一种高选择性富集镅的材料及方法与流程
[0001]
本发明提供一种环境样品中次锕系核素的回收方法,具体涉及从含有示踪量镅的溶液中高选择性富集镅的方法及材料。
背景技术:
[0002]
核能开发、核试验、极端核事故等不可避免地向环境中释放次锕系核素,而这些核素最终将进入生物圈,对人类健康构成威胁。次锕系核素不仅具有化学毒性,还具有放射性毒性,尤其是镅(am),进入体内后的内照射会对人造成持续的、不可逆转的辐射损伤。环境中微量的am即会给人的生存构成威胁。因此,有效地富集环境样品中的am是十分重要的。
[0003]
如何简化次锕系核素分离流程,提高分离效率,降低环境污染,成为人们关注的焦点。目前,固相萃取由于操作简单方便、适用环境广而受到关注。对于固相萃取,最重要的就是要合成对目标离子具有特异性识别能力的吸附材料
[1]
。离子印迹聚合物(iip)作为一种特异性的固相萃取材料,广泛用于分离和富集微量金属离子
[2-4]
。然而,iip尚未用于次锕系离子的吸附分离中,这主要是因为次锕系离子具有强放射性、高毒性,且不易获得纯品,无法用于iip的合成。
[0004]
替代模板法在分子印迹聚合物(mip)的制备中已经得到了广泛的应用
[5]
。1997年,andersson等
[6]
首次采用沙美利定类似物作为替代模板制备了mip之后,替代模板法越来越多地被用在mip的制备当中。主要集中在以下两种情况:a)模板分子太贵或者太危险,例如以三硝基苯酚作为三硝基甲苯的替代模板
[7]
,以n-壬酸香草酰胺作为辣椒素的替代模板
[8]
。b)模板分子在聚合过程中难以溶解或者易分解
[9]
。chang等
[10]
采用二羟基联苯作为替代模板合成了mip,并用于双酚a的分离和检测。离子印迹技术是分子印迹技术的一个分支,但替代模板法在iip的制备中还鲜有应用。
技术实现要素:
[0005]
针对目前存在的环境样品中放射性am难以回收处理的问题,本发明提供了一种能够对溶液中的示踪量am(~10-8
mol
·
l-1
)进行富集的材料及方法。鉴于三价镧系离子(ln
3+
)与三价锕系离子(an
3+
)具有非常相似的性质,本发明方法选用ln
3+
作为an
3+
的替代模板,利用替代模板法制备离子印迹聚合物,作为功能化吸附材料用于镅的富集,并联用电感耦合等离子体原子发射光谱法/质谱法(icp-aes/ms)进行离子浓度测定。
[0006]
本发明首先提供了一种用于富集镅的材料,该材料是通过下述方法制备得到的离子印迹聚合物(iip):
[0007]
1)合成膦酸类单体2-(二(4-乙烯基苄基)磷酰基)乙酸(bvbpac);
[0008]
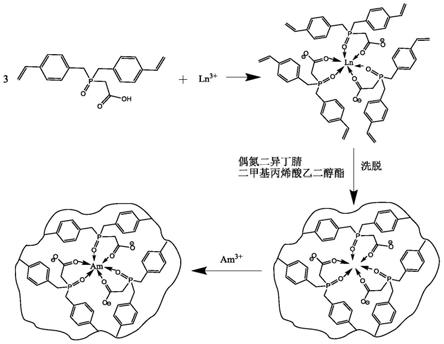
2)将单体bvbpac溶于有机溶剂中,加入有机碱和镧系金属离子化合物,搅拌反应;
[0009]
3)向步骤2)的混合液体中加入交联剂和引发剂,在惰性气氛保护下加热交联聚合,得到固体凝胶;
[0010]
4)将步骤3)聚合后的固体凝胶研磨筛分,洗涤去除镧系金属离子,干燥,得到iip。
[0011]
上述步骤1)合成膦酸类单体bvbpac的方法如下:
[0012][0013]
首先,4-乙烯基苄氯和镁反应合成格式试剂,然后加入亚磷酸二乙酯反应得到二-(4-乙烯基苄基)亚磷酸;将二-(4-乙烯基苄基)亚磷酸在碱性条件下与氯乙酸钠反应得到2-(二(4-乙烯基苄基)磷酰基)乙酸,反应温度优选为45~60℃。
[0014]
上述步骤2)中,所述镧系金属离子化合物中的镧系金属离子优选为la
3+
、nd
3+
、eu
3+
、lu
3+
;所述镧系金属离子化合物例如镧系金属的硝酸盐la(no3)3·
6h2o、nd(no3)3·
6h2o、eu(no3)3·
6h2o、lu(no3)3·
6h2o等。单体bvbpac跟作为模板的镧系金属离子的物质的量的比优选为3:1。
[0015]
上述步骤2)中,所述有机溶剂为极性有机溶剂,例如二甲基亚砜(dmso)。
[0016]
上述步骤2)中,所述有机碱例如三乙胺(et3n)、二甲胺、二乙胺等;单体bvbpac与有机碱的物质的量的比优选为1:1。
[0017]
上述步骤2)通常在室温下搅拌反应30min以上。
[0018]
上述步骤3)中,所述交联剂例如二甲基丙烯酸乙二醇酯(egdma)、二乙烯基苯(dvb)、三乙二醇二甲基丙烯酸酯(tegdan)、三烯丙基异三聚氰酸酯(tric)等,所述引发剂例如偶氮二异丁腈(aibn)、过氧化二苯甲酰(bpo)、过硫酸盐等。
[0019]
上述步骤3)中,交联聚合的温度优选为60~70℃,时间优选为18~24h。
[0020]
上述步骤4)中,研磨筛分优选粒子尺寸在200目左右;优选用0.1~0.5mol
·
l-1
的hcl溶液进行洗涤,洗涤次数优选为3~4次,每次2h左右,反复更换hcl溶液直到洗脱液中的镧系金属离子含量低于0.1mg
·
l-1
。
[0021]
上述方法以镧系金属离子为模板合成iip,该iip可以用于富集示踪量的镅。将该iip加入含镅的溶液中,振荡一段时间,即可对镅实现高效、高选择性的吸附,能够在大量金属离子干扰情况下对示踪量am
3+
(~10-8
mol
·
l-1
)进行富集回收。进一步的,优选在ph为弱酸性(ph=5~6)的条件下,于25℃振荡4h以上。
[0022]
本发明提供的从水溶液中富集示踪量镅的材料与方法相比于现有技术,其技术优势体现在:合成了新型膦酸类功能单体bvbpac,克服了离子印迹技术对于特殊模板的限制,使用替代模板法制备的iip实现了对镅的高效、高选择性富集,提高了吸附效率,节省了实验时间。此外,与icp-aes/ms联用,还实现了对金属离子浓度的快速准确检测。
附图说明
[0023]
图1是以bvbpac为功能单体制备ln-iip的过程图。
[0024]
图2是镧系离子印迹聚合物(iip)和非印迹聚合物(nip)洗脱模板离子前后的红外光谱图,其中:(a).la-iip和nip;(b).nd-iip和nip;(c).eu-iip和nip;(d).lu-iip和nip。
[0025]
图3是镧系离子印迹聚合物(iip)和非印迹聚合物(nip)透射电子显微镜图,其中:(a)la-iip,(b)nd-iip,(c)eu-iip,(d)lu-iip,(e)nip。
[0026]
图4是bvbpac与la
3+
,nd
3+
,eu
3+
,lu
3+
配位前后的
31
p nmr图。
[0027]
图5是bvbpac与la
3+
,nd
3+
,eu
3+
,lu
3+
配位前后的ft-ir图。
[0028]
图6是la-iip和nip对la
3+
的吸附动力学曲线,其中吸附剂:5.0mg;la
3+
浓度:20mg
·
l-1
;溶液体积:10.00ml;溶液ph:5.5。
[0029]
图7是la-iip和nip对la
3+
的吸附等温曲线,其中吸附剂:5.0mg;溶液体积:10.00ml;溶液ph:5.5;吸附时间:12h。
[0030]
图8是溶液ph对la-iip和nip吸附容量的影响,其中吸附剂:5.0mg;la
3+
浓度:20mg
·
l-1
;溶液体积:10.00ml;吸附时间:12h。
具体实施方式
[0031]
下面通过具体实验对本发明进行详细说明,但本发明并不局限于此。
[0032]
实施例一
[0033]
1.功能单体的合成及表征
[0034]
膦酸类单体2-(二(4-乙烯基苄基)磷酰基)乙酸(bvbpac)的合成路线如下:
[0035][0036]
二(4-乙烯基苄基)亚磷酸的合成
[0037]
在三口瓶中加入0.93g镁屑,缓慢滴加20ml溶有6.08g(40mmol)4-乙烯基苄氯的干燥乙醚,使体系保持微沸。滴加结束后继续在室温下搅拌1h,除去瓶中残留的镁屑。将20ml溶有2.1g(15mmol)亚磷酸二乙酯的乙醚滴加到上面合成的格氏试剂中,搅拌20min。将反应混合物倒入100ml稀盐酸中,搅拌20min。抽滤,滤液用乙醚萃取两次,将萃取液旋干后与滤渣合并。乙醚重结晶后得1.4g产物,产率33%。
[0038]
2-(二(4-乙烯基苄基)磷酰基)乙酸的合成
[0039]
将1.4g(5mmol)二-(4-乙烯基苄基)亚磷酸溶于10ml n,n-二甲基甲酰胺(dmf),加入1.1g(9mmol)氯乙酸钠,搅拌下加入0.52g(13mmol)naoh和0.52ml水的混合物,50℃反应1h。将反应混合物倒入100ml水中,搅拌20min,抽滤。滤液用盐酸酸化到ph<1,析出大量白色
固体,在冰箱中静置30min。抽滤,真空干燥得0.97g产物,产率57%。
[0040]
2-(二(4-乙烯基苄基)磷酰基)乙酸的结构表征
[0041]1h nmr(400mhz,dmso-d6):δ7.41(d,j=9.4hz,4h),7.23(d,j=7.5hz,4h),6.72(dd,j=17.6,11.0hz,2h),5.81(d,j=17.6hz,2h),5.24(d,j=10.8hz,2h),3.42-3.15(m,4h),2.73(d,j=14.0hz,2h);
[0042]
31
p nmr(400mhz,dmso-d6):δ37.36;
[0043]
元素分析:c 70.54%,h 6.24%(理论值:c 70.58%,h 6.22%);
[0044]
质谱:分子离子峰的m/z为339.1(m-h
+
)。
[0045]
2.iip,nip的合成及表征
[0046]
以bvbpac为功能单体制备ln-iip的过程见图1。在2.5ml的二甲基亚砜(dmso)溶液中,加入0.17g(0.5mmol)bvbpac和69.6μl et3n(0.5mmol),搅拌0.5h。加入0.25mmol la(no3)3·
6h2o,搅拌2h。然后加入0.755ml egdma(4mmol)和10mg aibn,通氮气20min。封管,油浴60℃加热18h,体系聚合为一块固体。取出后研磨,la-iip加入到100ml 0.1mol
·
l-1
的hcl溶液中搅拌洗涤,反复更换hcl溶液直到洗脱液中的la
3+
含量<0.1mg
·
l-1
。洗脱好的聚合物用水和丙酮反复洗涤,烘干,研磨筛分得到200目左右的颗粒,即la-iip。同样的方法合成nd-iip、eu-iip和lu-iip,只是将其中的la(no3)2·
6h2o分别更换为nd(no3)3·
6h2o、eu(no3)3·
6h2o、lu(no3)3·
6h2o。合成nip时,除了不加la(no3)2·
6h2o外,其余配方和操作与iip相同。
[0047]
傅里叶变换红外光谱(ft-ir)表征:
[0048]
对洗脱前后的iip,nip进行红外光谱表征,如图2所示。
[0049]
iip和nip的傅里叶变换红外光谱图非常相近,这是由于它们有着完全相同的组成,见图2中(a)。谱图中均有位于1726cm-1
的c=o伸缩振动峰和位于1163cm-1
附近的p=o伸缩振动峰。这表明功能单体bvbpac和交联剂egdma成功共聚。
[0050]
透射电子显微镜(tem)图像:
[0051]
将la-iip,nd-iip,eu-iip,lu-iip四种印迹聚合物和nip分别稀释在乙醇溶液中,超声分散,将其负载在铜网上面,做tem表征,如图3所示。
[0052]
由图3(a)可以看到,四种iip的tem图像均有明显的空穴分布,孔道结构复杂。而nip则没有明显的孔道结构,这表明iip成功地被模板离子印迹。iip相对nip有更复杂的孔道结构,便于传质,更有利于吸附金属离子。
[0053]
la
3+
与bvbpac配位的研究:
[0054]
31
p nmr实验:以氘代dmso为溶剂,通过
31
p nmr来研究四种镧系金属离子la
3+
与bvbpac的配位(图4)。如图4所示,在氘代dmso中,bvbpac中p=o基团中的p的化学位移为37.36ppm。在加入et3n后,化学位移向高场移动至39.91ppm,这可能是由于p=o与质子化的三乙胺上的h形成了氢键,使得o原子的电荷密度降低,从而诱导p原子的电荷密度降低。当体系中加入la
3+
,nd
3+
,eu
3+
,lu
3+
后,p的化学位移进一步位移至47.17ppm,45.00ppm,47.17ppm,51.76ppm,48.29ppm。这表明p=o与la
3+
,nd
3+
,eu
3+
,lu
3+
形成了配位键,使得o原子电荷密度进一步降低,而p原子上的电荷密度也随之降低。
[0055]
ft-ir实验:将功能单体bvbpac和印迹聚合物做红外光谱表征(图5)
[0056]
如图5所示,形成印迹聚合物之后,与bvbpac相比,位于1708cm-1
处的羧基c=o的伸
缩振动峰蓝移到1726cm-1
,且在1438cm-1
和1635cm-1
处出现了羧基的对称伸缩振动和不对称伸缩振动峰,两者间的差距为197cm-1
,说明c=o参与了配位。
[0057]
3.iip,nip的吸附实验
[0058]
研究离子印迹技术的参数:吸附容量(q),吸附效率(e)。
[0059][0060][0061]
其中,c0、c
e
分别吸附前后溶液中金属离子的浓度,v为吸附母液体积,m为吸附材料质量。
[0062]
(1)吸附动力学
[0063]
通过时间对吸附容量的影响(图6)可以看到,la-iip和nip的吸附平衡时间大约为4h。为了保证吸附平衡,后续实验的吸附时间均采用12h。
[0064]
为了研究吸附机理,我们采用拟一级动力学模型(式3)和拟二级动力学模型(式4)来对实验数据进行拟合。
[0065][0066][0067]
其中,q
e
(μmol
·
g-1
)和q
t
(μmol
·
g-1
)分别为平衡吸附容量和t(h)时刻下的吸附容量,k1(h-1
)和k2(μmol
·
g-1
·
h-1
)分别为拟一级动力学模型和拟二级动力学模型的吸附速率常数。从表1可以看到,iip和nip的吸附动力学曲线都更符合拟二级动力学模型。拟一级动力学模型假设吸附位点被占据的速率和未被占据位点的数量成正比;而拟二级动力学模型对应的是化学反应的机理
[11]
。这表明iip和nip的吸附过程都更接近于化学吸附,在吸附过程中识别位点的配位基团与la
3+
的作用是关键步骤。
[0068]
表1.吸附动力学拟合结果
[0069][0070]
注:q
e
(exp.)为实验测得的平衡吸附容量,q
e
(cal.)为计算得到的平衡吸附容量。
[0071]
(2)吸附等温曲线
[0072]
iip和nip的吸附容量随着平衡浓度的增加而增加(图7)。吸附等温数据用langmuir(式5)和freundlich(式6)两种模型来进行拟合。
[0073][0074][0075]
其中,c
e
(μmol
·
l-1
)是平衡浓度,q
m
(μmol
·
g-1
)是最大吸附容量,b(l
·
μmol-1
)是
langmuir常数,k
f
和n是freundlich常数。
[0076]
从图7和表2中可以看到,langmuir模型能够更好地拟合la-iip和nip的吸附数据。la-iip通过langmuir模型计算出的最大吸附容量为155.9μmol
·
g-1
,nip通过langmuir模型计算出的最大吸附容量为141.8μmol
·
g-1
。从图7可以看到,在相同的平衡浓度下,la-iip的吸附容量要明显大于nip,一方面是由于iip的印迹效应使得其中的位点对la
3+
的结合能力更强。另一方面则是因为在iip中,同时存在着特异性识别位点和非特异性结合位点,而nip中则只有非特异性结合位点。
[0077]
表2.langmuir和freundlich模型拟合结果
[0078][0079]
(3)ph对iip,nip吸附的影响
[0080]
实验首先研究了ph对la-iip吸附的影响。通过改变吸附母液的酸度,利用静态吸附法研究了ph值对于iip(nip)吸附容量的影响。在15ml离心管中,加入5ml吸附剂iip(nip),10ml不同ph值的吸附溶液,振荡12h,以4000rpm的转速离心10min,吸取上清液5ml进行测量。
[0081]
从图8可以看出,iip(nip)的吸附容量随着ph值的增大而增大。在ph≤2时,iip和nip均对la
3+
的吸附容量很低,这是由于识别位点中的羧酸基团被质子化而无法有效地与la
3+
配位。且此时h
+
会和la
3+
竞争磷酸基团上的活性位点,使得吸附容量进一步降低。在ph>2之后,iip的吸附容量要明显高于nip,这说明iip中的识别位点对la
3+
的结合能力要强于nip。由于la
3+
在ph>6之后就开始水解,生成氢氧化物吸附在iip(nip)上,在离心后被沉淀下来,这也使得该ph下iip和nip的吸附差别并不明显。为了保证母液中金属离子不被水解,故在后续实验中,对ph为5.5的母液进行吸附研究。
[0082]
(4)在大量金属离子存在下选择性提取示踪量am
3+
[0083]
之后利用这种替代模板法合成的la-iip去吸附示踪量的am
3+
,考察在大量金属离子干扰情况下对示踪量am
3+
(~10-8
mol
·
l-1
)的回收。选取的干扰离子m
n+
为cu
2+
、zn
2+
、ni
2+
、ca
2+
、mg
2+
、na
+
、k
+
,质量浓度比m
n+
:am
3+
=2000:1。结果发现,即便是在大量碱金属、碱土金属离子以及过渡金属离子存在的情况下,la-iip对示踪量am
3+
的吸附效率仍旧达到了97%,且对质量浓度为目标离子2000倍的干扰离子ni
2+
、ca
2+
、mg
2+
、na
+
、k
+
均完全不吸附,对cu
2+
、zn
2+
也只是有很微弱的吸附。
[0084]
实施例二
[0085]
以镧系离子nd
3+
作为am
3+
的替代模板,同样是以bvbpac为功能单体,利用上述凝胶-溶胶法合成nd-iip。接下来的配方和操作与la-iip的制备基本相同。之后对nd-iip(nip)进行结构表征,ft-ir表征见图2中(b),tem表征见图3中(b)。对其配位机理进行研究,见图4和图5。
[0086]
实施例三
[0087]
以同族镧系离子eu
3+
作为am
3+
的替代模板,以bvbpac为功能单体,利用凝胶-溶胶法合成eu-iip。接下里的配方和操作也是参考la-iip。之后对eu-iip(nip)进行结构表征,
ft-ir表征见图2中(c),tem表征见图3中(c)。对其配位机理进行研究,见图4和图5。
[0088]
实施例四
[0089]
以重镧系离子lu
3+
作为am
3+
的替代模板,以bvbpac为功能单体,利用凝胶-溶胶法合成lu-iip。接下里的配方和操作同样参考la-iip。之后对lu-iip(nip)进行结构表征,ft-ir谱图见图2中(d),tem谱图见图3中(d)。对其配位机理进行研究,见图4和图5。
[0090]
参考文献:
[0091]
[1]wan ibrahim,w.a.;abd ali,l.i.;sulaiman,a.;sanagi,m.m.;aboul-enein,h.y.,application of solid-phase extraction for trace elements in environmental and biological samples:a review.crit.rev.anal.chem.2014,44(3),233-254.
[0092]
[2]shakerian,f.;kim,k.-h.;kwon,e.;szulejko,j.e.;kumar,p.;dadfarnia,s.;haji shabani,a.m.,advanced polymeric materials:synthesis and analytical application of ion imprinted polymers as selective sorbents for solid phase extraction of metal ions.trac-trends anal.chem.2016,83,55-69.
[0093]
[3]liang,h.;chen,q.;ma,j.;huang,y.;shen,x.,synthesis and characterization of a new ion-imprinted polymer for the selective separation of thorium(iv)ions at high acidity.rsc advances 2017,7(56),35394-35402.
[0094]
[4]zhang,h.;liang,h.;chen,q.;shen,x.,synthesis of a new ionic imprinted polymer for the extraction of uranium from seawater.j.radioanal.nucl.chem.2013,298(3),1705-1712.
[0095]
[5]chen,l.;wang,x.;lu,w.;wu,x.;li,j.,molecular imprinting:perspectives and applications.chem.soc.rev.2016,45(8),2137-211.
[0096]
[6]andersson,l.i.;paprica,a.;arvidsson,t.,a highlyselective solid phase extraction sorbent for pre-concentration of sameridine made by molecular imprinting.chromatographia 1997,46(1/2),57-62.
[0097]
[7]xu,s.;lu,h.;li,j.;song,x.;wang,a.;chen,l.;han,s.,dummy molecularly imprinted polymers-capped cdte quantum dots for the fluorescent sensing of 2,4,6-trinitrotoluene.acs appl.mater.interfaces 2013,5(16),8146-54.
[0098]
[8]ma,x.;ji,w.;chen,l.;wang,x.;liu,j.;wang,x.,molecularly imprinted polymers with synthetic dummy templates for the preparation of capsaicin and dihydrocapsaicin from chili peppers.j.sep.sci.2015,38(1),100-7.
[0099]
[9]quaglia,m.;chenon,k.;hall,a.j.;de lorenzi,e.;sellergren,b.,target analogue imprinted polymers with affinity for folic acid and related compounds.j.am.chem.soc.2001,123(10),2146-2154.
[0100]
[10]chang,t.;yan,x.;liu,s.;liu,y.,magnetic dummy template silica sol
–
gel molecularly imprinted polymer nanospheres as magnetic solid-phase extraction material for the selective and sensitive determination of bisphenol a in plastic bottled beverages.food anal.meth.2017,10(12),3980-3990.
[0101]
[11]iftikhar,a.r.;bhatti,h.n.;hanif,m.a.;nadeem,r.,kinetic and thermodynamic aspects of cu(ii)and cr(iii)removal from aqueous solutions using rose waste biomass.j.hazard.mater.2009,161(2-3),941-7.
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1