一种分子量可控的多糖降解方法及低分子量多糖与流程
本发明涉及多糖降解技术领域,尤其涉及一种分子量可控的多糖降解方法及低分子量多糖。
背景技术:
天然多糖(polysaccharide)具有多种生物功能(抗肿瘤、抗氧化、降血糖、抗辐射等),且绿色安全无毒副作用,已成为生物医药的研究热点。研究表明,天然多糖的生物应答作用是由其化学结构决定的,多糖的分子量可以显著影响生物活性,分子量过大或者过小的多糖往往不具备生物活性。多糖分子量越大,体积越大,粘度越高,不利于跨越细胞膜进入体内或结合生物活性位点,发挥生物学活性,生物利用率较低。例如,针对绿藻多糖的构效关系研究表明,分子量的差异是其免疫调控活性的关键因素;对紫菜多糖的降解发现,低分子量的多糖具有较高的抑制癌细胞生长能力;真菌多糖因其溶解性差导致抗癌活性低下,通过降低分子量后提高了其水溶性,抗癌活性显著提高;紫球藻多糖经微波降解后发现,可以通过改变分子量达到改善高分子量紫球藻多糖无抗氧化活性的现象;冬虫夏草多糖经超声波降解后,分子量和特性黏度均降低,但未改变冬虫夏草多糖的单糖组成和糖苷键类型,对其生物活性研究表明降解后的多糖具有更加优良的抗肿瘤活性。分子量过低时又会影响其高级结构的形成,改变其生物活性,例如,分子量较大(90kda)的香菇多糖形成三螺旋构象比低分子量(小于50kda)单一柔性链具有更高的体内抗肿瘤活性。研究报道,分子量在10-1000kda范围内的多糖具有一定的生物活性,而相对分子质量小于5kda多糖会丧失生物活性。所以选用合适的方法可控的降低多糖分子量,保证多糖的分子量在生物活性的范围之内,又可以减小粘度,增加与受体的结合作用,提高多糖生物活性是提高天然多糖生物利用的可行方法之一。
技术实现要素:
针对上述存在的问题,本发明旨在提供一种分子量可控的多糖降解方法及低分子量多糖,采用抗坏血酸和fenton试剂的混合溶液作为降解液,辅助超声波对多糖溶液进行降解,通过控制降解液的浓度、超声波的功率和降解时间,获得不同分子量的多糖。
为了实现上述目的,本发明所采用的技术方案如下:
一种分子量可控的多糖降解方法,其特征在于,包括以下步骤,利用超声波+降解液对多糖溶液进行处理,获得不同分子量的多糖。
进一步的,所述降解液包括抗坏血酸和fetons试剂。
进一步的,利用超声波+降解液对多糖溶液进行处理的具体操作包括以下步骤,
s1:配制抗坏血酸和fetons试剂的混合溶液,作为降解液;
s2:在多糖溶液中加入降解液,同时利用超声波对其进行超声波降解;
s3:将降解后的多糖溶液用0.01mol/l的naoh中和至中性;
s4:取步骤s3中得到的中性多糖溶液的上清液,进行透析、醇沉,收集沉淀,得到低分子量多糖。
进一步的,所述fetons试剂由硫酸亚铁与h2o2混合而成,且fe2+与h2o2的浓度比为1:3。
进一步的,所述降解液中fetons试剂的浓度为0~10%,抗坏血酸的浓度为0~10mm。
进一步的,所述抗坏血酸、硫酸亚铁与h2o2均为分析纯。
进一步的,所述超声波的功率为0~600w,超声降解时间为0~100min。
进一步的,采用一种分子量可控的多糖降解方法降解得到的低分子量多糖。
本发明的有益效果是:
本发明中采用抗坏血酸和fenton试剂的混合溶液作为降解液,辅助超声波对多糖溶液进行降解,通过控制降解液的浓度、超声波的功率和降解时间,可以获得分子量差异显著的的多糖,同时还能够提高降解效率。
附图说明
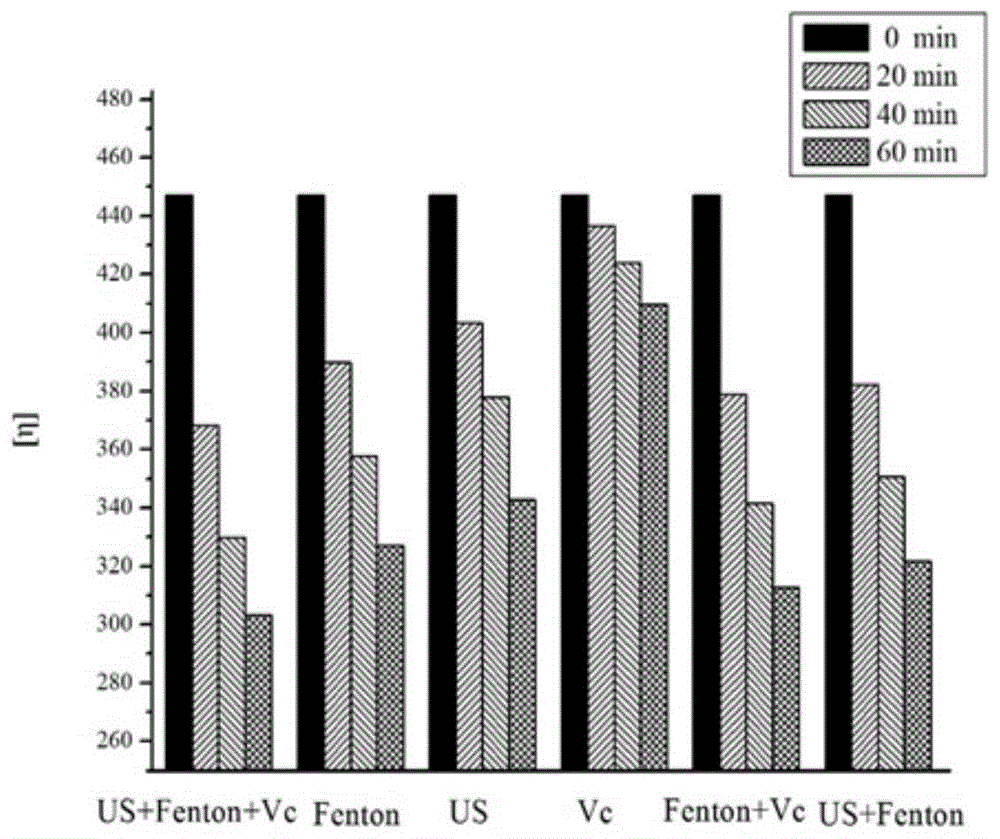
图1为本发明实施例一中不同降解方法对蕨麻多糖降解效果的影响;
图2为本发明实施例二中不同降解时间时蕨麻多糖溶液特性粘度随时间的变化规律;
图3为本发明实施例二中不同超声波功率时蕨麻多糖溶液特性粘度随时间的变化规律;
图4为本发明实施例二中不同抗坏血酸浓度时蕨麻多糖溶液特性粘度随时间的变化规律;
图5为本发明实施例二中不同fentons溶液浓度时蕨麻多糖溶液特性粘度随时间的变化规律;
图6为本发明实施例二中在旋转模式下,不同分子量蕨麻多糖溶液的表观粘度随剪切速率的变化规律;
图7为本发明实施例二中不同分子量蕨麻多糖溶液的触变环面积变化规律;
图8为本发明实施例二中不同分子量蕨麻多糖溶液的储能模量和损耗模量随角频率的变化规律。
具体实施方式
为了使本领域的普通技术人员能更好的理解本发明的技术方案,下面结合附图和实施例对本发明的技术方案做进一步的描述。
实施例一:
利用本发明中的多糖降解方法降解蕨麻多糖。具体的:
s1:配制抗坏血酸和fetons试剂的混合溶液,作为降解液;
所述fetons试剂由硫酸亚铁与h2o2混合而成,且fe2+与h2o2的浓度比为1:3;
所述降解液中fetons试剂的浓度为5%,抗坏血酸的浓度为2.5mm;所述抗坏血酸、硫酸亚铁与h2o2均为分析纯。
s2:在多糖溶液中加入降解液,同时利用超声波对其进行超声波降解;
在浓度为5mg/ml的蕨麻多糖溶液中加入步骤s1中配制好的降解液,同时利用超声波对蕨麻多糖溶液进行超声降解;所述超声波的功率为400w,超声降解时间为60min。
s3:将降解后的多糖溶液用0.01mol/l的naoh中和至中性;
s4:取步骤s3中得到的中性多糖溶液的上清液,进行逐级透析、醇沉,收集沉淀,得到低分子量多糖。
为了验证本发明中fenton试剂+抗坏血酸+超声降解方法对多糖溶液降解的效率,本实施例中还进行了对比实验,具体为:只使用fenton试剂、只使用超声、只使用抗坏血酸、使用fenton试剂+抗坏血酸、使用超声+fenton试剂对浓度为5mg/ml的蕨麻水提粗多糖溶液进行降解,在降解时间为0、20min、40min和60min时蕨麻多糖溶液的特性粘度情况如附图1所示。
其中,特性黏度是高分子物质的一个物理量,与溶液浓度无关,与高分子的分子量的大小有关,重均分子量越大,特性黏度越大;蕨麻多糖(pap)溶液的特性粘度采用乌氏粘度计(内径0.5-0.6mm)测定,测定温度控制在25±0.1℃,每次测定重复3次,3次测量结果的误差值不超过0.2s。蒸馏水为空白测定,测定不同浓度的多糖粘度时,用蒸馏水反复冲洗后再用待测液润洗测定结果按公式
其中,[η]为pap的特性粘度,ηsp为增比粘度(ηsp=ηr-1),ηr为相对粘度(ηr=t/t0,t为pap溶液流出的时间,t0为蒸馏水流出的时间,c为pap溶液的浓度(g/dl))。
从附图1中可以看出,抗坏血酸辅助超声波-fenton试剂联用的方法在20min内pap的特性粘度从447.1cm3/g下降到368.2cm3/g。这种下降量需要fenton试剂单独作用40min左右,超声波单独使用需要的时间更久。与此形成鲜明对比的是,单独使用抗坏血酸,特性粘度虽有下降,但是下降程度远远不如抗坏血酸辅助超声波-fenton试剂联用法。将抗坏血酸与fenton试剂联用,发现抗坏血酸有助于fenton降解pap多糖。不同方法比较分析发现,三者联用降解效率最高,说明抗坏血酸与fenton试剂可以有效的形成一个降解高分子聚合物的系统,超声波可以提高这种降解效率,抗坏血酸辅助超声波-fenton试剂联用法是一种最高效的降解方法。
实施例二:
为了验证本发明中多糖降解方法在改变降解工艺参数时,确实能够得到不同分子量的多糖,本实施例采用不同的工艺参数进行对比实验,具体为:
1)、降解时间对多糖降解的影响;
在浓度为5mg/ml的蕨麻多糖溶液中加入抗坏血酸和fetons试剂配制而成的降解液,同时利用超声波对蕨麻多糖溶液进行超声波降解;
具体的,所述fetons试剂由硫酸亚铁与h2o2混合而成,且fe2+与h2o2的浓度比为1:3;
所述降解液中fetons试剂的浓度为5%,抗坏血酸的浓度为2.5mm;所述抗坏血酸、硫酸亚铁与h2o2均为分析纯。
所述超声波的功率为600w。
在降解时间为0~120min时,pap特性粘度(测定方法比实施例一中测定方法相同)变化情况如附图2所示。
从附图2中可以很明显的看出,降解开始后pap特性粘度发生显著性降低,说明多糖的分子量变低,这是由于抗坏血酸-feton反应中产生了大量的自由基,超声波产生的空穴效应使pap迅速发生降解。随着降解时间的延长,pap特性粘度逐渐趋于平缓,40min后pap特性粘度变化不大,这可能是由于抗坏血酸-fenton反应基本完成。从附图2中还可以看出,为了保证抗坏血酸-fenton反应完全,故利用本发明中的多糖降解方法对蕨麻多糖进行降解时,60min为最佳降解时间。
2)、超声波功率对多糖降解的影响
在浓度为5mg/ml的蕨麻多糖溶液中加入抗坏血酸和fetons试剂配制而成的降解液,同时利用超声波对蕨麻多糖溶液进行超声波降解;
具体的,所述fetons试剂由硫酸亚铁与h2o2混合而成,且fe2+与h2o2的浓度比为1:3;
所述降解液中fetons试剂的浓度为5%,抗坏血酸的浓度为2.5mm;所述抗坏血酸、硫酸亚铁与h2o2均为分析纯;降解时间为60min;
在超声波功率为0、100w、200w、300w、400w、500w和600w情况下,pap特性粘度(测定方法比实施例一种测定方法相同)变化情况如附图3所示。
从附图3中可以明显的看出,加入超声波后可以显著提高pap的降解程度,表明超声波可以影响自由基和空化气泡的形成,从而提高高分子的降解效率。在超声波辅助的情况之下,pap特性粘度随超声功率的增加而下降,但超声功率一旦超过400w后,超声对pap特性粘度的影响不显著(p>0.05),这可能由于在降解的初始阶段,pap分子量相对较大,抗坏血酸辅助超声波-fenton试剂联用降解速率相对较快,当pap分子量降低到一定程度后变化趋向平缓。考虑能量节约原则,故采用本发明中多糖降解方法降解蕨麻多糖时最佳超声波功率为400w。
3)、抗坏血酸浓度对多糖降解的影响
在浓度为5mg/ml的蕨麻多糖溶液中加入抗坏血酸和fetons试剂配制而成的降解液,同时利用超声波对蕨麻多糖溶液进行超声波降解;
具体的,所述fetons试剂由硫酸亚铁与h2o2混合而成,且fe2+与h2o2的浓度比为1:3;
所述降解液中fetons试剂的浓度为5%;所述超声波功率为400w,降解时间为60min;
在抗坏血酸浓度为0、2.5mm、5mm、7.5mm和10mm情况下,pap特性粘度(测定方法比实施例一种测定方法相同)变化情况如附图4所示。
从图4中可以看出,加入抗坏血酸在一定程度上可以促进pap的降解;在不同浓度的抗坏血酸的作用下,pap特性粘度随浓度的增加而降低,但从整体水平来看,抗坏血酸对pap降解影响不十分显著,只呈现轻微下降趋,降解60min不同浓度的抗坏血酸对降解程度无显著差异(p>0.05)。
4)、fetons试剂浓度对多糖降解的影响
在浓度为5mg/ml的蕨麻多糖溶液中加入抗坏血酸和fetons试剂配制而成的降解液,同时利用超声波对蕨麻多糖溶液进行超声波降解;
具体的,所述fetons试剂由硫酸亚铁与h2o2混合而成,且fe2+与h2o2的浓度比为1:3;
所述降解液中抗坏血酸浓度为2.5mm;所述超声波功率为400w,降解时间为60min;
在fetons试剂浓度为0、2.5%、5%、7.5%和10%情况下,pap特性粘度(测定方法比实施例一种测定方法相同)变化情况如附图5所示。
从附图5中可以明显的看出,fenton试剂浓度对pap的降解程度具有显著影响:随fenton试剂浓度的增加,pap特性粘度降呈现显著下降趋势。这一结果与单独使用三种降解方法对降解效果的研究结果一致,表明fenton试剂对整体降解体系的影响最为显著,故可以通过改变fenton试剂浓度控制pap的降解程度。
在降解超声功率为400w、抗坏血酸浓度2.5mm的条件下,使用不同浓度的fenton试剂(2.5%、5%、10%)降解60min,降解结束后使用0.01mol/l的naoh将降解液中和至中性;过滤除去沉淀,取上清液;用36kd透析袋自来水流动透析3d,去离子水流动透析1d,减压浓缩,醇沉冻干后得到的多糖分别命名为papⅰ、papⅱ和papⅲ。
经sec-lls测试分析降解前蕨麻粗多糖(pap)和3种多糖降解产物(papⅰ、papⅱ和papⅲ)的分子量及其分布,结果如表1所示。
表1不同蕨麻多糖分子量及其分布情况
从表1中可以看出,随着降解程度的增加,多糖的分子量逐渐减小,分散系数逐渐增加。蕨麻多糖的分子量从未降解(pap)的3.108×106降低至8.143×104,表明本发明中超声波+降解液(抗坏血酸+fenton试剂)是一种有效的可以控制降解后多糖分子量的多糖降解方法。
为了进一步的验证本发明中多糖降解方法对蕨麻多糖的降解是否成功,本实施例中还进行了流变学研究,具体的,
分别将蕨麻多糖pap及其降解产物papⅰ、papⅱ和papⅲ用超纯水配成将50mg/ml的多糖溶液,80℃恒温130rad/min磁力搅拌4h后静置过夜。
采用mcr301型流变仪(antonpaar,gmbh,austria)对不同分子量的蕨麻多糖溶液进行稳态剪切测试。流变探头为平板pp50系统(内径=50mm),板间距离1mm,测试温度25℃。上样后,样品静止5min后开始测试,剪切速率扫描范围:0.01-100/s(以1.33倍的速率增加)。
在旋转模式下,考察不同分子量paps溶液的表观粘度随剪切速率的变化趋势,结果如附图6所示。
从附图6中可以看出,开始时多糖表观黏度随paps分子量的降低而降低(pap>papⅰ>papⅱ>papⅲ);随着剪切速率的升高,pap、papⅰ、papⅱ和papⅲ的表观黏度显著下降,出现剪切稀化现象,并且在相同剪切速率下,pap溶液的表观粘度值高于其他多糖溶液。有研究表明:多糖分子在较低的剪切速率下,糖链的解缠结和再缠结达成动态平衡,分子链网络密度没有太大改变;随着剪切速度不断提升,过大的剪切力就会破坏这种分子链网络结构,使得多糖分子在流动方向上的排列更加整齐,导致相邻分子链之间相互作用的减少,导致表观黏度下降。这种剪切稀化现象,pap和papⅰ表现的尤为明显,体现出较强的非牛顿流体的假塑性特征,说明此时pap和papⅰ多糖分子中含有较多的长的分子链。但是随着分子量的降低,这种特性显著减弱,这是由于分子量变小导致分子链之间相互作用变小,糖链可以缠绕的结构变少,导致糖链缠结缺乏牢固,所以流动曲线比较平缓。
将上述溶液流动性曲线经power-law模型τ=kγn拟合;
上述拟合公式用对数形式表示为lgτ=lgk+nlgγ,
式中,τ表示剪切应力(pa),k表示稠度系数(pa·sn),γ表示剪切速率(s-1),n表示流动指数,拟合结果如表2所示。
表2不同分子量蕨麻多糖的power-law模型拟合参数
流动指数(n),也称非牛顿指数,表明流体偏离牛顿流体的程度。当0<n<1时,流体的表观粘度随剪切速率的增大而降低,流体为假塑性流体;当n=1时,流体为牛顿流体;当n>1时,流体的表观粘度随剪切速率的增大而增大,此时流体为胀塑性流体。
从表2不同分子量paps的power-law模型拟合参数中可以看出,四种不同分子量的paps均是非牛顿流体,但是随分子量的减少流动指数逐渐增加,表明随分子量的减低paps流体性质由非牛顿流体趋向于牛顿流体。
进一步的,应用mcr301流变仪(antonpaar,gmbh,austria)经行触变性测试。流变探头为平板pp50系统(内径=50mm),板间距离1mm,测试温度25℃。旋转模式下设定三段参数:剪切速率分别为0.01-100/s,100/s,100-0.01/s。剪切速率从0.01/s以3.12/s的梯度增大到100/s,静止50s后,以3.12/s的梯度减小到0.01/s,结果如附图7所示。
对于非牛顿流体而言,触变性是一个重要的特征指标,可以反映在剪切速率变化的条件下,流体对于剪切应力的响应性。响应性大小由触变环表示,触变环面积越大,触变性越强,多糖分子结构被破坏所需要的能量也就越大。
从附图7中可以看出,四种paps均有一定的触变性。经计算,pap的触环面积为101.99pa/s,papⅰ的触环面积为59.91pa/s,papⅱ的触环面积为14.29pa/s,papⅲ的触环面积为8.95pa/s,由此结果看出,paps的分子量越高,触变性越强,说明适当降低多糖大分子的分子量可以提高体系的剪切稳定性。
进一步的,应用mcr301流变仪(antonpaar,gmbh,austria)经行动态剪切扫描。流变探头为同心圆筒cc27系统(内径=27mm),测试温度25℃。角频率变化范围:0.05-100rad/s,应变:0.1%(线性粘弹性范围最大0.5%),不同分子量paps溶液的储能模量(g′)和损耗模量(g″)随频率的变化规律结果如附图8所示。
从附图8中可以看出,不同分子量的paps溶液的g′和g″随角频率的增加而增大;而且在低频率剪切范围内g”≥g',表现液体粘性行为,体系的流动性强;在高频率剪切范围内g'≥g”,表现液体弹性行为,体系的流动性弱;g'=g”时,即为凝胶点。进一步观察可以看出,蕨麻多糖溶液体系的g'和g”的变化与多糖体系的分子量有关,分子量越大g'和g”的数值也越大,这可能是因为随着分子量的增大,分子链的增长和分子间作用力增多,溶液中分子间的缠结程度增巨,加剧糖链溶液分子的网络结构,增强了粘弹性。
实施例三:
利用本发明中的多糖降解方法降解黄参多糖。具体的:
s1:配制抗坏血酸和fetons试剂的混合溶液,作为降解液;
所述fetons试剂由硫酸亚铁与h2o2混合而成,且fe2+与h2o2的浓度比为1:3;
所述降解液中fetons试剂的浓度为5%,抗坏血酸的浓度为2.5mm;所述抗坏血酸、硫酸亚铁与h2o2均为分析纯。
s2:在多糖溶液中加入降解液,同时利用超声波对其进行超声波降解;
在浓度为5mg/ml的黄参多糖溶液中加入步骤s1中配制好的降解液,同时利用超声波对黄参多糖溶液进行超声降解;所述超声波的功率为400w,超声降解时间为60min。
s3:将降解后的黄参多糖溶液用0.01mol/l的naoh中和至中性;
s4:取步骤s3中得到的中性多糖溶液的上清液,进行逐级透析、醇沉,收集沉淀,得到低分子量多糖。
降解前后黄参多糖的分子量及其分布情况如下表3所示。
表3黄参多糖分子量及其分布情况
从表3中可以明显的看出,本发明中的多糖降解方法可以显著降低黄参多糖的分子量,表明本发明中的多糖降解方法是一种有效的多糖物质降解方法。
实施例四:
利用本发明中的多糖降解方法降解锁阳多糖。具体的:
s1:配制抗坏血酸和fetons试剂的混合溶液,作为降解液;
所述fetons试剂由硫酸亚铁与h2o2混合而成,且fe2+与h2o2的浓度比为1:3;
所述降解液中fetons试剂的浓度为2.5%,抗坏血酸的浓度为5mm;所述抗坏血酸、硫酸亚铁与h2o2均为分析纯。
s2:在多糖溶液中加入降解液,同时利用超声波对其进行超声波降解;
在浓度为5mg/ml的锁阳多糖溶液中加入步骤s1中配制好的降解液,同时利用超声波对锁阳多糖溶液进行超声降解;所述超声波的功率为600w,超声降解时间为60min。
s3:将降解后的锁阳多糖溶液用0.01mol/l的naoh中和至中性;
s4:取步骤s3中得到的中性多糖溶液的上清液,进行逐级透析、醇沉,收集沉淀,得到低分子量多糖。
降解前后锁阳多糖的分子量及其分布情况如下表4所示。
表4锁阳多糖分子量及其分布情况
从表4中可以明显的看出,本发明中的多糖降解方法可以显著降低锁阳多糖的分子量,表明本发明中的多糖降解方法是一种有效的多糖物质降解方法。
以上显示和描述了本发明的基本原理、主要特征和本发明的优点。本行业的技术人员应该了解,本发明不受上述实施例的限制,上述实施例和说明书中描述的只是说明本发明的原理,在不脱离本发明精神和范围的前提下,本发明还会有各种变化和改进,这些变化和改进都落入要求保护的本发明范围内。本发明要求保护范围由所附的权利要求书及其等效物界定。
- 还没有人留言评论。精彩留言会获得点赞!