化合物阻断hpd-l1蛋白抑制pbmc活化的能力。具体操作如下:采用澳赛尔斯生物技术有限公司购得的人pbmc,接种到96孔板中,每孔接种1ⅹ105个细胞。实验共分五组,一组为空白对照(只加入pbmc),一组为激活组(pbmc+anti-cd3/anti-cd28抗体),一组为抑制组(pbmc+anti-cd3/anti-cd28抗体+hpd-l1蛋白),其他两组分别为在抑制组的基础上加入1μm和10nm的化合物24。48小时后采用biolegend公司的ifn-γelisa检测试剂盒检测细胞上清液中ifn-γ的含量。实验结果显示,激活组中pbmc受到anti-cd3/anti-cd28抗体作用后,分泌ifn-γ显著增加,说明pbmc呈激活状态;抑制组中hpd-l1蛋白抑制pbmc分泌ifn-γ,说明pbmc的激活状态受到抑制。实验结果(图1)显示,化合物24具有显著的阻断pd-l1抑制人pbmc细胞分泌ifn-γ的作用。本发明中的其他化合物在该实验中也表现出相似的效果,例如,化合物5、9、10、25、31、37、43、47、56和74等在1μm和10nm浓度下具有显著的阻断pd-l1抑制人pbmc细胞分泌ifn-γ的作用。这表明本发明的化合物可阻断pd-1和pd-l1的相互作用,进而可增强t细胞免疫活性。实施例159化合物阻断肿瘤细胞抑制人t细胞增殖作用的实验人乳腺癌细胞mda-mb-231表面表达有pd-l1蛋白,当激活的人t细胞与mda-mb-231细胞接触后,会激活pd-1/pd-l1通路,进而抑制t细胞的激活,引起t细胞增殖变缓,并减少ifn-γ、il-2和tnf-α等细胞因子的释放。本实验的目的是检测化合物阻断mda-mb-231细胞抑制人t细胞增殖的能力。具体操作如下:将对数期生长的mda-mb-231细胞接种到96孔板中,待生长到合适细胞密度,将分选得到的人t细胞悬液加入上清中,用anti-cd3/anti-cd28抗体激活t细胞,加入待测化合物。48小时后通过流式细胞仪检测t细胞的增殖情况。实验共分六组,一组为空白对照组(只加入t细胞),一组为激活组(t细胞+anti-cd3/anti-cd28抗体),一组为抑制组(t细胞+anti-cd3/anti-cd28抗体和mda-mb-231),其他三组分别为在抑制组的基础上加入10μm、100nm和10nm的化合物24。实验结果(图2)显示,激活组中t细胞受到anti-cd3/anti-cd28抗体作用后,增殖显著增加,说明t细胞呈激活状态;抑制组中人乳腺癌细胞mda-mb-231与t细胞共培养后,t细胞增殖能力下降,说明t细胞的激活状态受到抑制;化合物24具有显著阻断肿瘤细胞抑制人t细胞增殖的能力。本发明中的其他化合物在该实验中也表现出相似的效果,例如,化合物5、9、10、25、31、37、43、47、56和74等在10μm、100nm和10nm浓度下具有显著的阻断肿瘤细胞抑制人t细胞增殖的能力。这表明本发明的化合物可促进t细胞的增殖,增强t细胞的免疫功能,进而可抑制pd-1/pd-l1介导的肿瘤免疫逃逸。实施例160化合物阻断肿瘤细胞抑制人t细胞分泌ifn-γ作用的实验如前所述,人乳腺癌细胞mda-mb-231表面表达有pd-l1蛋白,当激活的人t细胞与mda-mb-231细胞接触后,会激活pd-1/pd-l1通路,进而减少ifn-γ、il-2和tnf-α等细胞因子的释放。本实验的目的是检测化合物阻断mda-mb-231细胞抑制人t细胞分泌ifn-γ的能力。具体操作如下:将对数期生长的mda-mb-231细胞接种到96孔板中,待生长到合适细胞密度,将分选得到的人t细胞悬液加入上清中,用anti-cd3/anti-cd28抗体激活t细胞,加入待测化合物。48小时后采用biolegend公司的ifn-γelisa检测试剂盒检测上清中ifn-γ的表达量。实验共分六组,一组为空白对照组(只加入t细胞),一组为激活组(t细胞+anti-cd3/anti-cd28抗体),一组为抑制组(t细胞+anti-cd3/anti-cd28抗体+mda-mb-231),其他三组分别为在抑制组的基础上加入10μm、100nm和10nm的化合物24。实验结果(图3)显示,激活组中t细胞受到anti-cd3/anti-cd28抗体作用后,分泌ifn-γ显著增加,说明t细胞呈激活状态;抑制组中人乳腺癌细胞mda-mb-231与t细胞共培养后,t细胞分泌ifn-γ的水平下降,说明t细胞的激活状态受到抑制;化合物24具有显著的阻断肿瘤细胞抑制人t细胞分泌ifn-γ的能力。本发明中的其他化合物在该实验中也表现出相似的效果,例如,化合物5、9、10、25、31、37、43、47、56和74等在10μm、100nm和10nm的浓度下具有显著阻断肿瘤细胞抑制人t细胞分泌ifn-γ作用的能力。这表明本发明的化合物可解除pd-1/pd-l1对t细胞的抑制作用,恢复活化的t细胞分泌ifn-γ等细胞因子的能力,增强t细胞的免疫活性,进而增强t细胞的抗肿瘤免疫活性。实施例161化合物阻断hpd-l1蛋白抑制jurkat细胞分泌il-2作用的实验jurkat细胞活化后会分泌ifn-γ、il-2和tnf-α等细胞因子,而hpd-l1(人源pd-l1)蛋白与jurkat细胞表面的pd-1蛋白结合会抑制细胞的活化,降低细胞因子分泌。本实验的目的是检测化合物阻断hpd-l1蛋白抑制jurkat细胞活化的能力。具体操作如下:将jurkat细胞接种到96孔板中,每孔接种1ⅹ105个细胞。实验共分空白对照组(只加入jurkat细胞),激活组(jurkat细胞+anti-cd3抗体),抑制组(pbmc+anti-cd3抗体+hpd-l1蛋白),其他组为在抑制组的基础上加入10μm、1μm和100nm的阳性药bms-1016、化合物9和化合物74。48小时后采用biolegend公司的il-2elisa检测试剂盒检测细胞上清液中il-2的含量。实验结果显示,激活组中jurkat细胞受到anti-cd3抗体作用后,分泌il-2显著增加,说明jurkat细胞呈激活状态;抑制组中hpd-l1蛋白抑制jurkat细胞分泌il-2,说明jurkat细胞的激活状态受到抑制。实验结果(图4)显示,化合物9、74与阳性药bms-1016具有显著的阻断pd-l1抑制人jurkat细胞分泌il-2的作用。本发明中的其他化合物在该实验中也表现出相似的效果,例如,化合物5、9、10、25、31、37、43、47、56和74等在1μm和10nm浓度下具有显著的阻断pd-l1抑制jurkat细胞分泌il-2的作用。这表明本发明的化合物可阻断pd-1和pd-l1的相互作用,进而可增强t细胞免疫活性的功效。实施例162利用biacore分子相互作用仪测定化合物与hpd-l1蛋白的分子相互作用小分子与靶蛋白有相互作用是证明化合物是否在靶的重要依据,而小分子与蛋白质的结合力是验证小分子与蛋白是否有直接作用的重要指标。实验原理:biacore是基于一种表面等离子共振(spr)的物理光学现象的生物传感分析技术,不必使用荧光标记和同位素标记,从而保持了生物分子的天然活性。表面等离子体共振(surfaceplasmonresonance,spr)是一种光学现象,在传感芯片发生全反射界面上有一层约50nm厚的金属膜,偏振光入射到棱镜的一端,在棱镜与金属膜的界面会产生表面等离子波,当入射光波的传播常数与表面等离子波的传播常数匹配时,金属膜内的自由电子会产生共振,即表面等离子共振,引起表面等离子体共振的入射角称为spr角。该仪器检测spr角的变化,spr角又与芯片金属表面的折射率有关;分析时,先将一种生物分子即配体(蛋白、抗体等)偶联在生物传感器表面,再将含有另一种能与靶分子产生相互作用的生物分子(分析物)的溶液注入并流经生物传感器表面。生物分子间的结合引起生物传感器表面质量的增加,导致折射率的变化,通过监测spr的角度变化,可自动获得分析物的动力学结合和解离常数、亲和力及特异性等。实验方法:先将hpd-l1蛋白固定在cm5芯片上,将配置好的一系列浓度的化合物通过biacore分子相互作用仪流经固定有蛋白的芯片表面,经仪器分析得到相关数据。本实验运用biacore分子相互作用分析仪检测化合物9与hpd-l1是否有相互作用,实验结果见图5和表3。实验结果表明,化合物9与hpd-l1蛋白的kd值为0.5nm,说明化合物9与hpd-l1具有很强的结合力。通过此实验可以确定本发明的化合物直接作用于hpd-l1上,从而阻断pd-1和pd-l1的相互作用,进而产生增强免疫反应而发挥抗肿瘤作用。本发明的其他化合物也显示了与pd-l1蛋白具有很强的结合力。表3化合物9与hpd-l1蛋白的分子相互作用数据实施例163基于nfat报告基因的细胞水平pd-1/pd-l1阻断实验基于nfat报告基因的pd-1/pd-l1阻断实验是一种生物相关的、以moa(mechanismofaction)为基础的检测方法,可用于测定阻断pd-1/pd-l1相互作用的抗体、小分子化合物及其他生物制剂的效能和稳定性。该试验由两种基因工程细胞系:pd-1效应细胞(pd-1effectorcells),即稳定表达人pd-1受体和nfat诱导的荧光素酶报告基因的jurkatt细胞;pd-l1/aapc/cho-k1细胞(aapcpdl1chocellline),是一种稳定表达人pd-l1和一种以抗原非依赖方式激活同源tcr的细胞表面蛋白的cho-k1细胞。以上两株细胞委托吉满生物科技(上海)有限公司代为构建。具体操作如下:(1)细胞铺板取对数生长期pd-l1/aapc/cho-k1细胞,轻轻吸出培养基并加入2ml胰酶洗去细胞表面残余培养基,弃除胰酶之后加入1ml胰酶,37℃消化2-3min,待细胞消化完全,加入2ml完全培养基洗脱,1000rpm离心5min,弃上清,接着加入1ml完全培养基将细胞重悬;取20μl进行计数,调整细胞密度为5×105cells/ml,每孔100μl加入到96孔板中,37℃,5%co2培养箱中培养,孵育16h。(2)细胞给药受试样品为bms-1016(阳性对照)和本发明的化合物。取受试样品溶液用培养基稀释至检测浓度。取对数生长期pd-1效应细胞,将细胞转移至离心管,1000rpm离心5min,弃上清,接着加入1ml培养基将细胞重悬;取20μl进行计数,调整细胞密度为2×106cells/ml。将细胞培养板取出,移去上清,每孔加入50μl的密度为2×106cells/ml的pd-1效应细胞液。再将配制好的一系列浓度梯度(10μm-0.01μm)受试样品溶液以每孔50μl加至上述细胞培养板中,37℃,5%co2培养箱孵育6h。(3)检测结果提前取出一管bio-gloluciferase试剂,室温融化,每孔100μl加入到细胞培养板中,室温避光孵育2-3min。用varioukanflash多功能酶标仪的化学发光检测模块读取化学发光数值。实验结果见表4。结果表明,本发明的化合物在细胞水平可显著阻断pd-1和pd-l1的相互作用。例如。化合物5(ec50=2.238μm)、化合物9(ec50=3.644μm)、化合物24(ec50=1.754μm)、化合物55(ec50=0.6437μm)、化合物73(ec50=0.56μm)和化合物85(ec50=0.0.2101μm)。本发明的其他化合物在细胞水平上也可显著阻断pd-1/pd-l1的相互作用。表4化合物在细胞水平阻断pd-1/pd-l1相互作用的活性数据化合物编号ec5052.24μm93.64μm241.75μm550.64μm730.56μm850.21μm实施例164化合物的体内药代动力学研究本实验在雄性sd大鼠中进行,动物来源为上海西普尔-必凯实验动物有限公司。选取化合物8(化合物9的酯类前药)作为实验药物,检测其体内活性代谢产物化合物9。实验过程:给药前将大鼠称重,根据体重,计算给药量。通过静脉注射(剂量:2mg/kg)或灌胃口服(剂量:10mg/kg)给药。静脉注射给药前,给药后0.083h,0.25h,0.5h,1h,2h,4h,8h,24h。灌胃口服给药前,0.25h,0.5h,1h,2h,4h,6h,8h,24h采血,经颈静脉采血,每个样品采集约0.20ml,k2edta抗凝,采集后放置冰上。血液样本采集后置于冰上,并于1小时之内离心分离血浆(离心条件:6800g,6分钟,2-8℃)。血浆样本在分析前存放时则放于-80℃冰箱内。血浆中受试物的分析由美迪西普亚医药科技(上海)有限公司分析实验室完成,分析样品的同时进行质控样品的日内准确度评价,并要求超过66.7%的质控样品的准确度在80-120%之间。通过不同时间点的血药浓度数据,运用phoenixwinnonlin7.0计算药代动力学参数,实验结果见表5。表5化合物8大鼠体内药代动力学参数实验结果表明,化合物8可口服吸收,且在体内可顺利转化为活性代谢产物化合物9。本发明的其他化合物也都可以口服吸收,且具有较好的药代动力学性质。实施例165化合物对小鼠急性毒性试验单次给药毒性试验对初步阐明药物的毒性作用和了解其毒性靶器官具有重要意义。单次给药毒性试验所获得的信息对重复给药毒性试验的剂量设计和某些药物临床试验起始剂量的选择具有重要参考价值,拟用于人体的药物通常需要进行单次给药毒性试验。为验证本发明化合物的安全性,选取化合物8(化合物9的酯类前药)进行该实验,通过对c57bl/6小鼠(购自北京维通利华实验动物技术有限公司,体重18-22g,8周龄,适应性饲养1周后开始实验)单次口服给予较大剂量化合物,观察小鼠体重及正常生理活动有影响。实验分为溶媒对照组和实验组,每组10只,雌雄各半,实验组给药剂量为2g/kg。给药后密切观察小鼠存活情况,若小鼠出现死亡,解剖死亡小鼠寻找死因,并降低化合物浓度,重复次实验直至找到安全的化合物剂量;若小鼠生长正常,持续饲养两周后处死小鼠。实验结果:给药后动物生长状态良好,体重未出现明显变化,饮食正常,大便形状正常,毛发光泽,解剖观察体内脏器未发现病理性变化。以上结果表明,化合物8在2g/kg单次给药剂量下对小鼠未产生明显的毒性作用,证明其安全性良好。本发明的其他化合物也显示了很好的安全性。实施例166化合物对小鼠亚急性毒性试验亚急性毒性是评价药物长期作用于机体所产生的损害作用,对于评价药物的中长期毒性作用具有重要意义。选取化合物8(化合物9的酯类前药)进行该实验。实验分为溶媒对照组、高剂量组(300mg/kg)及低剂量组(150mg/kg),每组小鼠10只,雌雄各半。动物:c57bl/6小鼠(购自北京维通利华实验动物技术有限公司),体重18-22g,8周龄,适应性饲养1周后开始实验。具体操作如下:将适应性饲养1周的小鼠分组,每日观察小鼠生长状况、称重并给药,2周后脱颈处死小鼠,解剖小鼠,分离体内脏器,观察有无病变,取血检测各项血液指标有无异常。实验结果(图6、图7、图8)表明,各组小鼠生长状态良好,未发生死亡。高剂量组与低剂量组小鼠与溶媒对照组相比体重未发生明显变化,心、肝、肾重量无明显变化,脾脏重量略微减轻。血液各指标(ast、alt、bun、crea和ck)均未发生明显变化。以上实验结果表明,长期给予化合物8在300mg/kg剂量下具有很好的安全性。本发明的其他化合物也显示了很好的长期安全性。实施例167化合物对herg钾离子通道的影响快速激活人延迟整流外向钾电流(ikr)主要由herg离子通道介导,参与人类心肌细胞复极化。药物阻断这一电流将导致临床上出现qt间期延长综合征,易诱发急性心律失常甚至猝死。本实施例应用手动膜片钳的方法在转染herg钾通道的稳定细胞株上测试本发明化合物9和化合物74对herg钾电流的作用,从而确定受试物是否对herg离子通道具有抑制作用。将阳性对照药特非那定配制成最终浓度为0.001、0.01、0.1、和1μm,细胞外液中dmso最终浓度为0.3%的溶液,测定其对herg电流的抑制作用,结果见表6。将化合物9和化合物74配制成最终检测浓度为0.3、1、3、10和30μm,细胞外液中dmso最终浓度为0.3%的溶液,测定其对herg电流的抑制作用,结果见表7和表8。表6特非那定对herg电流的抑制作用表7化合物9对herg电流的抑制作用表8化合物74对herg电流的抑制作用在本次研究中,阳性对照品terfenadine对herg电流抑制作用具有浓度依赖性,其ic50值为0.054μm,这与文献报道的结果相符。在本实验检测浓度范围内(0.3、1、3、10和30μm),化合物9和化合物74在30μm时对herg钾通道电流的平均抑制率分别为15.03%和26.16%(n=2),这表明这两个化合物不抑制herg离子通道,因而对心脏的潜在副作用很小。本发明的其他化合物对心脏的潜在副作用也很小。实施例168小鼠黑色素瘤高转移细胞(b16f10)皮下植瘤模型为验证本发明化合物的体内药效,应用小鼠黑色素瘤高转移细胞(b16f10)皮下植瘤模型,选择化合物8(该化合物是化合物9的酯类前药)进行该实验。实验分为溶媒对照组、阳性药组(环磷酰胺80mg/kg)、化合物8(15mg/kg)组及化合物8(45mg/kg)组。动物:雌性c57bl/6小鼠(购自北京维通利华实验动物技术有限公司),体重18-22g,8周龄,适应性饲养1周后开始实验。具体操作如下:将对数生长期的b16f10细胞用胰酶消化离心收集细胞,pbs重悬2次后用pbs将细胞密度调整为2ⅹ106个/ml,每只小鼠皮下接种100μl细胞悬液。接种6小时后将小鼠随机分组,空白对照组8只,其余三组各10只,称重后给药;化合物使用0.5%羧甲基纤维素钠水溶液充分研磨成混悬液,每日给药一次,用游标卡尺每日测量肿瘤体积,连续给药18天后脱颈处死小鼠,剥离肿瘤组织称重。肿瘤体积的计算公式:v=0.5a×b2,a和b分别表示肿瘤的长径和短径。相对肿瘤增殖率t/c(%)的计算公式:t/c=trtv/crtv×100%(trtv:治疗组rtv;crtv:溶剂对照组rtv)。根据测量结果计算出相对肿瘤体积(rtv),rtv=vt/vo,其中vo为实验开始时的肿瘤体积,vt每一次测量时的肿瘤体积。肿瘤生长抑制率tgi(%)的计算公式:tgi=[1-(某组给药结束时平均瘤体积-该组开始给药时平均瘤体积)/(溶媒对照组治疗结束时平均瘤体积-溶媒对照组开始治疗时平均瘤体积)]×100%。药效评价指标如表9所示,肿瘤体积变化如图9所示。实验结果表明,化合物8在小鼠黑色素瘤高转移细胞皮下植瘤模型中表现出明显的肿瘤生长抑制作用;其中,在每天给药15mg/kg时,其肿瘤生长抑制率为45.77%,在每天给药45mg/kg时,其肿瘤生长抑制率为70.70%;并且,化合物8并未产生小鼠体重降轻的副作用,而环磷酰胺组小鼠体重显著下降。以上研究结果表明,化合物8具有显著的体内抗肿瘤活性。本发明的其他化合物也显示了相似的体内抗肿瘤疗效。表9化合物8的体内药效评价指标实施例169化合物在mc38-hpd-l1结肠癌细胞移植c57bl/6-hpd-l1荷瘤模型中的药效学评价受试化合物:化合物8(原始编号:y07200),使用0.5%cmc-na水溶液研磨为悬浊液;实验动物:c57bl/6-hpd-l1(人源化pd-l1的基因工程小鼠),6-8周龄,雌性;细胞系:mc38-hpd-l1(人源化pd-l1的基因工程细胞系);实验分组:实验分为溶媒对照组、化合物8(25mg/kg)组及化合物8(50mg/kg)组;具体操作如下:复苏细胞mc38-hpd-l1,收集对数生长期的细胞,去除培养液并用pbs洗两次后接种,接种量:1×106/100μl/只,接种位置:右侧flank位置;当肿瘤平均体积在50-60mm3左右,根据肿瘤体积随机分组,每组6只。分组当天定义为d0天,并于分组当天,根据实验方案开始给药。开始给药后,每周称量体重二次。每周测量瘤体积二次,瘤体积计算方式为:肿瘤体积(mm3)=0.5×(肿瘤长径×肿瘤短径2),待达到实验终点脱颈处死小鼠,剥离肿瘤组织称重,并进行相关检测实验。实验结果见图10和图11。实验结果表明,化合物8具有显著的体内抗肿瘤效应。在25mg/kg组其肿瘤生长抑制率为53.82%,在50mg/kg组其肿瘤生长抑制率为42.15%,说明化合物8可以有效抑制肿瘤生长。本发明的其他化合物也具有类似的抗肿瘤疗效。实施例170化合物对小鼠银屑病模型的影响为验证本发明化合物对自身免疫性疾病的作用,采用小鼠银屑病模型验证化合物的药效。实验动物:balb/c雌性小鼠,8周龄,购自浙江维通利华实验动物有限公司。受试药物配置:将1ml乙醚加入到10g羊毛脂和10g凡士林中,37℃下加热溶解。称取600mg化合物8(原始编号:y07200)溶于2mldmso,溶解后加入到羊毛脂与凡士林的混合液中,加入3ml乙醇搅拌均匀,继续加热若干分钟。将0.05g尼泊金甲酯和0.02g尼泊金丙溶于0.4ml乙醚后加入到混合物中,200μl乙醚刷洗1-2次。88℃加热60min。室温搅拌至凝,配制成300mg/kg化合物8软膏。同时配制不含化合物的对照软膏。造模给药:将小鼠随机分为空白组(control组)、模型组(model组)和给药组,每组10只。小鼠背部脱毛,露出2cm×3cm的皮肤区域。脱毛后适应3天。5%咪喹莫特(imiquimod,imq)乳膏62.5mg/只涂抹右耳及后背,每天一次,造模5天。同时给予给药组300mg/kg化合物8软膏,对照组以及模型组动物对照软膏,0.2g/只,每天2次,共给药5天。每天称重,测量小鼠右耳厚度,拍摄后背照片对其pasi打分。待给药结束后取血,皮肤和耳朵组织,进行下一步实验验证。实验结果见图12、图13、图14、图15、图16和图17。实验结果表明,化合物8可以有效地缓解小鼠银屑病的疾病进程,降低血清il-17a水平,缓解炎症症状,说明本发明的联苯类化合物可抑制银屑病动物模型中炎症因子的释放,且对银屑病具有显著疗效,这提示本发明的联苯类化合物可用于预防和治疗自身免疫性疾病、器官移植排斥、感染性疾病和炎症性疾病。本发明的其他化合物也具有类似的抗炎疗效。实施例171化合物对人肝微粒体代谢稳定性研究人肝微粒体代谢稳定性评价是药物研发中临床前评价候选化合物药代动力学性质的重要手段。本实验参照文献方法(pharmacolrep.2006,58,453-472)进行。实验温孵体系(体积为250μl,n=3)由肝微粒体,受试物工作溶液和磷酸盐缓冲液,将温孵体系于37℃共孵育一个小时,加入nadph溶液后计时开始,每个时间点以加入终止液终止反应,取样间点为0,5,15,30,60min,共5个点。阳性对照药为咪达唑仑(相同体系,相同条件)。阴性对照不加nadph,取样时间点为0和60min。此外,将中国专利申请2019102477713中的化合物1、9、10、11、13、14作为对比化合物,检测其在相同体系、相同条件下对人肝微粒体的代谢稳定性。用lc-ms/ms进行分析,通过受试物剩余量的百分率的自然对数与时间作图测得斜率的绝对值k,按以下公式进行计算:t1/2(半衰期)=ln2/k=0.693/k。实验结果如表10所示。表10化合物对人肝微粒体的代谢稳定性研究实验结果表明,本发明的化合物对人肝微粒体的代谢稳定性良好,t1/2均大于60分钟,其中化合物5、9、10、11、21、37、47、55、57、59、61、64、66、78、80、88、92、109和121的t1/2均大于120分钟。这表明本发明化合物具备良好的代谢稳定性,这可以降低药物的肝脏首过效应,显示出本发明化合物具备良好的药代动力学性质。另一方面,对比化合物如13和14等(中国专利申请2019102477713中的化合物)虽然具有显著的pd-1/pd-l1蛋白-蛋白相互作用的抑制活性,但是其代谢稳定性不够理想(t1/2均小于10分钟),因而严重影响其成药性。实施例172化合物的化学稳定性评价实验方法:取待测化合物用dmso溶解,配置浓度为10mm的溶液,将溶液放置于37℃恒温孵箱中,14天后取出。使用waters超高效液相色谱acquityuplctm检测第1天与第14天的化合物纯度,计算化合物的降解率。此外,将中国专利申请2019102477713中的化合物1、9、10、11、13、14作为对比化合物,检测其在相同条件下的化学稳定性。化合物降解率=(第1天化合物纯度-第14天化合物纯度)/第1天化合物纯度×100%,实验结果见表11。色谱条件:色谱柱:acquityuplctmbehc181.7μm2.1×50mmcolumn检测器:acquityuplc光电二极管矩阵(pda)检测器柱温:40℃检测波长:254nm流动相a:超纯水流动相b:含0.1%三氟乙酸的甲醇溶液梯度:20%流动相a,80%流动相b表11部分化合物的降解率化合物编号降解率5<0.01%7<0.01%8<0.01%9<0.01%100.02%24<0.01%25<0.01%260.02%37<0.01%42<0.01%47<0.01%51<0.01%56<0.01%58<0.01%61<0.01%81<0.01%840.02%85<0.01%对比化合物1>5%对比化合物9>5%对比化合物10>5%对比化合物11>5%对比化合物13>5%对比化合物14>5%实验结果表明,本发明的化合物化学稳定性良好,37℃恒温放置14天基本没有发生降解,这表明本发明化合物具备良好的化学稳定性。而中国专利申请2019102477713中的化合物(如对比化合物1、9、10、11、13和14)具有较差的化学稳定性,其降解率均大于5%,说明本发明化合物的化学稳定性明显优于对比化合物。实施例173分子对接研究分子对接是两个或多个分子之间通过几何匹配和能量匹配而相互识别的过程,分子对接在药物设计中已经体现出十分重要的应用价值。本实验运用软件,将本发明化合物9及相关化合物与已报道共晶结构(oncotarget.2017,8(42):72167-72181;pdb编号:6r3k)中pd-l1蛋白对接。实验方法:用软件中的proteinprepare模块处理共晶结构6r3k中的pd-l1蛋白,利用glidegrid模块根据晶体结构中化合物的结合位点定义格点文件;利用ligandpreparation模块对化合物进行处理;最后利用glide模块将经过处理的化合物与pd-l1蛋白进行对接,根据poseviewer中的结合模式、dockingscore及glideemodel评判对接结果;本次对接的化合物为本发明的化合物9与对比化合物x(将化合物9中的苯并噁二唑替换为苯环所得到的相应化合物),对接结果通过打分函数打分,结果见表12。表12分子对接打分结果实验结果表明,本发明化合物优势结构中的苯并噁二唑杂环可以很好地与hpd-l1蛋白发生相互作用,其中杂环的氧原子与hpd-l1蛋白中125位的精氨酸上的胍基产生关键的氢键相互作用;苯并噁二唑杂环与123位酪氨酸上的苯环产生关键的π-π相互作用,并且苯并噁二唑中噁二唑的强吸电子作用会加强π-π相互作用的强度,使该侧链与蛋白结合的强度更高,从而得到与hpd-l1蛋白结合能力更好的分子。通过打分函数打分结果,本发明如化合物9的打分为-11.627,而若将化合物9中的苯并噁二唑替换为苯环所得到的相应化合物的打分为-9.87,其打分显著低于化合物9,且本发明的其他化合物中苯并噁二唑均比替换成苯环得分更高。以上分子对接结果说明本发明化合物中的苯并噁二唑侧链对于化合物活性提升至关重要,即通过引入苯并噁二唑侧链,该类化合物阻断pd-1和pd-l1蛋白-蛋白相互作用的能力显著增加。实施例174片剂将实施例24中制得的化合物24(50g)、羟丙甲基纤维素e(150g)、淀粉(200g)、聚维酮k30适量和硬脂酸镁(1g)混合,制粒,压片。此外,可以根据药典2015版常规制剂法,将实施例1-156制得的化合物赋予不同的药物辅料制成胶囊剂、散剂、颗粒剂、丸剂、注射剂、糖浆剂、口服液、吸入剂、软膏剂、栓剂或贴剂等。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1