低粘度聚酰亚胺前驱体溶液及其制备方法、用途与流程
1.本发明属于聚合物及其复合材料制备领域,尤其涉及一种低粘度聚酰亚胺前驱体溶液及其制备方法、用途。
背景技术:
2.热固性聚酰亚胺树脂及其复合材料具有异常突出的耐热性和高力学强度,在航空航天领域常常用作承力结构材料和功能结构材料,但是热固性聚酰亚胺树脂复合材料成型工艺困能。为了提高树脂工艺性,nasa提出了pmr(单体原位聚合技术)法,实现高浓度聚酰亚胺前躯体溶液的制备,该树脂溶液非常适合浸渍纤维制备复合材料,此后这一方法被广泛应用于热固性聚酰亚胺树脂基复合材料的研发。从目前报道看,在pmr法中,常用于酯化四酸二酐的醇类试剂为甲醇和乙醇,但是一些常用的刚性对称四酸二酐(如pmda和sbpda)难以在乙醇或甲醇中酯化得到澄清溶液,也就无法完成高固含量聚酰亚胺前躯体溶液的制备。
技术实现要素:
3.本发明的目的在于克服现有技术中的不足,提供一种低粘度聚酰亚胺前驱体溶液及其制备方法、用途,以解决现有高浓度聚酰亚胺前躯体溶液制备方法中所存在的技术问题。
4.本发明的技术解决方案为:
5.根据第一方面,提供一种低粘度聚酰亚胺前驱体溶液的制备方法,该方法包括以下步骤:
6.1)将环状二酸酐在乙二醇单甲醚溶液中回流发生开环酯化反应得到含醚键二酯二酸溶液;
7.2)将单酸酐封端剂在乙二醇单甲醚溶液中回流发生开环酯化反应得到含醚键单酯单酸溶液;
8.3)将芳香族二胺加入到所述步骤1)制备的含醚键二酯二酸溶液中进行羧基氨基的成盐反应,得到胺基封端的聚酯铵盐溶液;
9.4)将步骤2)得到的含醚键单酯单酸溶液加入到步骤3)制备的胺封端聚酯铵盐溶液中进行反应,同时加入有机溶剂调节溶液浓度,得到聚酰亚胺前驱体溶液。
10.进一步地,所述步骤1)中,所述环状二酸酐为均苯四甲酸二酐、3,3’,4,4
’-
联苯四甲酸二酐、2,3,3’,4’,-联苯四甲酸二酐、3,3’,4,4
’-
二苯醚四甲酸二酐、2,3,3’,4’,-二苯醚四甲酸二酐、3,3’,4,4
’-
二苯甲酮四甲酸二酐、4,4-六氟异丙基邻苯二甲酸酐、4,4
’-
(4,4
’-
异丙基二苯氧基)双(邻苯二甲酸酐)、1,2,4,5-环己烷四甲酸二酐中的任意一种或任意几种以任意比例混合的混合物;
11.所述环状二酸酐与乙二醇单甲醚的摩尔比为1:(3~10),优选为1:6,回流反应时间为1~5h。
12.进一步地,所述步骤2)中,所述单酸酐封端剂包括降冰片烯酸酐、5-烯丙基降冰片烯酸酐、苯并降冰片烯酸酐、马来酸酐、4-苯乙炔基苯酐中的任意一种;
13.所述单酸酐封端剂与乙二醇单甲醚的摩尔比为1:(2~20),回流反应时间为3-6h。
14.进一步地,所述步骤3)中,所述芳香族二胺为间苯二胺、对苯二胺、4,4
’-
二氨基二苯甲烷、4,4-二氨基联苯、3,4-二氨基二苯醚、4,4-二氨基二苯醚、2,2
’-
二甲基-4,4
’-
二氨基联苯、3,3
’-
二甲基-4,4-二氨基联苯、4,4
’-
二氨基二苯砜、3,3
’-
二氨基二苯砜、4,4
’-
二氨基-二苯甲酮、3,3
’-
二氨基-二苯甲酮、2,2
’-
双(三氟甲基)-4,4
’-
二氨基联苯、4,4
’-
二氨基苯酰替苯胺、2,2
’-
双(4-氨基苯基)六氟丙烷、1,3-双(4-氨基苯氧基)苯、1,3-双(3-氨基苯氧基)苯、1,4-双(2-三氟甲基4-氨基苯氧基)苯、2,2-双[4-(4-氨基苯氧基)苯基]丙烷、2,2-双[4-(4-氨基苯氧基)苯基]六氟丙烷中的任意一种或任意几种以任意比例混合的混合物;
[0015]
所述反应温度为20~80℃;反应时间为体系澄清后0.5~3h。
[0016]
进一步地,所述步骤4)中,所述有机溶剂为甲醇、乙醇、正丙醇、异丙醇、正丁醇、乙二醇单甲醚、四氢呋喃、二氧六环、乙酸乙酯、丙酮、二氯甲烷、三氯甲烷、二氯乙烷、n,n二甲基甲酰胺、n,n-二甲基乙酰胺、n-甲基吡咯烷酮、环己酮、环戊酮、间甲酚中任意一种或任意几种以任意比例混合的混合物;
[0017]
所述有机溶剂的含量为使得所制备的聚酰亚胺前驱体溶液的质量分数为20%~70%;
[0018]
所述反应温度为20℃~70℃,反应时间为体系澄清后5~12h。
[0019]
进一步地,所述环状二酸酐、芳香族二胺、单酸酐封端剂的摩尔比为n:(n+1):2,其中n等于1~10。
[0020]
根据第二方面,提供一种采用上述制备方法得到的低粘度聚酰亚胺前驱体溶液。
[0021]
根据第三方面,提供一种基于上述低粘度聚酰亚胺前驱体溶液制备得到的纤维增强聚酰亚胺树脂基复合材料。
[0022]
根据第四方面,提供上述纤维增强聚酰亚胺树脂基复合材料的制备方法,包括:
[0023]
将聚酰亚胺前驱体溶液涂覆到纤维布上,加热得到预浸布;
[0024]
将预浸布多层叠放,进行交联反应即得。
[0025]
根据第五方面,提供上述纤维增强聚酰亚胺树脂基复合材料在制造耐高温印制线路基板材料、耐高温天线罩透波材料和耐高温承力结构材料中的应用。
[0026]
与现有技术相比,本发明的有益效果是:
[0027]
本发明利用含醚键醇酯化四酸二酐,不仅可以将刚性对称四酸二酐(如pmda和sbpda)成功酯化为澄清溶液,而且所制备的聚酰亚胺前躯体溶液相比于乙醇和甲醇法具有更低的溶液粘度,有利于制备高固含量低溶液粘度聚酰亚胺前躯体溶液。
[0028]
采用本发明方法制备聚酰亚胺前驱体溶液具有优异的溶解性和低溶液粘度,可以用于浸渍纤维制备预浸料,进而制备聚酰亚胺树脂基复合材料,能够用作耐高温印制线路基板材料、耐高温天线罩透波材料、耐高温承力结构材料。在航空、航天、微电子封装等高技术领域有重要应用价值。
附图说明
[0029]
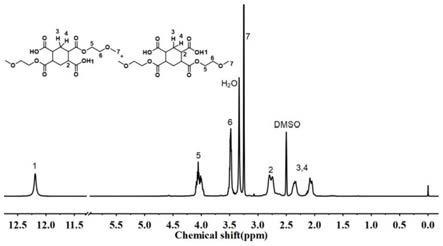
图1为实施例1中所得chde的核磁谱图;
[0030]
图2为实施例1中所得所得ne的核磁谱图;
[0031]
图3为实施例2中所得sbpde的核磁谱图。
具体实施方式
[0032]
下面对本发明的具体实施例进行详细说明。在下面的描述中,出于解释而非限制性的目的,阐述了具体细节,以帮助全面地理解本发明。然而,对本领域技术人员来说显而易见的是,也可以在脱离了这些具体细节的其它实施例中实践本发明。在此需要说明的是,为了避免因不必要的细节而模糊了本发明,仅仅示出了与根据本发明的方案密切相关的设备结构和/或处理步骤,而省略了与本发明关系不大的其他细节。所述方法如无特殊说明均为常规方法。所述原材料如无特殊说明均能从公开商业途径获得。本发明中固含量如无特别说明均为质量百分含量。
[0033]
实施例1
[0034]
1)在配有磁力搅拌、温度计和氮气出入口的三口瓶中,加入22.4172克(0.1000摩尔)1,2,4,5-环己烷四甲酸二酐和47.00克(0.6177摩尔)乙二醇单甲醚,加热至乙二醇单甲醚回流,反应一段时间得到无色澄清溶液,继续回流进行开环酯化反应1小时后停止加热,得到1,2,4,5-环己烷二酸二酯溶液(chde)。图1为所得chde的核磁谱图。
[0035]
2)在配有磁力搅拌的单口烧瓶中,加入6.3366克(0.0386摩尔)降冰片烯二酸酐和16.52克(0.2171摩尔)乙二醇单甲醚,加热回流,待反应液澄清后继续回流进行开环酯化反应2小时,停止加热,得到降冰片烯单酯单酸溶液(ne)。图2为所得ne的核磁谱图。
[0036]
3)将23.8981克(0.1193摩尔)3,4-二氨基二苯醚加入步骤1)所得1,2,4,5-环己烷二酸二酯溶液中,室温下搅拌反应,待溶液澄清后再继续搅拌进行羧基氨基的成盐反应1小时,得到胺基封端的聚酯胺盐溶液;
[0037]
4)将步骤2)所得降冰片烯单酯单酸溶液和25.44克乙醇加入到步骤3)所得胺基封端的聚酯胺盐溶液中,室温下搅拌进行羧基氨基的成盐反应8小时,得到固含量为50%的聚酰亚胺前驱体溶液。所得溶液的粘度列于表1中。
[0038]
将聚酰亚胺前驱体溶液通过适当方式涂敷在石英布上,经60℃/12h加热处理后,得到具有适当粘性的预浸布。将12层预浸布叠放在一起,然后放入模具中,在高温压机上经过310℃/2小时的热压固化得到复合材料构件,复合材料中石英纤维的体积分数控制在(50
±
2)%。该复合材料的性能列于表1中。
[0039]
实施例2
[0040]
1)在配有磁力搅拌、温度计和氮气出入口的三口瓶中,加入29.4220克(0.1000摩尔)3,3’,4,4
’-
联苯四甲酸二酐和55.00克(0.7228摩尔)乙二醇单甲醚,加热至乙二醇单甲醚回流,反应一段时间得到无色澄清溶液,继续回流进行开环酯化反应1小时后停止加热,得到3,3’,4,4
’-
联苯二酸二酯溶液(sbpde)。图3为sbpde的核磁谱图。
[0041]
2)在配有磁力搅拌的单口烧瓶中,加入7.6498克(0.0466摩尔)降冰片烯二酸酐和16.52克(0.2171摩尔)乙二醇单甲醚,加热回流,待反应液澄清后继续回流进行开环酯化反应2小时,停止加热,得到降冰片烯单酯单酸溶液(ne)。
[0042]
3)将24.6896克(0.1233摩尔)3,4-二氨基二苯醚加入步骤1)所得3,3’,4,4
’-
联苯二酸二酯溶液中,室温下搅拌反应,待溶液澄清后再继续搅拌进行羧基氨基的成盐反应1小时,得到胺基封端的聚酯胺盐溶液;
[0043]
4)将步骤2)所得降冰片烯单酯单酸溶液和25.77克乙醇加入到步骤3)所得胺基封端的聚酯胺盐溶液中,室温下搅拌进行羧基氨基的成盐反应8小时,得到固含量为50%的聚酰亚胺前驱体溶液。所得溶液的粘度列于表1中。
[0044]
将聚酰亚胺前驱体溶液通过适当方式涂敷在石英布上,经60℃/12h加热处理后,得到具有适当粘性的预浸布。将12层预浸布叠放在一起,然后放入模具中,在高温压机上经过310℃/2小时的热压固化得到复合材料构件,复合材料中石英纤维的体积分数控制在(50
±
2)%。该复合材料的性能列于表1中。
[0045]
对比例1
[0046]
1)在配有磁力搅拌、温度计和氮气出入口的三口瓶中,加入22.4172克(0.1000摩尔)1,2,4,5-环己烷四甲酸二酐和35.00克(0.7597摩尔)乙醇,加热至乙醇回流,在回流条件下进行开环酯化反应4小时后停止加热,得到1,2,4,5-环己烷二酸二酯溶液(chde),由于1,2,4,5-环己烷二酸二酯溶解性差,该溶液为浑浊液。
[0047]
2)在配有磁力搅拌的单口烧瓶中,加入6.3366克(0.0386摩尔)降冰片烯二酸酐和15.00克(0.3256摩尔)乙醇,加热回流,待反应液澄清后继续回流进行开环酯化反应2小时,停止加热,得到降冰片烯单酯单酸溶液(ne)。
[0048]
3)将23.8981克(0.1193摩尔)3,4-二氨基二苯醚加入步骤1)所得1,2,4,5-环己烷二酸二酯溶液中,室温下搅拌反应1小时,得到胺基封端的聚酯胺盐溶液,由于1,2,4,5-环己烷二酸二酯溶解性差,该溶液为浑浊液;
[0049]
4)将步骤2)所得降冰片烯单酯单酸溶液和24.64克乙醇加入到步骤3)所得胺基封端的聚酯胺盐溶液中,室温下搅拌进行羧基氨基的成盐反应8小时,得到固含量为50%的聚酰亚胺前驱体溶液,该溶液为浑浊液,所得溶液的粘度列于表1中。
[0050]
对比例2
[0051]
1)在配有磁力搅拌、温度计和氮气出入口的三口瓶中,加入29.4220克(0.1000摩尔)3,3’,4,4
’-
联苯四甲酸二酐和50.00克(1.0853摩尔)乙醇,加热至乙醇回流,在回流条件下进行开环酯化反应4小时后停止加热,得到3,3’,4,4
’-
联苯二酸二酯溶液(sbpde),由于3,3’,4,4
’-
联苯二酸二酯溶解性差,该溶液为浑浊液。
[0052]
2)在配有磁力搅拌的单口烧瓶中,加入7.6498克(0.0466摩尔)降冰片烯二酸酐和10.00克(0.2171摩尔)乙醇,加热回流,待反应液澄清后继续回流进行开环酯化反应2小时,停止加热,得到降冰片烯单酯单酸溶液(ne)。
[0053]
3)将24.6896克(0.1233摩尔)3,4-二氨基二苯醚加入步骤1)所得3,3’,4,4
’-
联苯二酸二酯溶液中,室温下搅拌反应1小时得到胺基封端的聚酯胺盐溶液,由于3,3’,4,4
’-
联苯二酸二酯溶解性差,该溶液为浑浊液;
[0054]
4)将步骤2)所得降冰片烯单酯单酸溶液和24.49克n,n-二甲基甲酰胺(dmf)加入到步骤3)所得胺基封端的聚酯胺盐溶液中,室温下搅拌进行羧基氨基的成盐反应8小时,得到固含量为50%的聚酰亚胺前驱体溶液,由于dmf的促溶作用,该前躯体为澄清均一溶液。所得溶液的粘度列于表1中。
[0055]
将聚酰亚胺前驱体溶液通过适当方式涂敷在石英布上,经60℃/12h加热处理后,得到具有适当粘性的预浸布。将12层预浸布叠放在一起,然后放入模具中,在高温压机上经过310℃/2小时的热压固化得到复合材料构件,复合材料中石英纤维的体积分数控制在(50
±
2)%。该复合材料的性能列于表1中。
[0056]
表1:聚酰亚胺前躯体溶液粘度及相应复合材料性能
[0057][0058]
其中,η:固体含量50%树脂溶液粘度;tg:玻璃化转变温度(损耗角正切曲线峰值对应温度,dma测定);e:溶液中有固体颗粒,粘度无限大;
--
:树脂溶液浑浊,无法浸渍纤维制备复合材料。弯曲强度和弯曲模量的测试标准为gb/t 3356-2014,剪切强度的测试标准为cb/t 30969-2014。
[0059]
如上针对一种实施例描述和/或示出的特征可以以相同或类似的方式在一个或更多个其它实施例中使用,和/或与其它实施例中的特征相结合或替代其它实施例中的特征使用。
[0060]
应该强调,术语“包括/包含”在本文使用时指特征、整件、步骤或组件的存在,但并不排除一个或更多个其它特征、整件、步骤、组件或其组合的存在或附加。
[0061]
这些实施例的许多特征和优点根据该详细描述是清楚的,因此所附权利要求旨在覆盖这些实施例的落入其真实精神和范围内的所有这些特征和优点。此外,由于本领域的技术人员容易想到很多修改和改变,因此不是要将本发明的实施例限于所例示和描述的精确结构和操作,而是可以涵盖落入其范围内的所有合适修改和等同物。
[0062]
本发明未详细说明部分为本领域技术人员公知技术。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1