16SrRNA基因序列分析鉴定临床疑难菌种的方法与流程
本发明属于生物技术领域,尤其涉及一种16srrna基因序列分析鉴定临床疑难菌种的方法。
背景技术:
16srrna基因序列分析是指通过克隆、测序或酶切、探针杂交获得样本中微生物的16srrna序列信息,再与16srrna数据库进行比对,从而对样本中的微生物进行种属鉴定及进化树分析,是一种快速有效的方法。16srrna存在于所有的生物中,其进化具有良好的时钟性质,在结构与功能上高度保守,被称为"细菌化石",故16srrna是细菌种属鉴定及系统分类研究中最常用的分子钟[1-2]。
生物细胞内,核糖体rna(rrna)与几十种蛋白质结合形成核糖体,继而沿着mrna模板移动,执行着蛋白质合成的功能,其在种系发生中发挥着不可或缺的作用。在漫长的进化过程中,rrna分子功能几乎保持恒定,某些部位分子排列顺序变化极其缓慢,形成“细菌化石”[7-9]。通过对恒定序列的测定,可揭示物种的亲缘关系,为系统发育提供线索;rrna同时具有高变性,通过对生物物种的特征性核酸序列的测定,可进行属种鉴定。细菌核糖体rna(rrna)按照沉降系数分类,分别有5s、16s及23srrna三种。16srrna为细胞所共有,在细菌rna含量中占比大,分子量适中约1.5kb,其既能体现不同菌属之间的差异,又可通过测序技术较易获得序列,故16srrna基因序列作为生物系统发育及鉴定指标最为合适[10-11]。对于临床上用常规方法尚不能鉴别的细菌,drancouurt等众多学者采用16srrna基因序列分析技术,均解开了谜题[1-2,7,10-12]。16srrna基因序列分析技术在疑难菌种的鉴定中有着重要的临床应用价值[13-15]。
目前,全球微生物感染呈现快速上升的趋势,细菌耐药率不断增高,新发感染病原微生物层出不穷,对临床微生物检验提出了更高的需求。临床微生物实验室需根据自身条件建立有效而可信的能鉴别各种细菌的方法,该法需速度快、敏感性、特异性高,且易于操作。传统的细菌鉴定主要依据形态和生理性状,需要进行培养及生化试验或免疫学检测。随着微生物学技术的发展,目前全自动细菌鉴定仪已基本替代手工方法,操作方便,鉴定范围超过98%的临床常见菌株,基本能够满足临床实验室对细菌鉴定的初步需求。然而亦可能存在如下问题:所依赖的表型表达不稳定;敏感度不高,某些方法只适用于单一菌种;某些方法的应用还受到血清变种的影响[3-6]。因此,对某些疑难罕见菌种,全自动细菌鉴定仪并不能给出理想的鉴定结果。因此,亟需建立了一种新的有效地鉴定疑难菌种的方法,并对其进行临床应用评价,满足临床对疑难菌种的鉴定需求。
技术实现要素:
本发明的目的在于运用16srrna基因序列分析技术,建立一种有效地鉴定疑难菌种的pcr方法,并对其反应体系及条件进行优化,将其应用于临床疑难菌种的鉴定。
具体的,本发明的技术方案如下:
本发明第一个方面公开了一种鉴定临床疑难菌种的方法,确切来讲,采用16srrna基因序列分析方法,包括:
s1:抽提待鉴定菌种的细菌dna,选择细菌通用引物,建立扩增细菌16srrna的pcr方法;
s2:优化pcr反应体系和反应条件得到扩增产物;
s3:将扩增产物与基因数据库进行序列比对,得到待鉴定细菌的菌种。
应当理解,本发明中的方法并不限于上述步骤,在s1之前、s1与s2之间、s2与s3之间,s3之后,本领域技术人员可以根据需要,增加其他额外的步骤,且均在本发明的保护范围之内。
优选的,所述细菌通用引物为27f和1492r通用引物。在本发明的一些具体实施例中,引物27f的核苷酸序列为:5’-agagtttgatcctggctcag-3’(seqidno:1);引物1492r的核苷酸序列为:5’-ggttaccttgttacgactt-3’(seqidno:2)。
优选的,pcr反应体系包括引物、pfubuffer、dntp、pfudna聚合酶、dna模板和ddh2o。
优选的,所述引物浓度为0.1-0.75μm/l;在本发明的一些具体实施例中,引物浓度为0.5μm/l。
优选的,所述pfudna聚合酶的用量为2.5-7.0u/体系;在本发明的一些具体实施例中,所述pfudna聚合酶的用量为5.0u/体系。
优选的,所述pcr反应条件为:95℃预变性5min,而后进行35个循环扩增(95℃变性30s,52-55℃退火10-40s,72℃延伸90s),随后72℃延伸7min。
应当理解,所述pcr反应条件并不限于上述条件,本领域技术人员可以根据需要,选择其他适宜的pcr反应条件,且均在本发明的保护范围之内。
在本发明的一些优选实施例中,pcr退火温度为52℃。
在本发明的一些优选实施例中,退火时间为30s。
优选的,在s3中,采用genebank数据库进行blast序列比对,得到待鉴定细菌的菌种。
优选的,所述细菌包括霍氏鲍特菌、马尔他布鲁菌和空肠弯曲菌。
本发明第二个方面公开了上述的方法在鉴定样本中微生物种类的应用。
在符合本领域常识的基础上,上述各优选条件,可任意组合,而不超出本发明的构思与保护范围。
本发明相对于现有技术具有如下的显著优点及效果:
本发明运用16srrna基因序列分析技术,建立了一种新的有效地鉴定疑难菌种的方法,扩大了对临床菌种的鉴定谱,满足临床对疑难菌种的鉴定需求。
附图说明
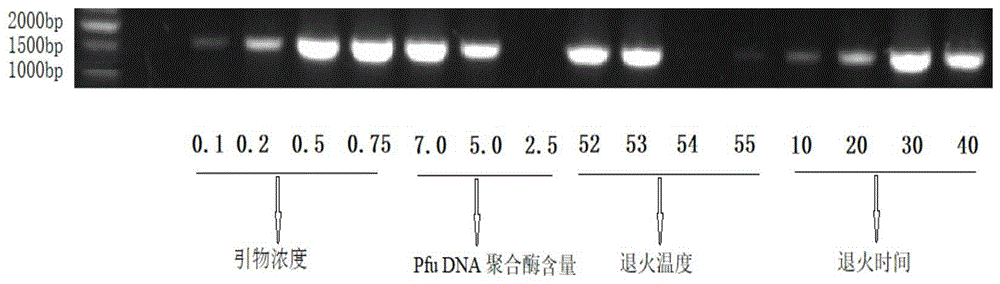
图1为本发明实施例中采用16srrna基因分析方法在不同的实验条件下pcr产物琼脂糖凝胶电泳图。
图2为本发明实施例中pcr产物双向测序图(bioeditsequencealignmenteditor软件分析后部分片段截图)。
图3为本发明实施例中产物序列与genebank数据库进行blast比对结果图。
图4为本发明实施例中产物序列与genebank数据库进行blast比对结果图。
图5为本发明实施例中pcr产物单向测序图(bioeditsequencealignmenteditor软件分析后部分片段截图)。
图6为本发明实施例中产物序列与genebank数据库进行blast比对结果图。
图7为本发明实施例中pcr产物双向测序图(bioeditsequencealignmenteditor软件分析后部分片段截图)。
图8为本发明实施例中产物序列与genebank数据库进行blast比对结果图。
具体实施方式
下面结合附图和实施例对本发明的技术方案进行详细描述,但并不因此将本发明限制在所述的实施例范围之中。
下列实施例中未注明具体条件的实验方法,按照常规方法和条件,或按照商品说明书选择。本发明所用试剂和原料均市售可得。
实施例1
本实施例公开了一种鉴定临床疑难菌种的方法,确切来讲,采用16srrna基因序列分析方法,包括:
s1:抽提待鉴定菌种的细菌dna,选择细菌通用引物,建立扩增细菌16srrna的pcr方法;
s2:优化pcr反应体系和反应条件得到扩增产物;
s3:将扩增产物与基因数据库进行序列比对,得到待鉴定细菌的菌种。
具体的,包括以下步骤:
一、基因组dna抽提
标本来自南通大学附属医院2015至2019年期间运用常规方法不能鉴别的细菌。取活化后的临床分离菌株4区划线接种在血平板上,35℃,补充5%co2的大气培养24~48h后收集菌落,根据omegabio-tek公司细菌基因组dna提取试剂盒说明书提取基因组dna。
二、16srrna基因序列分析
将引物稀释至10μm,并进行pcr扩增。pcr反应体系:1μl引物27f,1μl引物1492r,5μl10×pfubuffer,1μl10mmol/ldntp,1μl5u/μlpfudna聚合酶,3μldna模板,ddh2o定容至50μl;pcr反应条件:95℃预变性5min,而后进行35个循环扩增(95℃变性30s,52℃退火30s,72℃延伸90s),随后72℃延伸7分钟,最终产物降至25℃后取出琼脂糖凝胶电泳分析,回收目的条带,送invitrogen公司进行测序。将测序结果与16srrna数据库中的序列进行比较,确定其在进化树中的位置,从而鉴定样本中可能存在的微生物种类。
三、pcr体系和条件的优化
3.1引物浓度
pcr反应体系中,引物是pcr特异性反应的关键。引物浓度过低,会降低pcr扩增的产率;而引物浓度过高则会诱发错配,从而出现非特异性扩增。故设置了0.1、0.2、0.5及0.75μm/l4个浓度进行优化,上下游引物浓度一致。
3.2pfudna聚合酶的用量
适当增加pfudna聚合酶的用量,可提高pcr扩增效率,但过高可能导致非特异性条带的产生。比较了2.5、5.0、7.0u/体系时的扩增效率,选择最佳浓度。
3.3pcr退火温度
变性后温度快速冷却至40~60℃,可使引物和模板发生结合。退火温度一般取决于引物的长度、碱基组成及其浓度、靶基序列的长度。退火温度是影响pcr特异性的较重要因素。因此,在tm值附近选择退火温度,共设定了55、54、53及52℃4个温度进行实验。
3.4pcr退火时间
退火时间对pcr产物的稳定性有较大影响,故选择了10、15、20、30、40s5个时间进行比较,确定最佳退火时间。
四、实验结果
当引物浓度为0.1μm/l时,扩增效率变差,当引物浓度为0.5μm/l时,扩增效率稳定且无非特异性扩增;pfudna聚合酶的用量对pcr的扩增效率有较大影响,当每个体系中加入5.0upfudna聚合酶,扩增效率较高,且最经济;选择退火温度为52℃,退火时间为30s,pcr扩增效率较高,且结果稳定,结果如图1所示。
实施例2
本实施例研究部分临床疑难菌种的检测分析,结果如下所示:
2.1霍氏鲍特菌(bordetellaholmesii,b.holmesii)的检测分析结果
提取分离菌总dna为模板,利用所优化的pcr反应体系及条件进行扩增,将产物行双向测序(测序图见图2),而后与genebank数据库进行blast比对,结果表明该分离株16srrna序列与数据库中已报道的b.holmesii的16srrna序列近似,相似性达99%(图3),故此菌种鉴定为b.holmesii。将该分离株16srrna基因序列上传ncbigenbank,存取号为kt828544.1。国内尚未报道分离过b.holmesii,故将菌种上传中国普通微生物菌种保藏管理中心(cgmcc,菌种保藏号:cgmcc1.13721),菌种保藏证书见图4。
2.2马尔他布鲁菌(brucellamelitensis,b.melitensis)的检测分析结果
提取该分离菌总dna为模板,16srrna基因pcr扩增后将产物进行测序(测序图见图5),运用在线blast软件进行序列比对,与b.melitensis相似性达100%(图6),此菌种鉴定为马尔他布鲁菌。
2.3空肠弯曲菌(campylobacterjejuni,c.jejuni)的检测分析结果
提取该分离菌总dna为模板,16srrna基因pcr扩增后将产物进行测序(测序图见图7),运用在线blast软件进行序列比对,与c.jejuni相似性达100%(图8),此菌种鉴定为空肠弯曲菌。
16srrna序列兼具稳定性及高变性,随着当今分子生物学技术的飞速发展,16srrna基因序列分析已成为细菌准确鉴定的常用方法。运用16srrna基因序列分析,临床微生物学实验室可以实现快速、简便、准确地对微生物进行分类鉴定,并可从分子水平、遗传进化的角度更深入地研究分析细菌。发明人根据实验室条件,运用16srrna基因序列分析技术,建立了一种有效地鉴定疑难菌种的方法,扩大了对临床菌种的鉴定谱,满足临床对疑难菌种的鉴定需求。
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
参考文献
[1]drancourtm,bolletc,carlioza,etal.16sribosomaldnasequenceanalysisofalargecollectionofenvironmentalandclinicalunidentifiablebacterialisolates.jclinmicrobiol2000;38(10):3623-3630.
[2]woopc,lausk,tengjl,etal.thenandnow:useof16srdnagenesequencingforbacterialidentificationanddiscoveryofnovelbacteriainclinicalmicrobiologylaboratories.clinmicrobiolinfect2008;14(10):908-934.
[3]tangyw,ellisnm,hopkinsmk,etal.comparisonofphenotypicandgenotypictechniquesforidentificationofunusualaerobicpathogenicgram-negativebacilli.jclinmicrobiol1998;36(12):3674-3679.
[4]rodiciomdelr,mendozamdelc.identificationofbacteriathrough16srrnasequencing:principles,methodsandapplicationsinclinicalmicrobiology.enferminfeccmicrobiolclin2004;22(4):238-245.
[5]martinim,leeim,bottnerkd,etal.ribosomalproteingene-basedphylogenyforfinerdifferentiationandclassificationofphytoplasmas.intjsystevolmicrobiol2007;57(pt9):2037-2051.
[6]tortolie,bartolonia,
[7]shamputaic,rigoutsandl,portaelsf.moleculargeneticmethodsfordiagnosisandantibioticresistancedetectionofmycobacteriafromclinicalspecimens.apmis2004;112(11-12):728-752.
[8]baseint,gardinerbj,andujarvazquezetal.microbialidentificationusingdnatargetamplificationandsequencing:clinicalutilityandimpactonpatientmanagement.openforuminfectdis2018;5(11):ofy257.
[9]kolbertcp,persingdh.ribosomaldnasequencingasatoolforidentificationofbacterialpathogens.curropinmicrobiol1999;2(3):299-305.
[10]woopc,ngkh,lausk,etal.usefulnessofthemicroseq50016sribosomaldna-basedbacterialidentificationsystemforidentificationofclinicallysignificantbacterialisolateswithambiguousbiochemicalprofiles.jclinmicrobiol2003;41(5):1996-2001.
[11]woopc,chunglm,tengjl,etal.insilicoanalysisof16sribosomalrnagenesequencing-basedmethodsforidentificationofmedicallyimportantanaerobicbacteria.jclinpathol2007;60(5):576-579.
[12]cloudjl,nealh,rosenberryr,etal.identificationofmycobacteriumspp.byusingacommercial16sribosomaldnasequencingkitandadditionalsequencinglibraries.jclinmicrobiol2002;40(2):400-406.
[13]clarridgeje3rd.impactof16srrnagenesequenceanalysisforidentificationofbacteriaonclinicalmicrobiologyandinfectiousdiseases.clinmicrobiolrev2004;17(4):840-862.
[14]罗雯,万雅各,彭宣宪,etal.采用通用引物pcr配合sscp及rflp技术快速检测常见病原菌.中华微生物学和免疫学杂志2001;21(6):687-689.
[15]mignards,flandroisjp.16srrnasequencinginroutinebacterialidentification:a30-monthexperiment.jmicrobiolmethods2006;67(3):574-581。
- 还没有人留言评论。精彩留言会获得点赞!