一类氮杂黄酮衍生物及其作为抗肿瘤药物的应用的制作方法
本发明涉及药物应用领域,具体涉及一类氮杂黄酮衍生物及其在制备抗肿瘤药物中的应用。
背景技术:
近年来,随着我国经济与社会的快速发展,紧张的生活节奏和大面积的生态环境污染使我国癌症发病率居高不下,且有持续上升态势;恶性肿瘤(癌症)已经成为严重威胁我国居民健康的公共卫生问题之一,防控形势十分严峻。黄酮是一类天然存在的多酚类有机小分子化合物,它们是植物代谢过程中产生的多羟基次级代谢产物,存在于植物的根、茎、叶、花、种子和果实中,在自然界分布十分广泛。根据国内外众多的研究报导,黄酮类化合物具有多种良好的药理活性,例如抗肿瘤,抗病毒,抗氧化,抗菌,抗炎,降糖和降血脂等。因此,运用简便有效的合成方法获得更为广泛的黄酮类似物,可以进一步发掘黄酮衍生物的药理活性,推动更多黄酮类化合物在药物研发中的应用与发展。本发明运用生物电子等排替换、派生官能团及官能团的修饰等方法对天然产物黄酮进行衍生化研究,对所得衍生物进行药理活性测试,结果表明这些化合物具有显著的抗肿瘤作用,并且,是一类全新结构的具有抗肿瘤作用的化合物,进而可以发明作为抗肿瘤药物。
技术实现要素:
本发明涉及到一类氮杂黄酮衍生物及其作为抗肿瘤药物的用途,合成了一类全新结构的具有抗肿瘤作用的氮杂黄酮衍生物,体外抗肿瘤活性研究证明,本发明的氮杂黄酮衍生物对多种肿瘤细胞,包括人白血病细胞k562、人肝癌细胞hepg2和人结肠癌细胞hct-116具有很强的抗肿瘤活性,可望开发成新的抗肿瘤药物。
本发明提供的一类具有抗肿瘤活性的氮杂黄酮类衍生物,其结构式通式如下:
而且,r1为氢、甲基、苄基、乙酰基或苯甲酰基;r2为氢、氟、羟基、甲氧基、羧基或酯基;r3为氢、氟、羟基或甲氧基;a为5-8元的芳香环或芳香杂环、杂原子取代的芳基、环烷基、杂原子取代的环烷基。
而且,所述的一类氮杂黄酮衍生物的结构为:
如上述的一类具有抗肿瘤活性的氮杂黄酮衍生物在制备抗肿瘤药物中的应用。
而且,所述的抗肿瘤药物为治疗人白血病细胞k562,或为人肝癌细胞hepg2,或为人结肠癌细胞hct-116的药物。
本发明取得的优点和积极效果为:
本发明通过波普和质谱数据分析表明合成得到的氮杂黄酮衍生物为新化合物,且经体外抗肿瘤活性研究证明,本发明提供的氮杂黄酮衍生物对多种肿瘤细胞,包括人白血病细胞k562、人肝癌细胞hepg2和人结肠癌细胞hct-116具有很强的抗肿瘤活性,是一种优良的抗肿瘤新化合物,可望开发成新的抗肿瘤药物。
附图说明
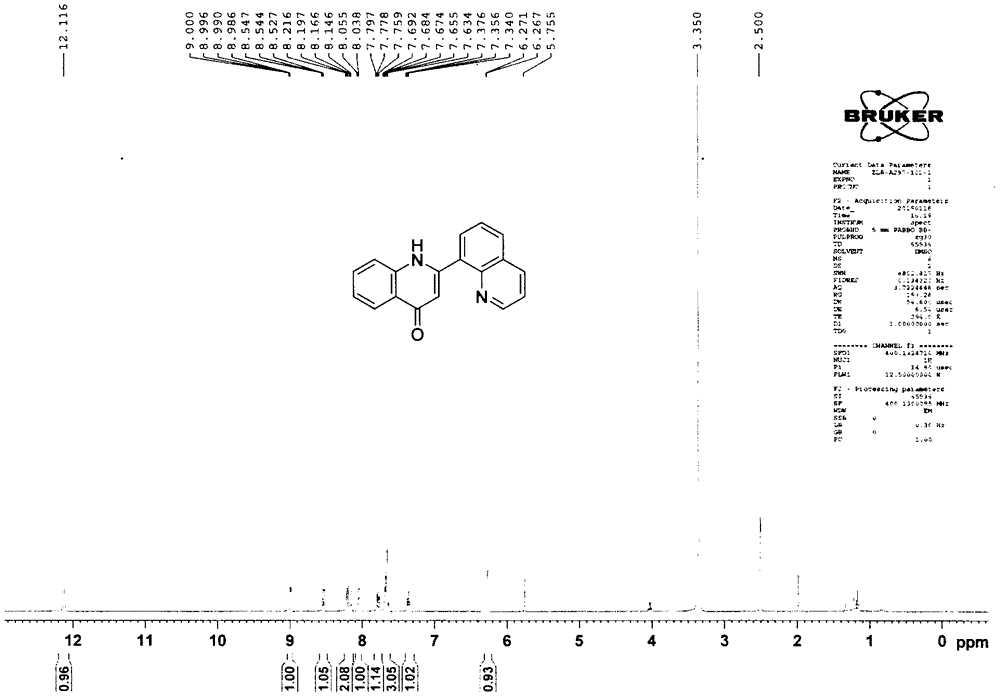
图1为本发明中化合物4-1在氘代dmso中的核磁氢谱图;
图2为本发明中化合物4-1在氘代dmso中的核磁碳谱图;
图3为本发明中化合物5-1在氘代dmso中的核磁氢谱图;
图4为本发明中化合物5-1在氘代dmso中的核磁碳谱图;
图5为本发明中化合物6-1在氘代dmso中的核磁氢谱图;
图6为本发明中化合物6-1在氘代dmso中的核磁碳谱图;
图7为本发明中化合物8-1在氘代dmso中的核磁氢谱图;
图8为本发明中化合物8-1在氘代dmso中的核磁碳谱图;
图9为本发明中化合物8-3在氘代dmso中的核磁氢谱图;
图10为本发明中化合物8-3在氘代dmso中的核磁碳谱图;
具体实施方式
下面详细叙述本发明的实施例,需要说明的是,本实施例是叙述性的,不是限定性的,不能以此来限定本发明的保护范围。
本发明中所使用的原料,如无特殊说明,均为常规的市售产品;本发明中所使用的方法,如无特殊说明,均为本领域的常规方法。
一类具有抗肿瘤活性的氮杂黄酮衍生物,其结构式通式如下:
较优地,所述r1为氢、甲基、苄基、乙酰基或苯甲酰基;r2为氢、氟、羟基、甲氧基、羧基或酯基;r3为氢、氟、羟基或甲氧基;a为5-8元的芳香环或芳香杂环、环烷基、杂原子取代的环烷基。
较优地,所述氮杂黄酮衍生物的名称及结构为表1所示:
表1氮杂黄酮衍生物的具体结构及名称
如上所述的一类具有抗肿瘤活性的氮杂黄酮衍生物的合成路线,具体为:
反应式1
较优地,化合物3通过化合物1与化合物2反应合成而来,合成时所用碱为三乙胺,溶剂为二氯甲烷,反应温度为25℃,反应时长为4小时。
较优地,化合物3在碱性条件下发生分子内claisen缩合反应得到化合物4,合成时所用碱为氢氧化钠,溶剂为1,4-二氧六环,反应温度为110℃,反应时长为2小时。
较优地,具体步骤如下:
在圆底烧瓶中,将化合物1、三乙胺溶解于无水二氯甲烷中,化合物1∶三乙胺∶无水二氯甲烷的比例mmol∶mmol∶ml为1∶2∶10.81;随后将化合物2在0℃下缓慢滴加入反应体系,化合物1∶化合物2的比例mmol∶mmol为1∶1;加毕25℃搅拌反应4h。待tlc检测反应完全,将反应体系用二氯甲烷稀释后缓慢倾入水中。萃取分离有机相,饱和氯化钠水溶液洗涤,无水硫酸钠干燥。减压浓缩得化合物3,无需纯化直接用于下一步反应。将化合物3溶解于1,4-二氧六环中,化合物3∶1,4-二氧六环的比例mmol∶ml为1∶2;然后加入氢氧化钠,化合物3∶氢氧化钠的比例mmol∶mmol为1∶3;加毕将反应体系转移至油浴加热110℃回流反应2h。tlc检测反应完全后,将反应体系冷却至室温,用乙酸乙酯稀释后缓慢倾入1m盐酸水溶液中。萃取分离有机相,饱和食盐水洗涤,无水硫酸钠干燥。减压浓缩后经柱层析(石油醚∶乙酸乙酯=4∶1)纯化,得化合物4-1~4-13。
反应式2
较优地,化合物5通过化合物4反应合成而来,合成时使用三溴化硼,溶剂为二氯甲烷,反应温度为25℃,反应时长为10小时。
较优地,具体步骤如下:
在圆底烧瓶中,将化合物4解于无水二氯甲烷中,化合物4∶无水二氯甲烷的比例mmol∶ml为1∶10;随后将三溴化硼在0℃下缓慢滴加入反应体系,化合物4∶三溴化硼的比例mmol:mmol为1∶4;加毕25℃搅拌反应10h。反应结束后,在0℃搅拌条件下缓慢加入冰块,析出固体,过滤干燥得化合物5-1~5-2。
反应式3
较优地,化合物6通过化合物4反应合成而来,合成时使用氢氧化钠,溶剂为乙醇与水,反应温度为25℃,反应时长为6小时。
较优地,具体步骤如下:
在圆底烧瓶中,将化合物4解于乙醇/水(v/v=1∶9)混合溶液中,化合物4∶乙醇/水混合溶液的比例mmol∶ml为1∶13.80;随后加入氢氧化钠,化合物4∶氢氧化钠的比例mmol∶mmol为1∶10;加毕25℃搅拌反应6h。反应结束后,将反应体系用盐酸(4m)水溶液调节ph为酸性,析出固体,过滤干燥得化合物6-1~6-2。
反应式4
较优地,化合物8通过化合物7反应合成而来,合成时所用碱为氢化钠,溶剂为n,n-二甲基甲酰胺,反应温度为60℃,反应时长为3小时。
较优地,具体步骤如下:
在圆底烧瓶中,将化合物7解于n,n-二甲基甲酰胺,化合物7∶乙n,n-二甲基甲酰胺的比例mmol∶ml为1∶8.10;随后缓慢加入氢化钠(60%分散于石蜡油),化合物7∶氢化钠的比例mmol∶mmol为1∶3;室温搅拌30分钟后,缓慢滴加碘甲烷或溴化苄,化合物7∶碘甲烷或溴化苄的比例mmol∶mmol为1∶3;加毕将反应体系转移至油浴加热60℃反应3h。反应结束后,将反应体系冷却至室温,用乙酸乙酯稀释后缓慢倾入1m盐酸水溶液中。萃取分离有机相,饱和食盐水洗涤,无水硫酸钠干燥。减压浓缩后经柱层析(石油醚∶乙酸乙酯=10∶1)纯化,得化合物8-1~8-2。
反应式5
较优地,化合物8通过化合物7反应合成而来,合成时所用碱为碳酸钾,溶剂为n,n-二甲基甲酰胺,反应温度为140℃,反应时长为12小时。
较优地,具体步骤如下:
在圆底烧瓶中,将化合物7解于n,n-二甲基甲酰胺,化合物7∶乙n,n-二甲基甲酰胺的比例mmol∶ml为1∶5.40;随后缓慢加入碳酸钾、乙酰氯或苯甲酰氯,化合物7∶碳酸钾∶乙酰氯或苯甲酰氯的比例mmol∶mmol∶mmol为1∶5∶5;加毕将反应体系转移至油浴加热140℃反应12h。反应结束后,将反应体系冷却至室温,用乙酸乙酯稀释后缓慢倾入水中。萃取分离有机相,饱和食盐水洗涤,无水硫酸钠干燥。减压浓缩后经柱层析(石油醚∶乙酸乙酯=5∶1)纯化,得化合物8-3~8-4。
如上述的一类具有抗肿瘤活性的氮杂黄酮衍生物在制备抗肿瘤药物中的应用。
较优地,所述的抗肿瘤药物为治疗人白血病细胞k562,或为人肝癌细胞hepg2,或为人结肠癌细胞hct-116的药物。
下面通过实施例具体说明。
实施例1
化合物4-1的合成
在圆底烧瓶中,将2-氨基苯乙酮(0.45g,3.70mmol)、三乙胺(1.03ml,7.40mmol)溶解于无水二氯甲烷(40ml)中,随后将喹啉-8-甲酰氯(0.71g,3.70mmol)在0℃下缓慢滴加入反应体系,加毕25℃搅拌反应4h。待tlc检测反应完全,将反应体系用二氯甲烷稀释后缓慢倾入水中。萃取分离有机相,饱和氯化钠水溶液洗涤,无水硫酸钠干燥。减压浓缩得n-(2-乙酰基苯基)喹啉-8-甲酰胺,无需纯化直接用于下一步反应。将n-(2-乙酰基苯基)喹啉-8-甲酰胺(1.08g,3.70mmol)溶解于1,4-二氧六环(7.40ml)中,然后加入氢氧化钠(0.45g,11.10mmol),加毕将反应体系转移至油浴加热110℃回流反应2h。tlc检测反应完全后,将反应体系冷却至室温,用乙酸乙酯稀释后缓慢倾入1m盐酸水溶液中。萃取分离有机相,饱和食盐水洗涤,无水硫酸钠干燥。减压浓缩后经柱层析(石油醚∶乙酸乙酯=4∶1)纯化得0.43g白色固体化合物4-1,收率42%。
1hnmr(400mhz,dmso-d6)δ12.12(s,1h),8.99(dd,j=4.0,1.6hz,1h),8.54(dd,j=8.0,1.2hz,1h),8.22-8.15(m,2h),8.05(d,j=6.8hz,1h),7.78(t,j=7.6hz,1h),7.69-7.63(m,3h),7.38-7.34(m,1h),6.27(d,j=1.6hz,1h);13cnmr(100mhz,dmso-d6)δ176.9,151.1,149.6,145.1,140.2,136.8,133.2,131.7,130.7,130.4,128.2,126.4,124.9,124.8,123.1,122.1,118.6,110.2.hrms(+esi-tof)m/z:[m+h]+calcd.forc18h13n2o273.1022;found273.1027.
实施例2
化合物5-1的合成
在圆底烧瓶中,将6-甲氧基-2-(1-萘基)喹啉-4(1h)-酮(100mg,0.33mmol)溶解于无水二氯甲烷(3.30ml)中,随后将三溴化硼(0.13ml,1.33mmol)在0℃下缓慢滴加入反应体系,加毕25℃搅拌反应10h。反应结束后,在0℃搅拌条件下缓慢加入冰块,析出黄色固体,过滤干燥得88mg化合物5-1,收率93%。
1hnmr(400mhz,dmso-d6)δ14.48(s,1h),10.62(s,1h),8.25(d,j=8.4hz,1h),8.15(d,j=8.0hz,1h),7.96(d,j=9.6hz,1h),7.87-7.85(m,2h),7.77-7.73(m,1h),7.70-7.58(m,4h),7.03(s,1h).13cnmr(100mhz,dmso-d6)δ167.8,157.1,151.7,134.4,133.2,131.5,130.3,130.1,128.8,128.1,127.1,126.5,125.5,124.5,122.2,121.6,106.8,104.5.hrms(+esi-tof)m/z:[m+h]+calcd.forc19h14no2288.1019;found288.1025.
实施例3
化合物6-1的合成
在圆底烧瓶中,将化合物4-12(100mg,0.29mmol)解于乙醇/水(4ml,v/v=1∶9)混合溶液中,随后加入氢氧化钠(116.48mg,2.91mmol),加毕25℃搅拌反应6h。反应结束后,将反应体系用盐酸(4m)水溶液调节ph为酸性,析出固体,过滤干燥得73mg白色固体化合物6-1,收率80%。
1hnmr(400mhz,dmso-d6)δ13.42(s,1h),8.84(s,1h),8.83-8.28(m,1h),8.19(d,j=8.4hz,1h),8.11(d,j=7.6hz,1h),7.93-7.89(m,2h),7.80(d,j=6.8hz,1h),7.73-7.60(m,3h),6.69(s,1h).13cnmr(100mhz,dmso-d6)δ174.4,166.6,152.7,142.8,133.2,132.7,131.4,130.9,130.1,128.7,128.1,127.7,127.1,126.8,126.7,125.4,124.7,122.4,119.9,110.2.hrms(+esi-tof)m/z:[m+h]+calcd.forc20h14no3316.0968;found316.0967.
实施例4
化合物8-1的合成
在圆底烧瓶中,将化合物7(100mg,0.37mmol)解于n,n-二甲基甲酰胺(3ml),随后缓慢加入氢化钠(44.22mg,1.11mmol,60%分散于石蜡油),室温搅拌30分钟后,缓慢滴加碘甲烷(68.84μl,1.11mmol),加毕将反应体系转移至油浴加热60℃反应3h。反应结束后,将反应体系冷却至室温,用乙酸乙酯稀释后缓慢倾入1m盐酸水溶液中。萃取分离有机相,饱和食盐水洗涤,无水硫酸钠干燥。减压浓缩后经柱层析(石油醚∶乙酸乙酯=10∶1)纯化得84mg白色固体化合物8-1,收率80%。
1hnmr(400mhz,dmso-d6)δ8.34-8.27(m,1h),8.13(dd,j=8.0,1.6hz,1h),8.08(d,j=8.0hz,1h),7.86-7.78(m,2h),7.72-7.58(m,3h),7.58-7.52(m,2h),7.52-7.48(m,1h),6.08(s,1h),3.38(s,3h).13cnmr(100mhz,dmso-d6)δ175.7,152.9,141.5,132.8,132.4,130.3,129.8,128.6,127.7,127.1,126.7,126.5,125.6,125.5,124.5,123.6,117.3,112.0,36.4.hrms(+esi-tof)m/z:[m+h]+calcd.forc20h16no286.1226;found286.1226.
实施例5
化合物8-3的合成
在圆底烧瓶中,将化合物7(200mg,0.74mmol)解于n,n-二甲基甲酰胺(4ml),随后缓慢加入碳酸钾(509.38mg,3.69mmol)、乙酰氯(0.26ml,3.69mmol),加毕将反应体系转移至油浴加热140℃反应12h。反应结束后,将反应体系冷却至室温,用乙酸乙酯稀释后缓慢倾入水中。萃取分离有机相,饱和食盐水洗涤,无水硫酸钠干燥。减压浓缩后经柱层析(石油醚∶乙酸乙酯=5∶1)纯化得71mg白色固体化合物8-3,收率31%。
1hnmr(400mhz,dmso-d6)δ8.21-8.12(m,3h),8.12-8.04(m,2h),7.92-7.88(m,1h),7.79-7.72(m,3h),7.72-7.63(m,1h),7.58-7.53(m,2h),2.52(s,3h);13cnmr(100mhz,dmso-d6)δ169.2,159.9,154.7,149.6,137.9,134.0,131.2,130.9,129.8,129.7,128.9,128.5,127.7,127.3,126.6,125.9,125.7,122.0,121.4,116.0,21.4.hrms(+esi-tof)m/z:[m+h]+calcd.forc21h16no2314.1176;found314.1180.
实施例6
本发明氮杂黄酮衍生物的抗肿瘤活性测定
取对数生长期的细胞,调整细胞密度为5×104cells/ml接种于96孔板上,每孔100μl,同时设置空白孔和对照孔,每个化合物浓度设置3个平行孔。于37℃、5%co2培养箱中培养(悬浮细胞培养2h,贴壁细胞培养24h)。分别加入终浓度为1.0、10μm的化合物,每孔0.5μl。空白孔为不含有细胞、dmso以及化合物的纯培养基孔,对照孔为含相同浓度dmso作用的细胞。按照以上方案处理后,将孔板置于37℃,5%co2恒温培养箱中培养48h。此后,每孔加入5mg/ml的mtt溶液20μl(用pbs配制,0.22μm滤膜过滤除菌),置于37℃,5%co2恒温培养箱中继续孵育4h,终止培养。悬浮细胞每孔直接加入100μl盐酸异丙醇,贴壁细胞小心移除孔内培养上清液,每孔加入100μldmso。37℃,培养箱内放置10min,使紫色结晶物充分溶解。用酶标仪(悬浮细胞选择580,620nm,贴壁细胞选择490,620nm)测定各孔的吸光度(od)值。根据测出的od值按照以下公式计算细胞存活率。
细胞存活率(%)=(实验组od-空白组od)/(对照组od-空白组od)×100%
活性测试结果如表2所示,结果显示,当利用1μm和10μm的化合物处理各细胞48h后,大部分受试细胞的存活率显著降低,其中化合物4-4、4-5、4-8、4-10、4-12、5-1、8-3和8-4的ic50值均低于1μm,本发明提供的大部分氮杂黄酮衍生物表现出显著的抗肿瘤活性。
表2氮杂黄酮衍生物的抗肿瘤活性测试结果
注:人白血病细胞k562、人肝癌细胞hepg2、人结肠癌细胞hct-116
化疗药物喜树碱(cpt)为阳性对照药物。
尽管为说明目的公开了本发明的实施例,但是本领域的技术人员可以理解:在不脱离本发明及所附权利要求的精神和范围内,各种替换、变化和修改都是可能的,因此,本发明的范围不局限于实施例和附图所公开的内容。
- 还没有人留言评论。精彩留言会获得点赞!