一种氨基二硫代过酸硫酯类化合物及其制备方法和应用
本发明涉及药物合成领域,主要涉及一种氨基二硫代过酸硫酯类化合物及其制备方法和应用,具体涉及一种含氮环的氨基二硫代过酸硫酯类化合物及其制备方法和在医学上的应用。
背景技术:
恶病质是一种进行性消耗综合征,伴随消瘦、贫血、精神衰颓等全身机能衰竭的现象,多由癌症和其他严重慢性病(如慢性肺阻、慢性心力衰竭、艾滋病等)引起,其中由肿瘤引起的恶病质被称之为肿瘤恶病质。肿瘤恶病质(cancercachexia)是各种恶性肿瘤患者的主要并发症,是导致众多肿瘤患者死亡的主要原因。肿瘤恶病质的防治已成为恶性肿瘤多学科综合治疗的重要组成部分,正受到越来越多的关注。肿瘤恶病质是由肿瘤细胞产物及机体释放的细胞因子共同引起的以全身代谢紊乱、进行性肌肉及脂肪消耗、体重下降、全身脏器进行性衰竭为特征的一种消耗综合征,最主要的特征是骨骼肌萎缩所带来的病人体重的明显下降。在晚期肿瘤患者中,肿瘤恶病质的发生率高达50%-80%,其中乳腺癌和白血病患者发生率为40%,肺癌、结肠癌和前列腺癌患者发生率为50%,而胃癌和胰腺癌患者发生率高达80%,约20%的肿瘤患者直接死于由恶病质引起的心肺功能衰竭,故肿瘤恶病质是导致众多肿瘤患者死亡的主要原因。肿瘤恶病质的发生不仅削弱了化疗、放疗的效果,缩短生存期,同时也会严重影响患者的生存质量。
肿瘤恶病质发病机制十分复杂,缺乏有效的治疗手段,目前并没有抗肿瘤恶病质相关的药物上市,临床上只能采取加强患者营养摄入的方式延缓肿瘤恶病质的恶化。当前的临床治疗主要是通过营养支持、刺激食欲、抑制炎症因子与细胞因子等方法,以达到增加体重、刺激食欲、克服恶病质的目的。例如,omega-3脂肪酸作为营养补充剂被应用于临床研究头颈癌肿瘤恶病质过程中的体重保护,但对病人体重和生存期并无太大改善作用。刺激食欲的激素治疗一般常用孕激素如甲地孕酮和甲羟孕酮等,同时还可以合并给予胃饥饿素和omega-3脂肪酸等,这些姑息治疗的治疗方案在临床对肿瘤恶病质患者有一定的症状改善作用,但是还是难以达到非常有效的治疗目的。研究者也尝试了多种针对炎症细胞因子的抗体,包括il-1α、il-6、tnf-α、myostatin等的特异抗体,但是结果发现抑制其中一种因子不能完全阻止恶病质的发展。infliximab(tnfα的单克隆抗体)和clazakizumab(il-6的单克隆抗体),分别被应用于胰腺肿瘤恶病质与非小细胞肺癌肿瘤恶病质的临床治疗研究,对患者的体重减少、骨骼肌萎缩及生活质量并没有明显改善。此外,胃饥饿素受体激动剂、选择性雄性激素受体(ar)激动剂、肾上腺素β受体阻滞剂和anti-myostatin多肽等都是研究的热点。近期原本最有希望的处于临床研究阶段的新药是anamorelin(阿拉莫林,一种胃饥饿素受体激动剂)和enobosarm(一种选择性ar激动剂),但是目前该2种药物的iii期临床研究都不令人满意。
总之,由于肿瘤恶病质是各种恶性肿瘤患者的主要并发症,不仅削弱了化疗、放疗的效果,缩短生存期,同时也会严重影响患者的生存质量。虽然肿瘤恶病质治疗的重要性已经获得了共识,但由于肿瘤恶病质的发病机制尚有诸多不明之处,肿瘤恶病质的治疗方法和新药的研发尚无突破性的进展,临床治疗成效有限,目前尚没有任何一个药物被批准作为特异性针对肿瘤恶病质的药物上市运用。因此,研发具有自主知识产权的抗肿瘤恶病质新型药物,是肿瘤治疗迫切需要解决的关键科技问题,也必将带来巨大的社会效益和经济效益。
技术实现要素:
本发明解决的技术问题是:肿瘤恶病质的治疗方法和新药的研发尚无突破性的进展,临床治疗成效有限,目前尚没有任何一个药物被批准作为特异性针对肿瘤恶病质的药物上市运用。本发明设计合成研究具有抗肿瘤恶病质疗效的新型化合物,研发具有自主知识产权的抗肿瘤恶病质新型药物,解决肿瘤治疗迫切需要解决的关键科技问题。
本发明的目的是:提供口服给药具有良好抗恶病质作用的新型氨基二硫代过酸硫酯类化合物,所述化合物在不同组织中通过影响不同的信号通路来缓解肿瘤恶病质骨骼肌萎缩与脂肪降解过程,起到显著的抗肿瘤恶病质的作用。实验结果显示,所述的化合物口服具有良好的抗恶病质活性,可进一步制备新型的抗恶病质药物。
为解决上述技术问题,本发明提供一种含氮环的氨基二硫代过酸硫酯类化合物,可制备抗恶病质药物。本发明所述的恶病质为实体肿瘤诱发的恶病质。
具体来说,针对现有技术的不足,本发明提供了如下技术方案:
一种氨基二硫代过酸硫酯类化合物或其在药学上可接受的盐,其特征在于,所述化合物的结构式如通式i所示:
其中,m=1~11,优选的,m=1~9;x为含氮脂肪杂环,所述脂肪杂环中的氮原子与硫代羰基的碳原子相连。
优选的,上述氨基二硫代过酸硫酯类化合物或其在药学上可接受的盐中,m=3~5,优选的,m=5。
优选的,上述氨基二硫代过酸硫酯类化合物或其在药学上可接受的盐中,所述脂肪杂环中的杂原子包括氧原子。
优选的,上述氨基二硫代过酸硫酯类化合物或其在药学上可接受的盐中,所述氮原子的数量为1~2个。
优选的,上述氨基二硫代过酸硫酯类化合物或其在药学上可接受的盐中,所述含氮脂肪杂环选自饱和脂肪含氮单杂环,或包含饱和脂肪含氮单杂环的稠环或螺环类似物。
优选的,上述氨基二硫代过酸硫酯类化合物或其在药学上可接受的盐中,所述饱和脂肪含氮单杂环的成环原子数为4-7个,优选的,所述饱和脂肪含氮单杂环选自四元饱和脂肪含氮单杂环、五元饱和脂肪含氮单杂环或六元饱和脂肪单杂环。
优选的,上述氨基二硫代过酸硫酯类化合物或其在药学上可接受的盐中,所述饱和脂肪含氮单杂环的成环原子数为5-6个,最优选成环原子数为5个。
优选的,所述饱和脂肪含氮单杂环选自吡咯烷,取代吡咯烷,哌啶、吗啉、氮杂环丁烷或哌嗪,更优选的,为吡咯烷。
优选的,上述氨基二硫代过酸硫酯类化合物或其在药学上可接受的盐中,与饱和脂肪含氮单杂环连接形成稠环或螺环的环状化合物的成环原子数为4-6个;
优选的,与饱和脂肪含氮单杂环连接形成稠环或螺环的环状化合物选自下述任一种:
优选的,上述氨基二硫代过酸硫酯类化合物或其在药学上可接受的盐中,所述含氮脂肪杂环含有取代基,所述取代基优选为羟基或1-4碳的烷基。
优选的,所述饱和脂肪含氮单杂环含有取代基,所述取代基优选为羟基或1-4碳的烷基。
优选的,上述氨基二硫代过酸硫酯类化合物或其在药学上可接受的盐中,所述含氮脂肪杂环选自吡咯烷、取代吡咯烷、哌啶、吗啉、氮杂环丁烷、哌嗪、吲哚啉、异吲哚啉、全氢吲哚、全氢异吲哚或2-氧杂-6-氮杂-螺[3,4]辛烷。
优选的,所述取代吡咯烷的取代基为羟基或1-4碳的烷基。
优选的,所述含氮脂肪杂环选自吡咯烷、全氢异吲哚、氮杂环丁烷或2-氧杂-6-氮杂-螺[3,4]辛烷;更优选的,所述含氮脂肪杂环为吡咯烷。
优选的,上述氨基二硫代过酸硫酯类化合物或其在药学上可接受的盐选自下述任一种结构式的化合物或这些化合物的含氮脂肪杂环被取代基取代后的化合物:
优选的,所述取代基选自羟基或1-8碳的烷基,优选为羟基或1-4碳的烷基。
本发明还提供一种氨基二硫代过酸硫酯类化合物或其在药学上可接受的盐的制备方法,其特征在于,包括下述步骤:
(1)向含氮脂肪杂环化合物中加入式ii所述化合物,混合后再加入二硫化碳和三乙胺;
(2)加入四溴化碳,混合;
(3)反应后得到所述氨基二硫代过酸硫酯类化合物或其在药学上可接受的盐。
优选的,上述制备方法中,所述包含脂肪族杂环的化合物与式ii化合物的摩尔比为1:(0.5~1.5);
所述包含脂肪族杂环的化合物与二硫化碳的摩尔比为1:(0.5~1.5);
所述包含脂肪族杂环的化合物与三乙胺的摩尔比为1:(0.5~1.5);
所述包含脂肪族杂环的化合物与四溴化碳的摩尔比为1:(1.5~2.5)。
优选的,上述制备方法中,步骤(1)和步骤(2)所述过程在冰浴条件下进行,步骤(3)的反应过程为室温条件。
优选的,上述制备方法中,所述步骤(1)在有机溶剂中进行,所述有机溶剂优选为二氯甲烷或四氢呋喃。
优选的,所述制备方法中,步骤(3)反应后所得产物还需经过水洗涤、饱和氯化钠洗涤和用无水硫酸钠干燥的过程。
优选的,所述制备方法中,步骤(3)的反应时间为1-3小时;优选的,反应过程还包括搅拌的步骤,搅拌速率为800-1200rpm。
优选的,所述制备方法中,步骤(3)反应后还需经过柱层析的过程,柱层析所用淋洗剂包括石油醚和二氯甲烷,体积比为(1-10):1;或者淋洗剂包括石油醚和乙酸乙酯,体积比为(3-25):1。
本发明还提供一种组合物,其特征在于,包含上述氨基二硫代过酸硫酯类化合物或其在药学上可接受的盐,和药学上可接受的添加剂,优选的,所述添加剂包含药学上可接受的载体、稀释剂或赋形剂。
本发明还提供上述氨基二硫代过酸硫酯类化合物或其在药学上可接受的盐,或上述组合物在制备抗恶质病药物,尤其是抗肿瘤恶质病药物中的应用。
本发明中,所用术语“硫代羰基”为-cs-,结构式为:
本发明所用术语“二硫代过酸硫酯类化合物”含三个硫原子,结构式为:
本文中所用术语“环状化合物”指的是碳环化合物或杂环化合物,碳环化合物指的是脂肪族环状化合物或芳香族环状化合物,杂环化合物至包含杂原子的脂肪族杂环或芳香族杂环。
本文中所用术语“杂环”指分子中含有杂环结构的有机化合物,构成环的原子除碳原子外,还至少包含一个杂原子,如氮原子、硫原子、氧原子,所述杂环可以为一个环、两个环或多个环。
本文所用术语“脂肪族环状化合物”指的是链状烃及其衍生物通过分子内缩合形成的环状烃及其衍生物,包括饱和环烃(也称饱和脂肪环)或不饱和环烃(不饱和脂肪环),饱和环烃如环烷烃或环烯烃,优选的,为具有三到七个碳原子及零个杂原子的饱和环烃或不饱和环烃,饱和环烃例如环丙烷、环丁烷、环戊烷或环己烷等;不饱和环烃例如环丙烯、环丁烯、环戊烯、环己烯、环丁二烯、环戊二烯等。所述脂肪环可以为一个环、两个环或多个环。
本文所用术语“芳香族环状化合物”指的是拥有共轭的平面环体系,具有离域π电子,具有芳香性的分子,如苯、苯的同系物,含取代基的苯环,稠环芳烃等。
本文中所用术语“脂肪杂环”也可称为脂肪族杂环或脂杂环,指没有芳香性的一类杂环化合物,可以为一个环、两个环或多个环,原子间成键可以为单键,形成饱和脂肪族杂环,原子间成键也可以包含双键,形成不饱和脂肪族杂环。
与“脂肪族杂环”对应的为“芳香族杂环”,为拥有芳香性的杂环化合物,具体是指对完全共轭的、单环的、平面多双键物质来说,具有离域π电子的分子。
本文所用术语“杂环基”指的是杂环中的杂原子失去氢原子后形成的基团,例如权利要求1中与硫代碳基相连的x也可称为杂环基。
本文所用术语“杂原子”指的是氮、氧或硫原子。
本发明所用术语“螺环”指的是两个或两个以上碳环或杂环共用一个碳原子所形成的多环有机化合物。可以为苯环与苯环共用、苯环与杂环共用、苯环与脂肪环共用、杂环与杂环共用、杂环与脂肪环共用、脂肪环与脂肪环共用。
本发明所用术语“稠环”指的是两个或两个以上碳环或杂环以共有环边所形成的多环有机化合物。可以为苯环与苯环的稠合、苯环与杂环的稠和、苯环与脂肪环的稠合、杂环与杂环的稠合、杂环与脂肪环的稠合、脂肪环与脂肪环的稠合。
本发明所用术语“成环原子数”指的是单个环的原子数。
本发明所用术语“取代基”指的是羟基、羧基、卤素、氰基、烷基(优选为c1~c4烷基)、烷氧基、烯基、芳基、卤代烷基、卤代烷氧基、杂环基烷基、杂环基羧基、羟基烷基、硝基等。
本发明所用术语“杂环基烷基”指的是经一个、两个或三个杂环基取代的烷基。
本发明所用术语“杂环基羧基”指的是由羧基与母体分子部分连接的杂环基。
本发明所用术语“羟基烷基”指的是经一个、两个或三个羟基取代的烷基。
本发明所用术语“本发明的化合物”的含义包含i式化合物及其药学上可接受的对应异构体、非对应异构体及盐。
本发明的化合物可以药学上可接受的盐存在,可为水溶性、油溶性或可分散的盐胡两性离子的形式存在,所述“药学上可接受的”指的是患者接触或使用过程中无过度毒性、刺激性反应或并发症,具备预期的用途。
本发明所述组合物除i式化合物及其药学上可接受的盐以外,还可包括适当剂量的一种或两种以上药学上可接受的载体、稀释剂或赋形剂,形成药物制剂,患者接触或使用过程中无过度毒性、刺激性反应或并发症,具备预期的用途。
本发明所述药物制剂可以为片剂、胶囊、流体、粉末或气雾等形式,还可包括药用惰性载体如乙醇、甘油、水等,如需要,也可加入合适的粘结剂、矫味剂、防腐剂、分散剂或着色剂等。
本发明所述药物制剂可适用于任何可行的途径给药,给药方式包括经口、经直肠、经鼻、局部(包括口腔、舌下或经皮)等途径给药。
本发明所用术语“室温”指的是15~30℃。
如无特殊说明,本发明中所用百分数都为质量百分数。
本发明中各英文及缩写的含义如下:
tlc:薄层色谱法,pe:石油醚,ea:乙酸乙酯,et3n:三乙胺,dcm:二氯甲烷,chloroform-d:氯仿,1hnmr:氢谱,13cnmr:碳谱,fbs:胎牛血清,hs:马血清,dmem培养液:含氨基酸和葡萄糖的培养液,pbs缓冲液:磷酸缓冲盐溶液,dmso:二甲基亚砜,he染色:苏木精-伊红染色法。
本发明的优点是:本发明的化合物进行了体内外实验发现能够缓解肿瘤恶病质引起的肌萎缩和脂解,并且在动物实验中显示其能够明显缓解肿瘤恶病质引起的体重和摄食量下降,说明氨基二硫代过酸硫酯类化合物具有抗肿瘤恶病质的作用,能够应用于肿瘤恶病质及其相关病症的治疗,是一种很理想的肿瘤恶病质治疗药物。
附图说明
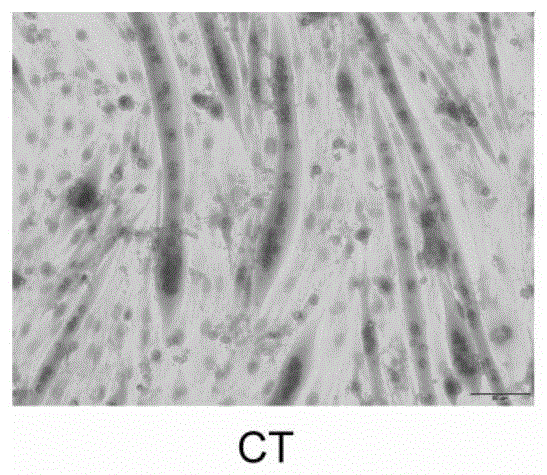
图1为实施例18中对照组的he染色图。
图2为实施例18中模型组的he染色图。
图3为实施例18中实验组1的he染色图。
图4为实施例18中实验组2的he染色图。
图5为实施例18中实验组3的he染色图。
图6为实施例18中实验组4的he染色图。
图7为实施例18中实验组5的he染色图。
图8为实施例18中实验组6的he染色图。
图9为实施例18中实验组7的he染色图。
图10为实施例18中实验组8的he染色图。
图11为实施例18中实验组9的he染色图。
图12为实施例18中实验组10的he染色图。
图13为实施例18中实验组11的he染色图。
图14为实施例18中实验组12的he染色图。
图15为实施例18中实验组13的he染色图。
图16为实施例18中实验组14的he染色图。
图17为实施例18中实验组15的he染色图。
图18为实施例18中实验组16的he染色图。
图19为实施例18中实验组17的he染色图。
图20为实施例18中实验组18的he染色图。
图21为实施例18中实验组19的he染色图。
图22为实施例18中实验组20的he染色图。
图23为实施例18中实验组21的he染色图。
图24为实施例18中实验组22的he染色图。
图25为实施例18中实验组23的he染色图。
图26为实施例18中实验组24的he染色图。
图27为实施例18中实验组25的he染色图。
图28为实施例18中实验组26的he染色图。
图29为实施例18中实验组27的he染色图。
图30为实施例18中实验组28的he染色图。
图31为实施例18中实验组29的he染色图。
图32为实施例18中实验组30的he染色图。
图33为实施例18中实验组31的he染色图。
图34为实施例18中实验组32的he染色图。
图35为实施例18中实验组33的he染色图。
图36为实施例18中实验组34的he染色图。
图37为实施例18中实验组35的he染色图。
图38为实施例18中实验组36的he染色图。
图39为实施例18中实验组37的he染色图。
图40为实施例18中实验组38的he染色图。
图41为实施例18中实验组39的he染色图。
图42为实施例18中实验组40的he染色图。
图43为实施例18中实验组41的he染色图。
图44为实施例18中实验组42的he染色图。
图45为实施例18中实验组43的he染色图。
图46为实施例18中实验组44的he染色图。
图47为实施例19中实验样品的甘油含量检测结果图。
图48为实施例20中小鼠的荷瘤体重变化曲线。
图49为实施例20中小鼠的去瘤体重变化曲线。
图50为实施例20中小鼠的肿瘤体积变化曲线。
图51为实施例20中小鼠的肿瘤实拍图。
图52为实施例20中小鼠的日均摄食量变化曲线。
图53为实施例20中小鼠的平均累积摄食量变化曲线。
图54为实施例20中小鼠的腓肠肌质量检测结果图。
图55为实施例20中小鼠的腓肠肌实拍图。
图56为实施例20中小鼠肌肉抓力检测结果图。
图57为实施例20中小鼠的附睾脂肪质量检测结果图。
图58为实施例20中小鼠的附睾脂肪实拍图。
具体实施方式
鉴于目前抗恶质病药物的性能还有待完善,本发明提供一种氨基二硫代过酸硫酯类化合物及其制备方法,以将其用于恶质病及相关病症的治疗。
一种优选的实施方式中,本发明提供具有良好抗恶病质作用的新型氨基二硫代过酸硫酯类化合物,具体涉及通式(i)结构的含氮环的氨基二硫代过酸硫酯类化合物:
其中:
m=1~11,优选的,m=1~9。
x为含氮脂肪杂环,以氮-碳键与硫代碳基碳原子相连。
优选地,其中x选自含氮四元脂肪环及其稠环或螺环类似物、含氮五元脂肪环及其稠环或螺环类似物、含氮六元脂肪环及其稠环或螺环类似物。
进一步优选地,x选自吡咯烷、取代吡咯烷、哌啶、吗啉、氮杂环丁环、哌嗪、吲哚啉、异吲哚啉、全氢吲哚、全氢异吲哚、2-氧杂-6-氮杂-螺[3,4]辛烷。
本发明中,化合物具有下述化合物1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17的结构:
本发明还提供所述的氨基二硫代过酸硫酯化合物在制备抗恶病质药物中的用途所述的氨基二硫代过酸硫酯化合物在制备抗恶病质药物中的用途。
本发明的另一目的是提供上述氨基二硫代过酸硫酯类化合物的制备方法,尤其涉及制备含氮环的氨基二硫代过酸硫酯类化合物的方法。
以化合物1为例,本发明的化合物的制备过程如下:
本发明中,所采用的药效学试验方法,是本领域技术人员所熟知的方法。
本发明中,所采用的c2c12细胞(小鼠成肌细胞)、3t3-l1细胞(小鼠脂肪细胞)和c26细胞(小鼠结肠癌细胞)是从中国科学院典型培养物保藏委员会细胞库购买获得。balb/c小鼠是通过上海灵畅生物科技有限公司公司购买获得的。
本发明中所用石油醚沸程为60~90℃,购自国药集团化学试剂有限公司。
fbs(胎牛血清),购自biologicalindustries。
horseserum(马血清),购自gibco。
高糖dmem培养基,购自hyclone。
rpmi-1640培养基,购自hyclone。
无酚红高糖dmem培养基,购自hyclone。
p/s双抗(青霉素-链霉素混合液)购自hyclone。
地塞米松,购自sigma–aldrich。
ibmx(3-异丁基-1-甲基黄嘌呤)广谱的磷酸二酯酶抑制剂,购自sigma–aldrich。
人重组胰岛素,购自上海今迈生物。
甘油检测试剂盒,购自北京普利莱基因技术有限公司。
含10%fbs的高糖dmem培养液,成分为:10%fbs+1%p/s+89%高糖dmem培养基。
含有10%fbs的1640培养液,成分为:10%fbs+1%p/s+89%rpmi-1640培养基。
2%hs分化液,成分为:2%hs+1%p/s+97%高糖dmem培养基。
在下面的实施例中,所用的各仪器的信息如下:
合成过程所用仪器:
旋转蒸发仪:厂家:buchi,型号:rotavaporr-200;
柱层析:所用硅胶(200-300目)和点样硅胶板购自国药集团化学试剂有限公司。
表征过程所用仪器:
荧光显微镜:厂家:olympus,型号:ix-73。1hnmr,13cnmr谱图所用核磁共振仪的厂家:美国varian,型号分别为varianmodelmercury400mhz,150mspectrometer。
下面通过具体实施例来进一步说明本发明所述氨基二硫代过酸硫酯类化合物及其制法和应用。
实施例1合成化合物1
冰浴条件下,在10ml无水二氯甲烷中加入吡咯烷(58μl,0.70mmol)和1-辛硫醇(122μl,0.70mmol),随后滴加二硫化碳(43μl,0.70mmol)和三乙胺(108μl,0.77mmol)。搅拌5min后加入四溴化碳(466mg,1.41mmol),撤去冰浴。室温搅拌反应2h后,tlc监测反应完全,其中,搅拌速率为1000rpm。反应液用水(3×10ml)洗涤,饱和氯化钠(2×10ml)洗涤,无水硫酸钠干燥,减压旋干溶剂。柱层析(pe:ch2cl2(体积比)=1:1)得白色固体121mg,产率:59.3%。
核磁表征结果为:1hnmr(400mhz,chloroform-d):δ3.97(t,j=7.0hz,2h),3.75(t,j=6.9hz,2h),2.86(t,j=7.4hz,2h),2.12(p,j=6.9hz,2h),2.00(p,j=6.9hz,2h),1.67(p,j=7.4hz,2h),1.39(p,j=6.9hz,2h),1.27(q,j=5.9,5.3hz,8h),0.88(t,j=6.6hz,3h)。
由此可确认该实施例所得到的白色固体化合物结构式如下所示:
实施例2:合成化合物2
冰浴条件下,在10ml无水二氯甲烷中加入吡咯烷(58μl,0.70mmol)和1-癸硫醇(146μl,0.70mmol),随后滴加二硫化碳(43μl,0.70mmol)和三乙胺(108μl,0.77mmol)。搅拌5min后加入四溴化碳(466mg,1.41mmol),撤去冰浴。室温搅拌2h后,tlc监测反应完全,其中,搅拌速率为1000rpm。反应液用水(3×10ml)洗涤,饱和氯化钠(2×10ml)洗涤,无水硫酸钠干燥,减压旋干溶剂。柱层析(pe:ch2cl2=1:1)得白色固体139mg,产率:62.1%。
核磁表征结果为:1hnmr(400mhz,chloroform-d):δ3.97(t,j=7.0hz,2h),3.75(t,j=6.9hz,2h),2.86(t,j=7.4hz,2h),2.12(p,j=6.8hz,2h),2.00(p,j=6.9hz,2h),1.68(q,j=7.3hz,2h),1.39(p,j=6.9hz,2h),1.27(d,j=5.8hz,12h),0.88(t,j=6.7hz,3h).13cnmr(150mhz,chloroform-d):δ193.02,56.64,50.53,38.69,31.89,29.54,29.49,29.31,29.21,28.63,28.58,26.51,24.20,22.68,14.12.
由此可确认该实施例所得到的白色固体化合物结构式如下所示:
实施例3:合成化合物3
冰浴条件下,在10ml无水二氯甲烷中加入吡咯烷(83mg,1.17mmol)和1-十一硫醇(219mg,1.17mmol),冰浴搅拌15分钟。滴加二硫化碳(71μl,1.17mmol)和三乙胺(178μl,1.29mmol)。搅拌5min后恒压滴入四溴化碳(776mg,2.34mmol)的二氯甲烷溶液(5ml),撤去冰浴,其中,搅拌速率为:1000rpm。室温反应2h,tlc监测反应完全。反应液用水(3×20ml),饱和氯化钠(10ml)洗涤,无水硫酸钠干燥,减压旋干溶剂。柱层析(ea:pe=1:25)得白色固体230mg,产率:59%。
核磁表征结果为:1hnmr(400mhz,chloroform-d)δ3.68(t,j=6.1hz,2h),3.39(t,j=6.0hz,2h),3.03(t,j=7.2hz,2h),1.90–1.77(m,2h),1.77–1.65(m,2h),1.51–1.37(m,2h),1.16(s,2h),1.00(s,14h),0.62(t,j=5.5hz,3h).13cnmr(151mhz,chloroform-d)δ192.86,54.20,49.92,35.98,31.28,28.97,28.88,28.70,28.59,28.36,28.23,25.40,23.66,22.05,13.48.
由此可确认该实施例所得到的白色固体化合物结构式如下所示:
实施例4:合成化合物4
冰浴条件下,在10ml无水二氯甲烷中加入吡咯烷(58μl,0.70mmol)和1-十二硫醇(168μl,0.70mmol),随后滴加二硫化碳(43μl,0.70mmol)和三乙胺(108μl,0.77mmol)。搅拌5min后加入四溴化碳(466mg,1.41mmol),撤去冰浴,其中,搅拌速率为:1000rpm。室温反应2h后,tlc监测反应完全。反应液用水(3×10ml)洗涤,饱和氯化钠(2×10ml)洗涤,无水硫酸钠干燥,减压旋干溶剂。柱层析(pe:ch2cl2=1:1)得白色固体141mg,产率:58.0%。
核磁表征结果为:1hnmr(400mhz,chloroform-d):δ3.97(t,j=7.0hz,2h),3.75(t,j=6.8hz,2h),2.86(t,j=7.4hz,2h),2.12(p,j=6.8hz,2h),2.00(p,j=6.6hz,2h),1.66(p,j=7.2hz,2h),1.38(d,j=7.8hz,2h),1.25(s,16h),0.88(t,j=6.6hz,3h).13cnmr(150mhz,chloroform-d):δ193.07,56.63,50.52,38.73,31.92,29.64,29.59,29.50,29.35,29.22,28.65,28.59,26.51,24.20,22.69,14.12.
由此可确认该实施例所得到的白色固体化合物结构式如下所示:
实施例5:合成化合物5
冰浴条件下,在10ml无水二氯甲烷中加入吡咯烷(83mg,1.17mmol)和1-十三硫醇(250mg,1.16mmol),冰浴搅拌(搅拌速率为:1000rpm)15分钟。滴加二硫化碳(71μl,1.17mmol)和三乙胺(178μl,1.29mmol)。搅拌5min后恒压滴入四溴化碳(776mg,2.34mmol)的二氯甲烷溶液(5ml),撤去冰浴,其中,搅拌速率为:1000rpm。室温反应2h,tlc监测反应完全。反应液用水(3×20ml),饱和氯化钠(10ml)洗涤,无水硫酸钠干燥,减压旋干溶剂。柱层析(ea:pe=1:25)得白色固体100mg,产率:27%。
核磁表征结果为:1hnmr(400mhz,chloroform-d)δ3.91(t,j=6.8hz,2h),3.62(t,j=6.8hz,2h),3.26(t,j=7.4hz,2h),2.11–2.00(m,2h),1.96(dd,j=13.5,6.7hz,2h),1.66(dd,j=14.7,7.2hz,2h),1.40(dd,j=14.8,8.5hz,2h),1.24(s,18h),0.85(t,j=6.3hz,3h).13cnmr(151mhz,chloroform-d)δ192.71,54.35,49.92,36.15,31.29,29.04,29.02,28.97,28.88,28.72,28.59,28.37,28.23,25.40,23.67,22.06,13.48.
由此可确认该实施例所得到的白色固体化合物结构式如下所示:
实施例6:合成化合物6
冰浴条件下,在10ml无水二氯甲烷中加入吡咯烷(58μl,0.70mmol)和1-十四硫醇(191μl,0.70mmol),随后滴加二硫化碳(43μl,0.70mmol)和三乙胺(108μl,0.77mmol)。搅拌5min后加入四溴化碳(466mg,1.41mmol),撤去冰浴,其中,搅拌速率为:1000rpm。室温反应2h后,tlc监测反应完全。反应液用水(3×10ml)洗涤,饱和氯化钠(2×10ml)洗涤,无水硫酸钠干燥,减压旋干溶剂。柱层析(pe:ch2cl2=1:1)得白色固体140mg,产率:53.1%。
核磁表征结果为:1hnmr(400mhz,chloroform-d):δ3.97(t,j=7.0hz,2h),3.75(t,j=6.9hz,2h),2.86(t,j=7.4hz,2h),2.12(p,j=6.9hz,2h),2.00(p,j=6.9hz,2h),1.68(q,j=7.4hz,2h),1.46–1.34(m,2h),1.26(s,20h),0.93–0.83(m,3h).13cnmr(150mhz,chloroform-d):δ192.99,56.63,50.53,38.69,31.93,29.69,29.65,29.58,29.49,29.36,29.21,28.64,28.57,26.51,24.20,22.69,14.13.
由此可确认该实施例所得到的白色固体化合物结构式如下所示:
实施例7:合成化合物7
冰浴条件下,在10ml无水二氯甲烷中加入吡咯烷(58μl,0.70mmol)和1-十六硫醇(216μl,0.70mmol),随后滴加二硫化碳(43μl,0.70mmol)和三乙胺(108μl,0.77mmol)。搅拌5min后加入四溴化碳(466mg,1.41mmol),撤去冰浴,其中,搅拌速率为:1000rpm。室温反应2h后,tlc监测反应完全。反应液用水(3×10ml)洗涤,饱和氯化钠(2×10ml)洗涤,无水硫酸钠干燥,减压旋干溶剂。柱层析(pe:ch2cl2=1:1)得白色固体161mg,产率:56.8%。
核磁表征结果为:1hnmr(400mhz,chloroform-d):δ3.97(t,j=7.0hz,2h),3.75(t,j=6.9hz,2h),2.86(t,j=7.4hz,2h),2.12(p,j=6.9hz,2h),2.00(p,j=6.9hz,2h),1.66(p,j=7.5hz,2h),1.40(d,j=7.4hz,2h),1.25(s,26h),0.88(t,j=6.5hz,3h).13cnmr(150mhz,chloroform-d):δ193.06,56.63,50.52,38.73,31.94,29.70,29.66,29.59,29.50,29.37,29.22,28.65,28.59,26.51,24.20,22.70,14.12.
由此可确认该实施例所得到的白色固体化合物结构式如下所示:
实施例8:合成化合物8
冰浴条件下,在10ml无水二氯甲烷中加入吡咯烷(58μl,0.70mmol)和1-十八硫醇(238μl,0.70mmol),随后滴加二硫化碳(43μl,0.70mmol)和三乙胺(108μl,0.77mmol)。搅拌5min后加入四溴化碳(466mg,1.41mmol),撤去冰浴,其中,搅拌速率为:1000rpm。室温反应2h后,tlc监测反应完全。反应液用水(3x10ml)洗涤,饱和氯化钠(2×10ml)洗涤,无水硫酸钠干燥,减压旋干溶剂。柱层析(pe:ch2cl2=1:1)得白色固体154mg,产率:50.9%。
核磁表征结果为:1hnmr(400mhz,chloroform-d):δ3.97(t,j=7.0hz,2h),3.75(t,j=6.9hz,2h),2.86(t,j=7.4hz,2h),2.12(p,j=6.8hz,2h),2.00(p,j=6.9hz,2h),1.66(p,j=7.4hz,2h),1.38(q,j=7.1hz,2h),1.25(s,28h),0.88(t,j=6.6hz,3h).13cnmr(150mhz,chloroform-d):δ193.06,56.63,50.52,38.73,31.94,29.71,29.67,29.59,29.50,29.37,29.33,29.22,28.65,28.59,26.51,24.20,22.70,14.12.
由此可确认该实施例所得到的白色固体化合物结构式如下所示:
实施例9:合成化合物9
冰浴条件下,在10ml无水二氯甲烷中加入异吲哚啉(132μl,1.17mmol)和1-十二硫醇(237mg,1.17mmol),冰浴搅拌15分钟。滴加二硫化碳(71μl,1.17mmol)和三乙胺(178μl,1.29mmol)。搅拌5min后恒压滴入四溴化碳(776mg,2.34mmol)的二氯甲烷溶液(5ml),撤去冰浴,其中,搅拌速率为:1000rpm。室温反应2h,tlc监测反应完全。反应液用水(3×20ml),饱和氯化钠(10ml)洗涤,无水硫酸钠干燥,减压旋干溶剂。柱层析(ea:pe=1:25)得白色固体270mg,产率:58%。
核磁表征结果为:1hnmr(400mhz,chloroform-d)δ7.38–7.26(m,4h),5.24(s,2h),5.07(s,2h),2.89(t,j=7.1hz,2h),1.79–1.63(m,2h),1.40(s,2h),1.25(s,16h),0.88(t,j=6.2hz,3h).13cnmr(151mhz,chloroform-d)δ193.52,134.19,127.38,122.11,61.32,54.98,38.05,31.29,29.02,29.01,28.96,28.87,28.72,28.58,28.05,27.95,22.07,13.50.
由此可确认该实施例所得到的白色固体化合物结构式如下所示:
实施例10:合成化合物10
冰浴条件下,在10ml无水二氯甲烷中加入吲哚啉(130μl,1.17mmol)和1-十二硫醇(237mg,1.17mmol),冰浴搅拌15分钟。滴加二硫化碳(71μl,1.17mmol)和三乙胺(178μl,1.29mmol)。搅拌5min后恒压滴入四溴化碳(776mg,2.34mmol)的二氯甲烷溶液(5ml),撤去冰浴,其中,搅拌速率为:1000rpm。室温反应2h,tlc监测反应完全。反应液用水(3×20ml),饱和氯化钠(10ml)洗涤,无水硫酸钠干燥,减压旋干溶剂。柱层析(ea:pe=1:25)得浅黄色油状产物125mg,产率:27%。
核磁表征结果为:1hnmr(400mhz,chloroform-d)δ7.12(t,j=7.4hz,1h),7.06(d,j=6.7hz,1h),6.91(d,j=7.6hz,1h),6.73(t,j=7.0hz,1h),3.70(t,j=8.4hz,2h),3.01(t,j=8.2hz,2h),2.69(t,j=7.3hz,2h),1.62–1.54(m,2h),1.38(s,2h),1.25(s,16h),0.88(s,3h).13cnmr(151mhz,chloroform-d)δ151.76,128.69,126.84,123.85,118.35,108.67,56.19,34.68,31.32,29.04,28.99,28.92,28.75,28.69,28.33,28.19,27.78,22.09,13.53.
由此可确认该实施例所得到的白色固体化合物结构式如下所示:
实施例11:合成化合物11
冰浴条件下,在10ml无水二氯甲烷中加入2-氧杂-6-氮杂-螺[3,4]辛烷(132mg,1.17mmol)和1-十二硫醇(237mg,1.17mmol),冰浴搅拌15分钟。滴加二硫化碳(71μl,1.17mmol)和三乙胺(178μl,1.29mmol)。搅拌5min后恒压滴入四溴化碳(776mg,2.34mmol)的二氯甲烷溶液(5ml),撤去冰浴,其中,搅拌速率为:1000rpm。室温反应2h,tlc监测反应完全。反应液用水(3×20ml),饱和氯化钠(10ml)洗涤,无水硫酸钠干燥,减压旋干溶剂。柱层析(ea:pe=1:3)得白色固体180mg,产率:40%。
核磁表征结果为:1hnmr(400mhz,chloroform-d)δ4.43(ddd,j=19.4,17.9,6.1hz,4h),3.96(s,1h),3.75(s,2h),3.55(t,j=6.7hz,1h),2.61(t,j=6.9hz,2h),2.19(t,j=6.8hz,1h),2.06(t,j=7.0hz,1h),1.42(s,2h),1.15(s,2h),1.01(s,16h),0.64(t,j=6.3hz,3h).13cnmr(151mhz,chloroform-d)δ193.52,79.55,63.48,57.81,54.22,48.50,45.78,43.13,38.04,35.42,33.17,31.17,29.00,28.94,28.85,28.71,28.57,28.03,27.92,22.05,13.49.
由此可确认该实施例所得到的白色固体化合物结构式如下所示:
实施例12:合成化合物12
冰浴条件下,在10ml无水二氯甲烷中加入顺式-全氢异吲哚(147mg,1.17mmol)和1-十二硫醇(237mg,1.17mmol),冰浴搅拌15分钟。滴加二硫化碳(71μl,1.17mmol)和三乙胺(178μl,1.29mmol)。搅拌5min后恒压滴入四溴化碳(776mg,2.34mmol)的二氯甲烷溶液(5ml),撤去冰浴,其中,搅拌速率为:1000rpm。室温反应2h,tlc监测反应完全。反应液用水(3×20ml),饱和氯化钠(10ml)洗涤,无水硫酸钠干燥,减压旋干溶剂。柱层析(ea:pe=1:25)得淡黄色油状产物360mg,产率:76%。
核磁表征结果为:1hnmr(400mhz,chloroform-d)δ3.66(dd,j=12.9,7.4hz,1h),3.56(dd,j=13.2,6.3hz,1h),3.50–3.39(m,1h),3.32(dd,j=11.1,5.7hz,1h),2.56(t,j=7.3hz,2h),2.08(ddd,j=33.1,11.8,5.9hz,2h),1.36(dd,j=14.4,7.5hz,4h),1.27–1.06(m,8h),0.95(s,16h),0.58(t,j=6.4hz,3h).13cnmr(151mhz,chloroform-d)δ193.54,59.76,53.96,38.06,37.53,35.14,31.30,29.03,29.01,28.96,28.88,28.73,28.59,28.03,27.95,25.07,24.92,22.07,21.98,21.70,13.51.
由此可确认该实施例所得到的白色固体化合物结构式如下所示:
实施例13:合成化合物13
冰浴条件下,在10ml无水二氯甲烷中加入3-羟基吡咯烷(174mg,2mmol)和十二硫醇(404mg,2mmol),随后滴加二硫化碳(152mg,2mmol)和三乙胺(204mg,2.2mmol)。搅拌5min后加入四溴化碳(1326mg,4mmol),撤去冰浴,其中,搅拌速率为:1000rpm。室温反应2h后,tlc监测反应完全。反应液用水(3×10ml)洗涤,饱和氯化钠(2×10ml)洗涤,无水硫酸钠干燥,减压旋干溶剂。柱层析(pe:ch2cl2=6:1)得白色固体150mg,产率:22%。
核磁表征结果为:1hnmr(400mhz,chloroform-d)δ4.60(s,1h),4.25–3.77(overlap,4h),2.85(t,j=7.1hz,2h),2.30–1.79(overlap,3h),1.72–1.61(m,2h),1.44–1.35(m,2h),1.25(s,16h),0.88(t,j=6.3hz,3h).13cnmr(151mhz,chloroform-d)δ194.12,71.41,68.99,64.58,54.18,48.39,38.73,31.92,29.64,29.59,29.50,29.35,29.22,28.67,28.58,22.69,14.12.
由此可确认该实施例所得到的白色固体化合物结构式如下所示:
实施例14:合成化合物14
冰浴条件下,在10ml无水二氯甲烷中加入哌啶(100mg,1.17mmol)和1-十二硫醇(237mg,1.17mmol),冰浴搅拌15分钟。滴加二硫化碳(71μl,1.17mmol)和三乙胺(178μl,1.29mmol)。搅拌5min后恒压滴入四溴化碳(776mg,2.34mmol)的二氯甲烷溶液(5ml),撤去冰浴,其中,搅拌速率为:1000rpm。室温反应2h,tlc监测反应完全。反应液用水(3×20ml),饱和氯化钠(10ml)洗涤,无水硫酸钠干燥,减压旋干溶剂。柱层析(ea:pe=1:25)得白色固体210mg,产率:50%。
核磁表征结果为:1hnmr(400mhz,chloroform-d)δ4.33(s,2h),3.99(s,2h),2.86(t,j=6.1hz,2h),1.89–1.58(m,8h),1.40(s,2h),1.27(s,16h),0.89(s,3h).13cnmr(151mhz,chloroform-d)δ195.98,54.55,51.21,38.16,31.27,29.00,28.98,28.94,28.85,28.70,28.56,27.94,25.62,24.86,23.55,22.04,13.48.
由此可确认该实施例所得到的白色固体化合物结构式如下所示:
实施例15:合成化合物15
冰浴条件下,在10ml无水二氯甲烷中加入吗啉(100mg,1.17mmol)和1-十二硫醇(237mg,1.17mmol),冰浴搅拌15分钟。滴加二硫化碳(71μl,1.17mmol)和三乙胺(178μl,1.29mmol)。搅拌5min后恒压滴入四溴化碳(776mg,2.34mmol)的二氯甲烷溶液(5ml),撤去冰浴,其中,搅拌速率为:1000rpm。室温反应2h,tlc监测反应完全。反应液用水(3×20ml),饱和氯化钠(10ml)洗涤,无水硫酸钠干燥,减压旋干溶剂。柱层析(ea:pe=1:25)得白色固体210mg,产率:50%。
核磁表征结果为:1hnmr(400mhz,chloroform-d)δ4.36(s,2h),4.07(s,2h),3.79(s,4h),2.86(t,j=7.2hz,2h),1.73–1.58(m,2h),1.40(s,2h),1.26(s,16h),0.89(t,j=6.4hz,3h).13cnmr(151mhz,chloroform-d)δ197.65,65.65,52.62,50.54,38.11,31.27,29.00,28.98,28.93,28.84,28.70,28.55,27.99,27.92,22.04,13.49.
由此可确认该实施例所得到的白色固体化合物结构式如下所示:
实施例16:合成化合物16
冰浴条件下,在10ml无水二氯甲烷中加入n-甲基哌嗪(180mg,1.8mmol)和十二硫醇(363mg,1.8mmol),随后滴加二硫化碳(139mg,1.8mmol)和三乙胺(200mg,1.98mmol)。搅拌5min后加入四溴化碳(1200mg,3.6mmol),撤去冰浴,其中,搅拌速率为:1000rpm。室温反应2h后,tlc监测反应完全。反应液用水(3×10ml)洗涤,饱和氯化钠(2×10ml)洗涤,无水硫酸钠干燥,减压旋干溶剂。柱层析(pe:ch2cl2=3:1)得白色固体270mg,产率:40%。
核磁表征结果为:1hnmr(400mhz,chloroform-d)δ4.21(brs,4h),2.85(t,j=7.2hz,2h),2.53(s,4h),2.34(s,3h),1.76–1.55(m,2h),1.39(s,2h),1.25(s,16h),0.87(d,j=6.8hz,3h).13cnmr(151mhz,chloroform-d)δ197.65,54.45,45.62,38.75,31.92,29.65,29.63,29.58,29.49,29.35,29.21,28.60,28.57,22.69,14.13.
由此可确认该实施例所得到的白色固体化合物结构式如下所示:
实施例17:合成化合物17
在10ml无水四氢呋喃溶液中加入氮杂环丁烷盐酸盐(187mg,2mmol)和氢氧化钾(112mg,2mmol),室温搅拌2h后吸出上层清液,加入10ml二氯甲烷中,并在冰浴条件下加入十二硫醇(0.48ml,2mmol)。随后滴加二硫化碳(0.12ml,2mmol)和三乙胺(0.31ml,2.2mmol)。搅拌5min后加入四溴化碳(1300mg,4mmol),撤去冰浴,其中,搅拌速率为:1000rpm。室温反应2h后,tlc监测反应完全。反应液用水(3×10ml)洗涤,饱和氯化钠(2×10ml)洗涤,无水硫酸钠干燥,减压旋干溶剂。柱层析(pe:ch2cl2=8:1)得浅黄色固体270mg,产率:40%。
核磁表征结果为:1hnmr(400mhz,chloroform-d)δ4.35(t,j=7.6hz,4h),2.83(t,j=7.5hz,2h),2.49–2.39(m,2h),1.72–1.62(m,2h),1.38(brs,2h),1.25(s,16h),0.88(s,3h).13cnmr(151mhz,chloroform-d)δ193.88,56.11,53.94,39.10,31.92,29.65,29.58,29.50,29.35,29.20,28.68,28.54,22.69,15.77,14.12.
由此可确认该实施例所得到的白色固体化合物结构式如下所示:
实施例18
氨基二硫代过酸硫酯类化合物缓解小鼠结肠癌细胞(c26)诱导的小鼠成肌细胞(c2c12)萎缩实验
采用直径测量法评价小鼠成肌细胞(c2c12)分化后的肌管直径。实验使用的染色方法为苏木精—伊红染色法,简称he染色法。苏木精染液为碱性,带正电荷,很容易以离子键结合细胞核中带负电荷且呈酸性的脱氧核糖核酸(dna)而染蓝色;伊红为酸性染料,在水中离解成带负电荷的阴离子,很容易与细胞质中蛋白质的氨基正电荷结合而染红色。染色后的细胞置于高倍显微镜下采图样(放大倍数为:400),利用imagej软件可统计肌管细胞直径。
通过以上方法可评价药物对细胞肌萎缩模型的作用。具体方法如下:
将c2c12细胞接种于24孔板中,培养体系为含有10%fbs的高糖dmem培养液,并加入1%的双抗,置于5%co2,37℃环境中。待细胞生长密度达到50%~60%时,将培养体系换成含有2%hs的高糖dmem培养液,并加入1%的双抗,2%hs分化液,每48h换液一次,第5或第6天分化成熟。另外,将c26细胞接种于t75瓶子中,培养体系为含有10%fbs的高糖dmem培养液,并加入1%的双抗,置于5%co2,37℃环境中。传代时600万细胞/瓶传代,20ml培养液培养48h后,取上清,1000rpm离心3min,取上清,4000rpm离心10min,取上清,即得到c26上清液。将c26上清和2%hs分化液1:1(体积比)混合,作为肌萎缩诱导液。除对照组加2%hs分化液以外,其他各组均加入等量的肌萎缩诱导液,一组作为模型组,其余组作为给药实验组。同时向细胞中按以下梯度浓度加入氨基二硫代过酸硫酯类化合物。
表1实施例18中成肌细胞(c2c12)萎缩实验的给药样品
其中,序号对应附图的编号,即,附图1-46对应表1中第1-46序号的样品。μm指的是μmol/l。
肌管直径测量方法如下:
作用48h后,用固定液(无水乙醇:甲醛:冰醋酸体积比=20:2:1)固定1h以上,利用苏木精—伊红染色法进行染色,并置于高倍显微镜下采集图像,利用imagej统计肌管直径。按如下公式计算肌萎缩逆转率:
肌萎缩逆转率=(给药组肌管平均值-模型组肌管平均值)/(对照组肌管平均值-模型组肌管平均值)×100%
结果与结论:
附图1-46是氨基二硫代过酸硫酯类化合物缓解c26细胞培液诱导的c2c12成熟肌管细胞萎缩的he染色代表性图片,附表2为肌管统计结果表。如图1-46和表2所示,氨基二硫代过酸硫酯类化合物对肌细胞萎缩有明显的逆转作用,且呈浓度依赖性关系。从结果中可以发现长链取代的二硫代过酸硫酯基本都具有较好的逆转肌细胞萎缩作用,但其活性与链长关联密切,其中化合物4效果最为显著,在无细胞毒性的12.5μm时,肌管萎缩逆转率达到92.83%。该化合物作为进一步研究开发的候选化合物。但当链长为18个碳原子(化合物8)时活性较低。
表2肌管统计结果表
实施例19
化合物4缓解3t3-l1脂肪细胞脂解的实验结果
采用甘油检测评价细胞内脂肪含量。甘油激酶将甘油磷酸化为3-磷酸甘油;3-磷酸甘油被甘油磷酸氧化酶氧化产生过氧化氢;在过氧化氢酶的作用下生色底物转化为苯醌亚胺,其光密度值和甘油浓度成正比。
通过以上方法可评价药物对细胞脂解模型的作用,方法如下:
将3t3-l1细胞接种于6孔板中,培养体系为含有10%fbs的高糖dmem培养液,并加入1%的双抗,置于5%co2,37℃环境中。待细胞长满后继续融合3天,开始分化操作。第一次分化的培养体系为含有0.5mmibmx、5mg/ml胰岛素、1μm地塞米松和10%fbs的高糖dmem培养液,并加入1%的双抗,分化72h;第二次分化的培养体系为含有5mg/ml胰岛素和10%fbs的高糖dmem培养液,并加入1%的双抗,分化72h;第三次分化的培养体系为含有10%fbs的高糖dmem培养液,并加入1%的双抗,分化72h,分化成功后明显可见细胞内含有大量油滴。另外,将c26细胞接种于t75瓶子中,培养体系为含有10%fbs的高糖dmem培养液,并加入1%的双抗,置于5%co2,37℃环境中。传代时600万细胞/瓶传代,向长满c26细胞的t75瓶子中加入15ml无酚红高糖dmem培养液培养48h后,取上清,1000rpm离心3min,取上清,4000rpm离心10min,取上清,得到c26上清液。将c26上清和无酚红高糖dmem培养液1:1混合,作为脂解诱导液。除对照组加无酚红高糖dmem培养液以外,其他各组均加入等量的脂解诱导液,一组作为模型组,其余组作为给药实验组。同时向细胞中按以下浓度12.5μm,25μm,50μm,100μm加入化合物4储备液。
表3实施例19中脂肪细胞脂解的给药样品
甘油检测方法如下:作用48h后,利用北京普利莱基因技术有限公司的甘油检测试剂盒检测上清中甘油的含量。
结果与结论:参照附图47。
附图47是和化合物4的甘油检测结果。如图47显示,化合物4呈浓度依赖性减少c26细胞培液造成的3t3-l1成熟脂肪细胞甘油释放。对照组、模型组、实验组1、实验组2、实验组3和实验组4的细胞甘油含量分别为287.44μm、441.95μm、379.44μm、347.14μm、313.67μm和310.95μm。
实施例20
化合物4治疗肿瘤恶病质动物模型实验结果,方法如下:
将c26细胞接种于t75培养瓶中,培养体系为含有10%fbs的1640培养液,并加入1%的双抗,置于5%co2,37℃环境中扩大培养。1000rpm,3min离心收细胞并用冰pbs缓冲液洗去培养液,并配制成1000万/ml的细胞悬液。细胞悬液接种于balb/c小鼠左右两侧腋下各100万保种。待肿瘤体积增加至800cm3时取出肿瘤,按3.5ml冰生理盐水/g匀浆得到肿瘤组织悬液。待接种小鼠按体重分组,细胞悬液接种于balb/c小鼠左侧腋下,接种量为100ul/只。接种后第二天即开始给药。化合物4先溶解于dmso中,再用预热的pbs溶液(37℃)混合溶解成均一稳定的溶液,终浓度为1mg/ml(3%dmso+2%无水乙醇+1%聚氧乙烯蓖麻油),给药量为5mg/kg,给药途径为灌胃口服(ig)。每天监测小鼠的体重、体温、肿瘤大小和摄食量。16天后模型组小鼠体重下10%左右,即认为进入恶病质晚期阶段。检测小鼠四肢肌肉抓力,脱颈处死小鼠后取得腓肠肌、附睾脂肪及肿瘤样本,称量样本重量。
其中,肌肉抓力的检测方法为:右手将小鼠平稳放在yls-13a大小鼠抓力测定仪上,使其前肢紧紧抓住抓盘,左手向前稳住抓盘。当左手慢慢放开抓盘时,右手立即向后缓慢拉动小鼠的尾巴直至前肢脱离抓盘。每只小鼠重复8次,以8次均值作为每只小鼠的骨骼肌力量指标。
结果与结论:参照附图48至58。图中三条曲线分别代表的是:健康组,c26肿瘤模型组和给药组(化合物4组,其中化合物4的给药剂量为5mg/kg)。
附图48至51是小鼠存活期间的荷瘤体重(图48),去瘤体重(图49),肿瘤体积(图50)和肿瘤实拍图(图51),其中,荷瘤体重和去瘤体重是8只小鼠的平均体重,肿瘤体积为8只小鼠肿瘤的平均体积,肿瘤实拍图为8只小鼠的肿瘤实拍图。如附图48所示,健康组小鼠体重在持续增长;而c26肿瘤模型组小鼠荷瘤体重在实验开始至第11天开始下降,至实验结束继续减少,如附图49所示,去瘤体重同样如此;而化合物4组能够明显缓解小鼠体重下降,至实验结束荷瘤体重和去瘤体重均高c26肿瘤模型组,差异均有统计学意义(p<0.01)。但是,如附图50和51所示,化合物4组与c26肿瘤模型组相比,肿瘤体积略有减小,但差异不是很大,说明化合物4对c26肿瘤无明显抑制作用。
附图52至53是小鼠存活期间的日均摄食量和平均累积摄食量,为8个小鼠的平均计算结果。c26肿瘤模型组小鼠的摄食量较健康组出现了明显的下降,化合物4组摄食量高于c26肿瘤模型组,可改善食欲。
附图54至55是小鼠腓肠肌质量,腓肠肌实拍图,所述腓肠肌质量是8个小鼠的平均值,腓肠肌实拍图为8个小鼠/组的结果。如图54、55所示,c26肿瘤模型组小鼠腓肠肌质量显著小于健康组,且差异有统计学意义(p<0.001),化合物4可明显缓解腓肠肌萎缩(p<0.05)。附图56为小鼠四肢肌肉抓力,c26肿瘤模型组小鼠肌肉抓力显著小于健康组和化合物4组,且差异有统计学意义(p<0.001),化合物4可明显增强肌肉抓力(p<0.05)。
附图57至58是小鼠附睾脂肪质量,附睾脂肪实拍图。如图所示,c26肿瘤模型组小鼠附睾脂肪质量显著小于健康组和化合物4组,且差异有统计学意义(p<0.01),化合物4可明显增加附睾脂肪质量(p<0.05)。
以上结果说明,化合物4能在不影响肿瘤大小的情况下缓解肿瘤恶病质导致的体重下降、肌肉萎缩、脂肪降解和体温下降,并改善食欲。
以上所述是本发明的优选实施方式,需要指出的是,对于本发明所涵盖技术领域的普通技术人员,在不脱离本发明方法的前提下,还可以做出若干补充和改善,这些补充和改善也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!