一种利用转移氢化法由氧化苦参碱制备苦参碱的方法与流程
本发明涉及天然药物化学技术领域,涉及一种由氧化苦参碱转化制备苦参碱的方法,特别是涉及一种利用转移氢化法由氧化苦参碱制备苦参碱的方法。
背景技术:
氧化苦参碱,又名苦参素,分子式为c15h24n2o2,分子量为264.36。白色针形棱柱形晶体或白色结晶性粉末,无臭、味苦。熔点207℃-208℃,易溶于水、甲醇、乙醇、氯仿、苯,难溶于乙醚。药效学研究表明,氧化苦参碱具有利尿,抗乙型和丙型肝炎病毒,免疫调节等作用,此外,对肺癌、胃癌细胞诱导的血管内皮细胞增殖具有抑制作用。
苦参碱具有很高的药用价值,分子式为c15h24n2o,白色针状或棱柱晶体,无臭,味苦。现代药理研究表明,苦参碱具有抗菌、抗病毒、抗寄生虫、抗心律失常和抗癌等作用,临床常用于治疗细菌性痢疾和肠炎。目前,苦参碱主要从豆科植物苦参、苦豆子和山豆根中提取分离,在制备过程中为了便于分离和提高苦参碱的得率,需要将氧化苦参碱(c15h22n2o2)转化为苦参碱。
目前工业上常采用加入亚硫酸盐的方法来还原氧化苦参碱,但是亚硫酸盐还有可能在还原氧化苦参碱的同时,与苦参碱、氧化苦参碱等分子上的羰基发生副反应,产生的副产物可能对后续的分离工艺带来影响;同时,羰基与亚硫酸盐产生的副产物通常水溶性不好,在固液分离操作时将该副产物分离,从而带来苦参碱产率的降低。
cn106810555a的发明专利申请公开了一种利用金属还原剂由氧化苦参碱制备苦参碱的方法,该方法是先将纯度为65%-100%的氧化苦参碱溶于含水50%-100%的强极性溶剂中,向溶液中加入金属还原剂和电解质,于40℃-100℃反应0.5小时-8小时,减压浓缩至50℃热测相对密度为1.02~1.06,浓缩液采用极性2.0-4.5的不溶于水的有机溶剂进行萃取,萃取液经蒸发、结晶、离心分离、干燥,得到白色晶体苦参碱。本发明工艺方法操作简便,成本低廉,易于工业化生产。转化收得率可达96%以上,产物纯度可达98%以上。对于苦参碱和氧化苦参碱生产企业,可以实现两个主要产品之间的灵活转化,以适应市场对氧化苦参碱和苦参碱的需求变化。但是该方法在反应的同时,产生的金属还原物作为固体废弃物,存在污染环境的问题。
催化转移氢化反应是指在催化剂作用下,氢由氢的给体(氢源)转移到有机化合反应底物的反应。通过催化转移氢化反应,可以将氢源上的氢加成到氧化苦参碱的n-o键上,实现由氧化苦参碱制备苦参碱的目的。该反应采用的催化剂通常是固体催化剂,反应后易于从反应体系中分离。选择的氢源,反应中除了目标产物,其它产物可能是气体,随时从体系中溢出,使反应向正反应方向进行,提高反应效率;同时,合适氢源所产生的气体还可能是不影响环境的气体,从而起到环保的作用。因此,催化转移氢化反应是目前绿色化学反应工艺中最受青睐的反应之一。
技术实现要素:
本发明的目的是提供一种利用转移氢化法由氧化苦参碱制备苦参碱的方法,对于生产苦参碱和氧化苦参碱的企业来说,对于生产苦参碱的企业来说,可以提高反应效率,保护环境。
为了达到上述目的,本发明通过以下技术方案实现:
一种利用转移氢化法由氧化苦参碱制备苦参碱的方法,其特征在于,包括如下步骤:
a.制备氧化苦参碱的极性溶液;
b.向溶液中加入氢源和催化剂,于40℃-100℃回流5小时-48小时;
c.减压浓缩至50℃热测相对密度为1.02-1.06;
d.浓缩液采用极性2.0-4.5的不溶于水的有机溶剂进行萃取;
e.萃取液经蒸发、结晶、离心分离、干燥,得到白色晶体苦参碱。
其中,步骤a中所述氧化苦参碱的极性溶液是将原料氧化苦参碱溶于含水10%-100%的极性溶剂中制成的溶液,或者是将原料苦豆子用含水10%-100%的极性溶剂提取制成的提取液。
步骤a中所述原料氧化苦参碱纯度为40%-100%,或者是原料苦豆子的含水10%-100%的极性溶剂的提取液。这是本发明的显著优势之一,采用本发明,即使使用较低纯度的氧化苦参碱原料,也可以得到纯度达到95.0%以上的苦参碱产品,同时,本发明还可以直接应用于苦豆子生物碱的生产过程,将氧化苦参碱转化为苦参碱,有利于工艺过程中对单体生物碱的分离,提高苦参碱的得率。
步骤a中所述含水10%-100%的极性溶剂为水,或水和甲醇、乙醇中的任意一种或两种的混合液,用量为氧化苦参碱的30倍-100倍。其中加入一定量的甲醇、乙醇可以达到很好的增溶作用,有利于氧化苦参碱原料,尤其是纯度较低氧化苦参碱原料的溶解;同时,甲醇、乙醇的质子化效应还有利于缩短反应时间。
步骤a中所述苦豆子提取用含水10%-100%的极性溶剂为水,或水和甲醇、乙醇中的任意一种或两种的混合液,用量为苦豆子的18倍-30倍。
步骤b中氢源的加入量与步骤a中的氧化苦参碱加入量的摩尔比为1.5-5:1,反应温度为40℃-100℃,反应时间为5小时-48小时。
所述氢源为环己烯、1,3-环己烯、1,4-环己烯、苄醇、苯乙醇、异丙醇、辛醇、甲酸、甲酸铵盐、水合肼、氨硼烷等中的任意一种或多种。
步骤b中所述催化剂为钯碳或镍催化剂中的任意一种,加入量与步骤a中氧化苦参碱量的质量比为0.05-0.2:1。
进一步,所述镍催化剂为镍-铝合金、雷尼镍、kt-02型镍催化剂中任意一种。
步骤b反应原理方程式如下:
步骤c中所述减压浓缩至50℃热测相对密度为1.02-1.06的主要目的有两个:其一是适度缩小溶液体积以利于下一步的萃取操作;其二是尽可能除去有机溶剂,以保证萃取效果、收率及所得到的苦参碱产品纯度。
步骤d中所述极性2.0-4.5的不溶于水的有机溶剂为三氯甲烷、二氯甲烷、苯、甲苯、二甲苯中的任意一种或多种,采用的有机溶剂对苦参碱有较好的溶解度,可以取得最佳的萃取效果。
步骤e中所述白色晶体苦参碱的纯度为95.0%以上。
与现有技术相比,本发明具有以下优点。
1.反应体系易于分离:采用本发明的利用转移氢化法由氧化苦参碱制备苦参碱的方法,催化剂通常是固体催化剂,反应后易于从反应体系中分离;的氢源,如水合肼、甲酸、甲酸铵等,反应中除了目标产物,其它产物都是气体,随时从体系中溢出。
2.反应效率高:采用本发明的利用转移氢化法由氧化苦参碱制备苦参碱的方法,选用的合适氢源,如水合肼、甲酸、甲酸铵等,反应中除了目标产物,其它产物都是气体,随时从体系中溢出,使反应向正反应方向进行,提高反应效率。
3.产品纯度高:采用高效液相归一化法检测反应物,以干燥品计算,所得苦参碱纯度可达95.0%以上
4.环保:采用本发明的利用转移氢化法由氧化苦参碱制备苦参碱的方法,可以避免产生金属还原法带来的固体废弃物对环境造成的污染,环保效益显著。
工艺简单,生产成本较cn106810555a公开的一种利用金属还原剂由氧化苦参碱制备苦参碱的方法更为低廉,适用于工业化生产,经济效益显著。
附图说明
图1a为实施例1-实施例4中苦参碱、氧化苦参碱对照品的高效液相色谱(hplc)图。
图1b为由实施例1得到的苦参碱样品的高效液相色谱(hplc)图。
图2为实施例2得到的苦参碱样品的高效液相色谱(hplc)图。
图3为实施例3得到的苦参碱样品的高效液相色谱(hplc)图。
图4为实施例4得到的苦参碱样品的高效液相色谱(hplc)图。
图5a为实施例5中苦参碱、氧化苦参碱对照品的高效液相色谱(hplc)图。
图5b为实施例5加氢反应前苦豆子提取液样品的高效液相色谱(hplc)图。
图5c为实施例5加氢反应后苦豆子提取液样品的高效液相色谱(hplc)图。
图6为由实施例4得到的苦参碱样品的1h-nmr谱。
图7为由实施例5得到的苦参碱样品的13c-nmr谱。
具体实施方式
下面结合具体实施例对本发明的技术方案做进一步说明。
实施例1
将氧化苦参碱纯品5g溶解于50%甲醇中,转移至反应釜中,加入1.7g1,3-环己烯,0.5g镍-铝合金,于50℃下反应5小时后,停止反应,减压浓缩至比重为1.04,用二氯甲烷萃取;将萃取液减压回收二氯甲烷完全,加入丙酮热溶、冷却析晶,离心分离得到苦参碱精品4.5g,高效液相归一化法测得纯度大于95.14%,氧化苦参碱转化为苦参碱的转化收得率以摩尔数计算为94.53%。
对所得产品进行高效液相色谱(hplc)分析,hplc色谱条件为:以十八烷基键合相硅胶为固定相,甲醇-0.2%磷酸溶液(13:87,v/v)为流动相,流速为1ml/min,检测波长为205nm,柱温为25℃-50℃。
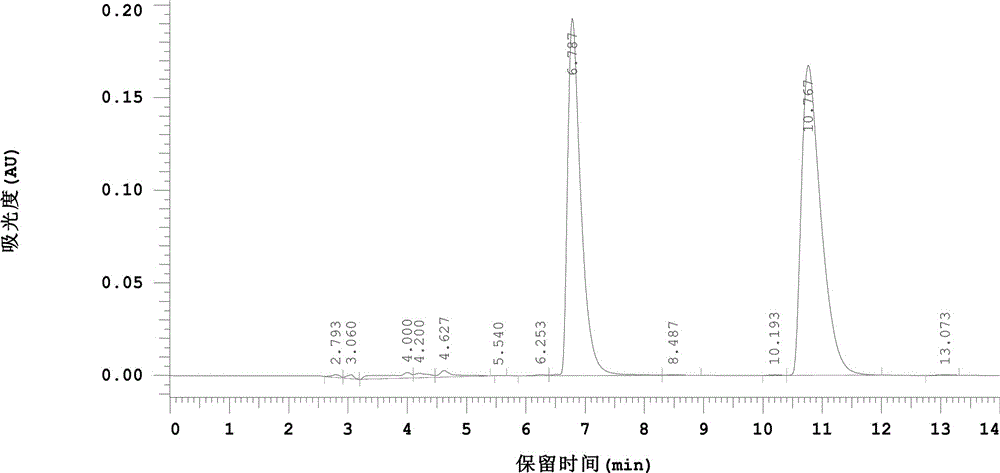
图1a所示为实施例1-实施例4中苦参碱、氧化苦参碱对照品的高效液相色谱(hplc)图,图1b为由实施例1得到的苦参碱样品的高效液相色谱图。保留时间在6.7min左右的为苦参碱色谱峰,保留时间在10.7min左右的为氧化苦参碱的色谱峰,证明由实施例1得到的苦参碱样品的纯度。
经对由实施例1得到的苦参碱样品,用高效液相色谱归一化法(对应图1b)进行纯度测定,具体结果见表1。
表1、实施例1所得苦参碱样品的高效液相色谱归一化法结果
由表1可见,经计算,测得苦参碱的纯度为95.14%。
实施例2
将氧化苦参碱纯品10g,溶解于30%乙醇中,转移至反应釜中,加入7.0g异丙醇,2g雷尼镍,于80℃下反应36小时后,停止反应,减压浓缩至比重为1.04,用甲苯萃取;将萃取液减压回收甲苯完全,加入丙酮热溶、冷却析晶,离心分离得到苦参碱精品9.2g,高效液相归一化法测得纯度大于95.16%,氧化苦参碱转化为苦参碱的转化收得率以摩尔数计算为95.94%。
对所得产品进行高效液相色谱(hplc)分析,hplc色谱条件同实施例1。
图2所示为由实施例2得到的苦参碱样品的高效液相色谱(hplc)图。保留时间在6.6min左右的为苦参碱色谱峰,证明由实施例2得到的苦参碱样品的纯度。
经对由实施例2得到的苦参碱样品,用高效液相色谱归一化法(对应图3)进行纯度测定,具体结果见表2。
表2、实施例2所得苦参碱样品的高效液相色谱归一化法结果
由表2可见,经计算,测得苦参碱的纯度为95.16%。
实施例3
将氧化苦参碱粗品50g(含氧化苦参碱65%),溶解于60%乙醇中,转移至反应釜中,加入20g氨硼烷,5gkt-02型镍催化剂,于80℃下反应36小时后,停止反应,减压浓缩至比重为1.04,用甲苯萃取;将萃取液减压回收甲苯完全,加入丙酮热溶、冷却析晶,离心分离得到苦参碱精品28.5g,高效液相归一化法测得纯度大于96.23%,氧化苦参碱转化为苦参碱的转化收得率以摩尔数计算为97.48%。
对所得产品进行高效液相色谱(hplc)分析,hplc色谱条件同实施例1。
图3所示为由实施例3得到的苦参碱样品的高效液相色谱(hplc)图。保留时间在6.7min左右的为苦参碱色谱峰,证明由实施例3得到的苦参碱样品的纯度。
经对由实施例3得到的苦参碱样品,用高效液相色谱归一化法(对应图3)进行纯度测定,具体结果见表3。
表3、实施例3所得苦参碱样品的高效液相色谱归一化法结果
由表3可见,经计算,测得苦参碱的纯度为96.23%。
实施例4
将生产过程中得到的含氧化苦参碱50%的水溶液1000g,加入95%乙醇500ml,混匀,转移至反应釜中,加入500g水合肼(含水合肼85%),100g雷尼镍,于80℃下反应48小时后,停止反应,减压浓缩至比重为1.04,用二甲苯萃取;将萃取液减压回收二甲苯完全,加入丙酮热溶、冷却析晶,离心分离得到苦参碱精品409.5g,高效液相归一化法测得纯度98.16%,氧化苦参碱转化为苦参碱的转化收得率以摩尔数计算为96.39%。本实施例中产物除苦参碱外,仅产生氮气,具有明显的环保作用。
对所得产品进行高效液相色谱(hplc)分析,hplc色谱条件同实施例1。
图4所示为由实施例4得到的苦参碱样品的高效液相色谱(hplc)图。保留时间在6.7min左右的为苦参碱色谱峰,证明由实施例4得到的苦参碱样品的纯度。
经对由实施例4得到的苦参碱样品,用高效液相色谱归一化法(对应图4)进行纯度测定,具体结果见表4。
表4、实施例4所得苦参碱样品的高效液相色谱归一化法结果
由表4可见,经计算,测得苦参碱的纯度为98.16%。
实施例5
将苦豆子100kg置于多功能提取罐中,加入800l含30%乙醇的水溶液,回流提取3次。提取完成后,将苦豆子提取液合并,提取液中含总固形物为42.579kg,检测其中含有的氧化苦参碱664.8g(含量为1.6%,g/g),苦参碱437.6g(含量为1.0%,g/g)。在多功能提取罐中加入700g甲酸,100g钯碳,于80℃下反应48小时后,停止反应,用高效液相检测反应后的色谱图,可以观察到氧化苦参碱已全部转化。反应液中含总固形物为43.064kg,通过高效液相法测定其中的苦参碱含量为973.1g(含量为2.3%,g/g),计算氧化苦参碱转化为苦参碱的转化率为85.75%。本实施例中,通过转移氢化法,可以将氧化苦参碱转化为苦参碱,苦参碱易溶于极性2.0-4.5的不溶于水的有机溶剂,有利于苦豆子主要生物碱的分离;产物除苦参碱外,仅产生二氧化碳,具有明显的环保作用;尤其是,苦参碱的含量大大提高,通过后期的分离纯化,可以得到高纯度的苦参碱,可见本方法不仅可以保护环境,而且可以提高苦参碱的纯度。
对所得产品进行高效液相色谱(hplc)分析,为了尽可能得将苦豆子提取液中所有生物碱分离,hplc色谱条件为:以十八烷基键合相硅胶为固定相,甲醇-0.2%磷酸溶液(7:93,v/v)为流动相,流速为1ml/min,检测波长为205nm,柱温为25℃-50℃。
图5a所示为实施例5中苦参碱、氧化苦参碱对照品的高效液相色谱(hplc)图;图5b所示为实施例5加氢反应前苦豆子提取液的高效液相色谱(hplc)图;图5c所示是实施例5加氢反应后苦豆子提取液的高效液相色谱(hplc)图。保留时间在19.3min左右的为苦参碱色谱峰,保留时间在24.1min左右的为氧化苦参碱的色谱峰。证明按照实施例5中所述的工艺方法,可将苦豆子提取液中的氧化苦参碱还原为苦参碱,提高苦参碱的得率。
图6和图7所示为由实施例5得到的苦参碱样品的核磁共振图谱,证明产物为苦参碱。其中,图6为1h-nmr图谱(cdcl3-d,400mhz),图7为13c-nmr图谱(cdcl3-d,100mhz)。具体信号归属如下:
1h-nmr(cdcl3-d,400mhz):4.40(dd,1h,j=12,4hz,h-17e),3.82(dt,1h,j=8,8hz,h-11),3.05(t,1h,j=12hz,h-17a),2.81(m,2h,h-2e,h-10e),2.43(dt,1h,j=4,16hz,h-14e),2.27(ddd,1h,j=4,8hz,h-14a),2.09(m,2h,h-4e,h-7),1.98(m,3h,h-2a,h-10a,h-8e),1.66(m,1h,h-5),1.58(m,6h,h-9e,h-6,h-8a,h-12,h-13e),1.43(m,5h,h-3,h-4a,h-9a,h-13a)。
13c-nmr(cdcl3-d,100mhz):57.28(c-2),20.84(c-3),27.23(c-4),35.37(c-5),43.27(c-6),63.83(c-7),26.52(c-8),21.25(c-9),57.36(c-10),53.25(c-11),27.81(c-12),19.07(c-13),32.93(c-14),169.48(c-15),41.48(c-17)。
上述实施例仅为本发明较佳的实施方式,本发明的实施方式并不受上述实施例的限制,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明技术原理的前提下,可以设计出很多其他的修改或优化实施方式,这些修改或优化实施方式将落在本申请公开的范围之内。
- 还没有人留言评论。精彩留言会获得点赞!