香豆素-氰基吡啶衍生物在比率检测二氧化硫中的应用的制作方法
本发明涉及香豆素衍生物,具体属于一种香豆素-氰基吡啶衍生物在比率检测二氧化硫中的应用。
背景技术:
监测细胞环境中的细胞通讯和新陈代谢可以更好地研究生理和病理过程。因此,可视化细胞之间的相互作用对于维持生物体的健康具有重要意义。近年来,对活性硫(rss)的研究越来越多,气态信号分子二氧化硫(so2)衍生物成为研究的重点。so2在人类健康中起着非常重要的作用,许多生理活性都与之密切相关。内源性so2可以调节心肌细胞中钙离子的平衡,并在心肌保护中发挥作用。同时,内源性so2还具有调节血压,调节心脏功能,改善血管重塑,抗氧化剂和调节脂质代谢等生理作用。此外,高浓度的二氧化硫还会对心血管系统的功能产生破坏作用。研究表明,线粒体是产生内源性二氧化硫的主要场所。tst(硫代硫酸盐硫转移酶)催化gsh和na2s2o3(硫代硫酸盐)内源性产生so2,并与活细胞中的亚硫酸盐/亚硫酸氢盐(so32-/hso3-)平衡。线粒体产生的二氧化硫被传递到整个细胞,包括由数百种碳水化合物,脂质和蛋白质组成的细胞膜。细胞膜在细胞生理过程中起着重要作用,例如信号转导、胞吐作用、内吞作用和细胞凋亡。但是高空间分辨率和高时间跟踪细胞表面上的生物分子对于以前的监测方法仍然是一个挑战。二氧化硫是人体内重要的信号分子之一,需要通过细胞膜排出细胞来发挥相应的作用,并维持生物体的正常生理活动。到目前为止,有关于细胞膜上二氧化硫检测的相关报道少之又少,并且目前尚不清楚体内so2的信号通路。因此,研究人员迫切需要开发有效且方便的工具来动态监控内源性二氧化硫。
针对以上存在的问题,设计选择性好、灵敏度高、细胞膜与线粒体分别靶向、具有低细胞毒性的荧光探针用于检测活细胞和组织中内源性so2水平变化,已成为当前生物医学发展中具有挑战性的前沿课题之一。
在本发明中,合成了一种基于香豆素-氰基吡啶的化合物,通过探针与so2在反应前后双通道荧光变化,实现对细胞中线粒体和细胞膜的分别高效靶向,从而可视化研究了so2信号通路。
技术实现要素:
本发明的目的是提供一种香豆素-氰基吡啶衍生物在比率检测二氧化硫中的应用,且检测方法简单,操作方便,选择性好,灵敏度高。
本发明提供的一种香豆素-氰基吡啶衍生物在比率检测二氧化硫中的应用,所述的香豆素-氰基吡啶衍生物,中文名称为(z)-4-(1-氰基-2-(7-(二乙氨基)-2-氧代-2h-铬-3-基)乙烯基)-1-甲基吡啶-1-阳离子,英文名称为(z)-4-(1-cyano-2-(7-(diethylamino)-2-oxo-2h-chromen-3-yl)vinyl)-1-methylpyridin-1-ium,命名为cpa-i;或者中文名称为(z)-4-(1-氰基-2-(7-(二乙氨基)-2-氧代-2h-铬-3-基)乙烯基)-1-壬基吡啶-1-阳离子,英文名称(z)-4-(1-cyano-2-(7-(diethylamino)-2-oxo-2h-chromen-3-yl)vinyl)-1-nonylpyridin-1-ium,命名为cpa-ii;结构式为:
本发明提供的一种香豆素-氰基吡啶衍生物的合成方法,步骤为:
(1)将4-(二乙氨基)-2-羟基苯甲醛和丙二酸二乙酯在无水乙醇中混合,往混合物中缓缓加入哌啶,将混合物在80-85℃加热反应7-10小时后,减压除去溶剂,加入乙酸和等体积的浓盐酸,118-125℃反应7-10小时;冷却至室温后,将反应混合物倒入冰水中,其中反应混合物总体积与冰水的体积比为1:1-1.5;加入naoh溶液将ph提高至4-6,立即形成棕色沉淀;将混合物过滤,水洗,真空干燥,得到黄棕色固体即为7-二乙氨基香豆素,其中4-(二乙氨基)-2-羟基苯甲醛、丙二酸二乙酯、哌啶和乙酸的摩尔比为1:1.5-2.5:1-1.2:30-40;
(2)在0℃下,按体积比为1:1将三氯氧磷缓慢滴加到dmf中,搅拌30-50分钟,得到红色液体;将7-二乙氨基香豆素的dmf溶液倒入红色液体中,得到猩红色悬浮液,在60-70℃加热10-13小时;反应完成后将反应混合物倒入冰水中,其中反应混合物总体积与冰水的体积比为1:6-6.5;加入naoh溶液将ph提高至4-6,出现沉淀;将混合物过滤,水洗,真空干燥,然后在无水乙醇中重结晶,即得7-(二乙氨基)-2-氧代-2h-色烯-3-甲醛;其中7-二乙氨基香豆素和三氯氧磷的摩尔比为1:3-3.5;
(3)将7-(二乙氨基)-2-氧代-2h-色烯-3-甲醛和4-吡啶乙腈在无水乙醇中混合,在100-110℃反应,反应期间通过薄层色谱法(tlc)监测,待反应完成后,将反应产物冷却至室温,减压除去溶剂,所得残渣用乙醚与正己烷分别洗涤2-3次,将无水乙醇添加到获得的残余物中,在冰箱中放置过夜;之后过滤沉淀物,使用冷乙醇洗涤,并在真空下干燥,得到黑色固体即(z)-3-(7-(二乙氨基)-2-氧代-2h-铬-3-基)-2-(吡啶-4-基)丙烯腈;其中7-(二乙氨基)-2-氧代-2h-色烯-3-甲醛和4-吡啶乙腈的摩尔比为1:1.4-2;
(4)将(z)-3-(7-(二乙氨基)-2-氧代-2h-铬-3-基)-2-(吡啶-4-基)丙烯腈和碘甲烷溶解在乙腈中,在82-86℃搅拌6-8小时,然后冷却至室温,减压除去溶剂,以体积比25:1的二氯甲烷和甲醇作洗脱剂经硅胶柱色谱分离,纯化即得(z)-4-(1-氰基-2-(7-(二乙氨基)-2-氧代-2h-铬-3-基)乙烯基)-1-甲基吡啶-1-阳离子(cpa-i),其中(z)-3-(7-(二乙氨基)-2-氧代-2h-铬-3-基)-2-(吡啶-4-基)丙烯腈和碘甲烷的摩尔比为1:9-11;
或者,
将(z)-3-(7-(二乙氨基)-2-氧代-2h-铬-3-基)-2-(吡啶-4-基)丙烯腈和碘壬烷溶解在乙腈中,在82-86℃搅拌6-8小时,然后冷却至室温,减压除去溶剂,以体积比25:1的二氯甲烷和甲醇作洗脱剂经硅胶柱色谱分离,纯化即得(z)-4-(1-氰基-2-(7-(二乙氨基)-2-氧代-2h-铬-3-基)乙烯基)-1-壬基吡啶-1-阳离子(cpa-ii),其中(z)-3-(7-(二乙氨基)-2-氧代-2h-铬-3-基)-2-(吡啶-4-基)丙烯腈和碘壬烷的摩尔比为1:9-11。
作为优选:
所述的步骤(1)中4-(二乙氨基)-2-羟基苯甲醛、丙二酸二乙酯、哌啶和乙酸的摩尔比为1:2:1:35。
所述的步骤(2)中7-二乙氨基香豆素和三氯氧磷的摩尔比为1:3.2。
所述的步骤(3)中7-(二乙氨基)-2-氧代-2h-色烯-3-甲醛和4-吡啶乙腈的摩尔比为1:1.5。
所述的步骤(4)中(z)-3-(7-(二乙氨基)-2-氧代-2h-铬-3-基)-2-(吡啶-4-基)丙烯腈和碘甲烷或碘壬烷的摩尔比为1:10。
本发明合成的cpa-i与cpa-ii可用于比率快速检测so2,也可在检测细胞中二氧化硫中应用。
本发明提供的cpa-i比率检测二氧化硫的方法,步骤为:
(1)、配制ph=5、浓度为10mm的pbs缓冲溶液,用亚硫酸钠配制2mm的二氧化硫水溶液,将香豆素-氰基吡啶衍生物cpa-i溶于dmso配制成2mm的溶液;
(2)、取2ml的纯pbs缓冲溶液、10μlcpa-i的dmso溶液加到一个荧光比色皿中,在荧光分光光度仪上检测,随着待测样二氧化硫的加入,486nm处的荧光强度逐渐增强,644nm处的荧光强度逐渐降低;
(3)、用蒸馏水配制2mm的亚硫酸钠溶液,将纯pbs缓冲溶液加到2ml荧光比色皿中,逐渐加入二氧化硫溶液的体积为0、10、20、30、40、50、60、70、80、90、100μl,同时在荧光光谱仪上测定644nm处荧光强度为540、492.6、452.4、415.4、368.5、325.9、287.6、243.2、200.8、170.7、142.4,以二氧化硫浓度为横坐标,以荧光强度f为纵坐标绘制图,得到二氧化硫浓度的工作曲线;线性回归方程为:f644=-4.051c+533.39,c的单位为10-6mol/l。
本发明提供的cpa-ii比率检测二氧化硫的方法,步骤为:
(1)、配制ph=5、浓度为10mm的pbs缓冲溶液,用亚硫酸钠配制2mm的二氧化硫水溶液,将香豆素-氰基吡啶衍生物cpa-ii溶于dmso配制成2mm的溶液;
(2)、取2ml的ch3cn/pbs(v/v=3:7,ph=5)溶液、10μlcpa-ii的dmso溶液加到一个荧光比色皿中,在荧光分光光度仪上检测,随着待测样二氧化硫的加入,481nm处的荧光强度逐渐增强,648nm处的荧光强度逐渐降低;
(3)、用蒸馏水配制2mm的亚硫酸钠溶液,按体积比7:3将pbs缓冲溶液和ch3cn溶液加到2ml荧光比色皿中,逐渐加入二氧化硫溶液的体积为0、10、20、30、40、50、60、70、80、90、100、110、120、130、140、150μl,同时在荧光光谱仪上测定648nm处荧光强度为860.8、798.5、740.7、675.4、619.1、581.1、549、516.6、469.2、430.8、393.8、345.7、308.1、263.3、221.2、193.9,以二氧化硫浓度为横坐标,以荧光强度f为纵坐标绘制图,得到二氧化硫浓度的工作曲线;线性回归方程为:f648=-4.311c+821.308,c的单位为10-6mol/l。
与现有技术相比,本发明具有如下优点和效果:
1、本发明香豆素-氰基吡啶衍生物合成简单,成本低廉;
2、本发明香豆素-氰基吡啶衍生物不仅能分别实现细胞膜与线粒体高效靶向,而且可实现比率检测二氧化硫,检测结果灵敏度高,选择性好;
3、本发明检测手段简单,只需要借助荧光光谱仪即可实现;
4、本发明采用双通道检测,检测信号明显且专一性强。
附图说明
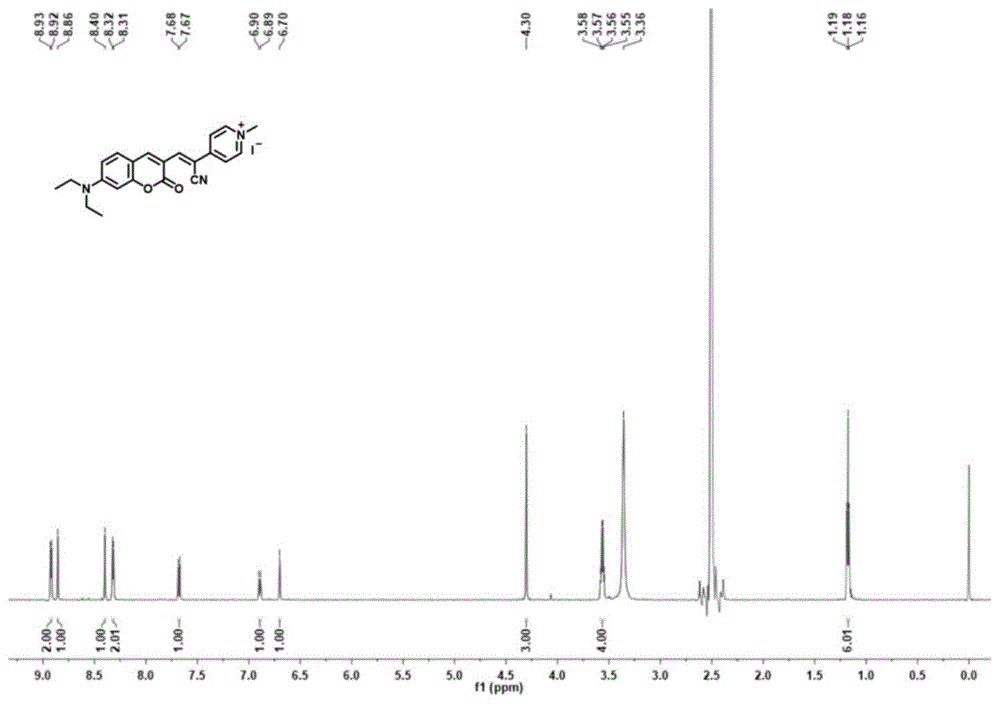
图1实施例1制备的香豆素-氰基吡啶衍生物cpa-i的核磁氢谱图
图2实施例1制备的香豆素-氰基吡啶衍生物cpa-i的核磁碳谱图
图3实施例1制备的香豆素-氰基吡啶衍生物cpa-i的质谱图
图4实施例1制备的香豆素-氰基吡啶衍生物cpa-ii的核磁氢谱图
图5实施例1制备的香豆素-氰基吡啶衍生物cpa-ii的核磁碳谱图
图6实施例1制备的香豆素-氰基吡啶衍生物cpa-ii的质谱图
图7香豆素-氰基吡啶衍生物cpa-i与二氧化硫作用的荧光发射图
图8香豆素-氰基吡啶衍生物cpa-i与各种分析物的荧光柱状图
图9香豆素-氰基吡啶衍生物cpa-i测定二氧化硫的工作曲线
图10香豆素-氰基吡啶衍生物cpa-i测定外源二氧化硫细胞成像图
图11香豆素-氰基吡啶衍生物cpa-i测定内源二氧化硫细胞成像图
图12香豆素-氰基吡啶衍生物cpa-ii与二氧化硫作用的荧光发射图
图13香豆素-氰基吡啶衍生物cpa-ii与各种分析物的荧光柱状图
图14香豆素-氰基吡啶衍生物cpa-ii测定二氧化硫的工作曲线
图15香豆素-氰基吡啶衍生物cpa-ii测定外源二氧化硫细胞成像图
图16香豆素-氰基吡啶衍生物cpa-ii测定内源二氧化硫细胞成像图
图17共定位细胞成像图
具体实施方式
下面结合实施例和附图对本发明做进一步说明,但本发明不受下述实施例的限制。
实施例1
cpa-i与cpa-ii的制备和表征
将丙二酸二乙酯(6.09g,38mmol),4-(二乙氨基)-2-羟基苯甲醛(3.67g,19mmol)和哌啶(1.88ml)与无水乙醇(60ml)混合,然后将混合物在85℃反应7小时。反应完成后,在减压下蒸发溶剂。接下来,加入乙酸(38ml)和浓hcl(38ml)的混合物,在120℃下反应8小时。接下来,冷却至25℃后,将反应混合物倒入100ml0℃的水中。加入naoh溶液(40%)将ph提高至5,立即形成棕色沉淀。将混合物过滤并用水洗涤4次,在真空中干燥,获得黄棕色固体即7-二乙氨基香豆素。(3.92g,产率:95%)。1hnmr(600mhz,dmso-d6)δ7.82(d,j=9.3hz,1h),7.42(d,j=8.8hz,1h),6.68(dd,j=8.8,2.5hz,1h),6.51(d,j=2.3hz,1h),5.99(d,j=9.3hz,1h),3.42(q,j=7.0hz,4h),1.12(t,j=7.0hz,6h).13cnmr(151mhz,dmso-d6)δ156.80,144.96,129.78,109.22,108.62,97.04,44.49,12.77.
在0℃下将三氯氧磷(6ml)缓慢加入到dmf(6ml)中,搅拌30分钟,得到红色液体。将7-二乙氨基香豆素(4.37g,20.12mmol)的dmf(35ml)混合溶液倒入红色液体中,得到猩红色悬浮液,将反应混合物在65℃加热13小时。接下来,将反应混合物加入300ml的0℃水中。加入naoh(40%)溶液以将ph提高至5,出现沉淀。过滤混合物,用水洗涤4次,干燥并在乙醇中重结晶,得到7-(二乙氨基)-2-氧代-2h-色烯-3-甲醛(3.0g,产率:58%)。1hnmr(600mhz,dmso-d6)δ9.89(s,1h),8.41(s,1h),7.68(d,j=9.0hz,1h),6.82(d,j=9.0hz,1h),6.60(s,1h),3.51(q,j=7.0hz,4h),1.15(t,j=7.0hz,6h).13cnmr(151mhz,dmso-d6)δ187.60,161.21,158.92,153.89,146.61,133.55,113.58,110.94,108.14,96.83,45.01,12.83.
将7-(二乙氨基)-2-氧代-2h-色烯-3-甲醛(0.25g,1.01mmol)和4-吡啶乙腈(0.18g,1.52mmol)在10ml无水乙醇中混合,将混合物在110℃加热反应,反应期间通过薄层色谱法(tlc)监测,待反应完成后,将反应产物冷却至室温,减压除去溶剂,所得残渣用乙醚与正己烷分别洗涤3次,将无水乙醇添加到获得的残余物中,并将混合物在冰箱中放置过夜,以沉淀出所需化合物。之后过滤沉淀物,然后使用冷乙醇洗涤2次,并在真空下干燥,得到黑色固体即(z)-3-(7-(二乙氨基)-2-氧代-2h-铬-3-基)-2-(吡啶-4-基)丙烯腈(0.23g,产率:65%)。
将(z)-3-(7-(二乙氨基)-2-氧代-2h-铬-3-基)-2-(吡啶-4-基)丙烯腈(0.07g,0.2mmol)和碘甲烷(0.28g,2mmol)溶解在乙腈中,混合物在85℃搅拌回流7小时,然后冷却至室温,减压除去溶剂,以体积比25:1的二氯甲烷和甲醇作洗脱剂经硅胶柱色谱分离,纯化得深紫色粉末即cpa-i(56.91mg,产率:79%)。1hnmr(600mhz,dmso)δ8.92(d,j=6.5hz,2h),8.86(s,1h),8.40(s,1h),8.32(d,j=6.8hz,2h),7.68(d,j=9.1hz,1h),6.89(d,j=9.1hz,1h),6.70(s,1h),4.30(s,3h),3.56(q,j=7.0hz,4h),1.18(t,j=7.0hz,6h).(图1)13cnmr(151mhz,dmso)δ160.55,158.06,154.11,149.54,146.03,145.12,144.77,133.02,122.79,116.98,111.51,111.18,108.79,102.10,97.22,47.55,45.24,12.93.(图2)esi-msm/z:[cpa-i+h]+calcd.for360.1707,found360.1700.(图3).
将(z)-3-(7-(二乙氨基)-2-氧代-2h-铬-3-基)-2-(吡啶-4-基)丙烯腈(0.07g,0.2mmol)和碘壬烷(0.51g,2mmol)溶解在乙腈中,混合物在85℃搅拌回流7小时,然后冷却至室温,减压除去溶剂,以体积比25:1的二氯甲烷和甲醇作洗脱剂经硅胶柱色谱分离,纯化得深紫色粉末即cpa-ii(70.84mg,产率:75%)。1hnmr(600mhz,dmso)δ9.02(d,j=6.1hz,2h),8.85(s,1h),8.40(s,1h),8.33(d,j=5.9hz,2h),7.68(d,j=9.1hz,1h),6.89(d,j=9.1hz,1h),6.70(s,1h),4.55(t,j=7.2hz,2h),3.56(dd,j=13.5,6.6hz,4h),1.92-1.88(m,2h),1.31-1.22(m,12h),1.17(t,j=7.0hz,6h),0.86(t,j=6.8hz,3h).(图4)13cnmr(151mhz,dmso)δ160.55,158.10,154.16,149.96,145.33,145.15,144.78,133.07,123.20,116.99,111.54,111.20,108.82,102.08,97.24,60.36,45.25,31.70,31.13,29.23,29.05,28.88,25.85,22.57,14.45,12.93.(图5)esi-msm/z:[cpa-ii+h]+calcd.for472.2959,found472.2953(图6).
实施例2
配制ph=5、浓度为10mm的pbs缓冲溶液,配制2mmcpa-i的dmso溶液,配制2mm二氧化硫水溶液;取2mlch3cn/pbs(v/v=3:7,ph=5)溶液、10μlcpa-i的dmso溶液加入到一个荧光比色皿中,取二氧化硫的水溶液,逐渐用微量进样器加到此比色皿中,加样的同时在荧光分光光度仪上检测,随着二氧化硫的加入,486nm的荧光强度逐渐增强,644nm的荧光强度逐渐降低。荧光发射图见图7。
实施例3
配制ph=5、浓度为10mm的pbs缓冲溶液,配制2mmcpa-i的dmso溶液,配制2mm二氧化硫水溶液;在荧光比色皿中,各加入2ml的纯pbs缓冲溶液和10μlcpa-i的dmso溶液,再分别加入30倍当量(除另有说明)的分析物:cpa-i,k+,mg2+,na+,no3-,i-,h2s,ch3coo-,n3-,h2po4-,no2-,so42-,cys(100当量),hcy(100当量),gsh(100当量),so32-(10当量)的水溶液,在荧光分光光度仪上检测,绘制不同分析物对应的644nm处荧光强度柱状图(见图8)。二氧化硫使得检测体系在644nm处荧光强度明显降低,其它的分析物基本没有引起检测体系荧光强度的变化。
实施例4
配制ph=5、浓度为10mm的pbs缓冲溶液,配制2mmcpa-i的dmso溶液,配制2mm二氧化硫水溶液;在11个比色皿中各加入2ml的纯pbs缓冲溶液和10μlcpa-i的dmso溶液,然后分别加入二氧化硫溶液的体积为0、10、20、30、40、50、60、70、80、90、100μl,同时在荧光光谱仪上测定644nm处荧光强度为540、492.6、452.4、415.4、368.5、325.9、287.6、243.2、200.8、170.7、142.4,以二氧化硫浓度为横坐标,以荧光强度f为纵坐标绘制图,得到二氧化硫浓度的工作曲线;线性回归方程为:f644=-4.051c+533.39,c的单位为10-6mol/l,见图9。
实施例5
配制ph=7.4、浓度为10mm的pbs缓冲溶液,配制2mmcpa-i的dmso溶液,配制2mm二氧化硫水溶液;把10μlcpa-i的dmso溶液加入到2ml的pbs缓冲溶液中;将探针溶液加入hela细胞培养液中,使得其浓度为10μm,与hela细胞在37℃下,反应15min,体系在荧光成像仪下有微弱蓝色荧光,与明显的红色荧光;然后再加入外源的二氧化硫,使其浓度为100μm,体系在荧光成像仪下显示蓝色荧光增强,红色荧光淬灭,见图10。
实施例6
配制ph=7.4、浓度为10mm的pbs缓冲溶液,配制2mmcpa-i的dmso溶液,配制2mm二氧化硫水溶液,配制ph=7.4、浓度为500μm的na2s2o3水溶液;把10μlcpa-i的dmso溶液加入到2ml的pbs缓冲溶液中;将探针溶液加入hela细胞培养液中,使得其浓度为10μm,与hela细胞在37℃下,反应15min,体系在荧光成像仪下有微弱蓝色荧光,与明显的红色荧光;然后再加入1mlna2s2o3水溶液,体系在荧光成像仪下显示蓝色荧光逐渐增强,红色荧光逐渐淬灭,见图11。
实施例7
配制ph=5、浓度为10mm的pbs缓冲溶液,配制2mmcpa-ii的dmso溶液,配制2mm二氧化硫水溶液;取2ml的ch3cn/pbs(v/v=3:7,ph=5)溶液、10μlcpa-ii的dmso溶液加入到一个荧光比色皿中,取二氧化硫的水溶液,逐渐用微量进样器加到此比色皿中,加样的同时在荧光分光光度仪上检测,随着二氧化硫的加入,481nm的荧光强度逐渐增强,648nm的荧光强度逐渐降低。荧光发射图见图12。
实施例8
配制ph=5、浓度为10mm的pbs缓冲溶液,配制2mmcpa-ii的dmso溶液,配制2mm二氧化硫水溶液;在荧光比色皿中,各加入2ml的ch3cn/pbs(v/v=3:7,ph=5)溶液和10μlcpa-ii的dmso溶液,再分别加入30倍当量(除另有说明)的分析物:cpa-i,k+,mg2+,na+,no3-,i-,h2s,ch3coo-,n3-,h2po4-,no2-,so42-,cys(100当量),hcy(100当量),gsh(100当量),so32-(15当量)的水溶液,在荧光分光光度仪上检测,绘制不同分析物对应的648nm处荧光强度柱状图(见图13)。二氧化硫使得检测体系在648nm处荧光强度明显降低,其它的分析物基本没有引起检测体系荧光强度的变化。
实施例9
配制ph=5、浓度为10mm的pbs缓冲溶液,配制2mmcpa-ii的dmso溶液,配制2mm二氧化硫水溶液;在16个比色皿中各加入2ml的ch3cn/pbs(v/v=3:7,ph=5)溶液和10μlcpa-ii的dmso溶液,,然后分别加入二氧化硫溶液的体积为0、10、20、30、40、50、60、70、80、90、100、110、120、130、140、150μl,同时在荧光光谱仪上测定648nm处荧光强度为860.8、798.5、740.7、675.4、619.1、581.1、549、516.6、469.2、430.8、393.8、345.7、308.1、263.3、221.2、193.9,以二氧化硫浓度为横坐标,以荧光强度f为纵坐标绘制图,得到二氧化硫浓度的工作曲线;线性回归方程为:f648=-4.311c+821.308,c的单位为10-6mol/l,见图14。
实施例10
配制ph=7.4、浓度为10mm的pbs缓冲溶液,配制2mmcpa-ii的dmso溶液,配制2mm二氧化硫水溶液;把10μlcpa-ii的dmso溶液加入到2ml的pbs缓冲溶液中;将探针溶液加入hela细胞培养液中,使得其浓度为10μm,与hela细胞在37℃下,反应15min,体系在荧光成像仪下有微弱蓝色荧光,与明显的红色荧光;然后再加入外源的二氧化硫,使其浓度为150μm,体系在荧光成像仪下显示蓝色荧光增强,红色荧光淬灭,见图15。
实施例11
配制ph=7.4、浓度为10mm的pbs缓冲溶液,配制2mmcpa-ii的dmso溶液,配制2mm二氧化硫水溶液,配制ph=7.4、浓度为500μm的na2s2o3水溶液;把10μlcpa-ii的dmso溶液加入到2ml的pbs缓冲溶液中;将探针溶液加入hela细胞培养液中,使得其浓度为10μm,与hela细胞在37℃下,反应15min,体系在荧光成像仪下有微弱蓝色荧光,与明显的红色荧光;然后再加入1mlna2s2o3水溶液,体系在荧光成像仪下显示蓝色荧光逐渐增强,红色荧光逐渐淬灭,见图16。
实施例12
配制ph=7.4、浓度为10mm的pbs缓冲溶液,配制2mmcpa-i的dmso溶液,配制2mm二氧化硫水溶液;把10μlcpa-i的dmso溶液加入到2ml的pbs缓冲溶液中;将探针溶液加入hela细胞培养液中,使得其浓度为10μm,与hela细胞在37℃下,反应15min;将体系在荧光成像仪下观测;紧接着将10μmdio加入刚刚的体系中,在37℃下,反应30min。将体系在荧光成像仪下观测,同时对得到的荧光图片进行共定位率计算,见图17。
配制ph=7.4、浓度为10mm的pbs缓冲溶液,配制2mmcpa-ii的dmso溶液,配制2mm二氧化硫水溶液;把10μlcpa-ii的dmso溶液加入到2ml的pbs缓冲溶液中;将探针溶液加入hela细胞培养液中,使得其浓度为10μm,与hela细胞在37℃下,反应15min;将体系在荧光成像仪下观测;紧接着将0.2μmmitotrackergreen加入刚刚的体系中,在37℃下,反应20min。将体系在荧光成像仪下观测,同时对得到的荧光图片进行共定位率计算,见图17。
- 还没有人留言评论。精彩留言会获得点赞!