一种手性1-叔丁基-3-甲基-6-甲基哌嗪-1,3-二甲酸酯的制备方法与流程
[0001]
本发明属于有机化合物合成技术领域,具体涉及一种手性1-叔丁基-3-甲基-6-甲基哌嗪-1,3-二甲酸酯的制备方法。
背景技术:
[0002]
哌嗪类化合物是杂环化合物中重要的含氮杂环,哌嗪衍生物具有抗抑郁、消炎止痛、抗肿瘤等活性而受到重视,另外,哌嗪环的1,4-位是碱性基团,具有较好的水溶性和碱性,因此将其引入药物分子中,能增加药物的水溶性,有效地调节药物的酸碱平衡,从而增强药物分子在动植物体内的生物活性。
[0003]
cn110183471a公开了一种新型哌嗪类衍生物及制备方法及应用,该化合物为(r)-6-甲基-3-(4-(异丙基磺酰基)苯基)-6-氯-1h-吡咯并[2,3-b]吡啶)-4,5,6,7-四氢吡唑并[1,5-a]哌嗪,该技术以1((r)-2-(苄基氨基)丙-1-醇)和2(3-醛基-4-溴吡唑)为起始原料,制备得到的新型哌嗪类衍生物能有效抑制人乳腺癌细胞的增殖,并且与阿霉素联合使用具有较好的协同抗人乳腺癌细胞的作用,且该化合物制备工艺条件较温和,克服了哌嗪类化合物合成条件苛刻、产率较低、不适合工业化生产的缺点。
[0004]
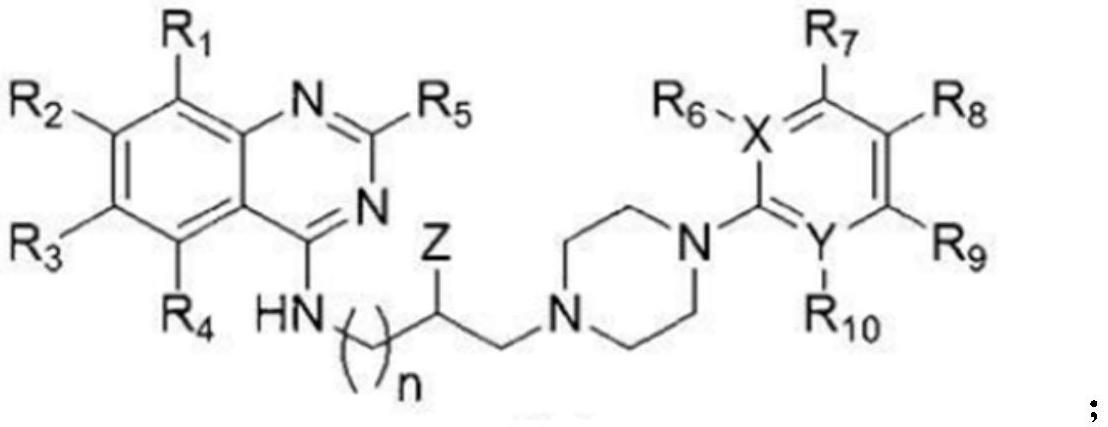
cn111253325a公开了一种氨基喹唑啉芳基哌嗪类化合物及其药物组合和应用,该发明提供的氨基喹唑啉芳基哌嗪类化合物具有如下所示的化合物或其药学上可接受的盐:
[0005][0006]
其中,r1~r
10
各自独立表示氢、卤素、氨基、硝基、羟氨基、胍基、脲基、三氟甲基、芳磺酰基、取代芳基、c1~c
10
烷氧基、c1~c
10
氨烷基、c1~c
10
烷基或c1~c
10
酰胺基;n为0~5的整数;x=c、n、o或s;y=c或n;z=h、oh、卤素或酯基。该化合物在体外具有一定的细胞毒作用,能明显抑制乳腺癌干细胞成球、抑制肿瘤细胞的克隆形成、抑制肿瘤细胞的迁移和侵袭;在体内能显著抑制乳腺癌的肺转移、抑制乳腺肿瘤以及非小细胞肺癌肿瘤的生长,并且具有良好的安全性,没有引起动物体重、血象以及各种主要器官的异常。
[0007]
现有技术中还公开了以cbz-丝氨酸为起始原料制备(3r,6r)-1-叔丁基-3-甲基-6-甲基哌嗪-1,3-二甲酸酯的合成路线,具体如下:
[0008][0009]
上述制备方法需要经过8个反应过程,且包含两次脱cbz保护氨基的过程,在脱cbz保护氨基的过程中需使用钯碳作为催化剂,钯碳价格昂贵,增加了生产成本,不利于工业生产。
[0010]
由于哌嗪衍生物在抗抑郁、消炎止痛、抗肿瘤等方面具有广泛的应用,因此,开发一种原料易得、工艺路线简单、生产成本较低的制备方法,实现哌嗪类化合物的高纯度制备,是本领域亟待解决的问题。
技术实现要素:
[0011]
针对现有技术存在的不足,本发明的目的在于提供一种手性1-叔丁基-3-甲基-6-甲基哌嗪-1,3-二甲酸酯的制备方法,所述制备方法以手性boc-d-丙氨酸和手性l-丝氨酸甲酯盐酸盐为起始原料,经过缩合反应、脱氨基保护反应、关环反应、还原反应、氨基保护反应、氧化反应、关环反应和开环反应,得到高收率的目标产物;所述制备方法中的原料易得,反应条件温和,适于工业化生产。
[0012]
为达此目的,本发明采用以下技术方案:
[0013]
本发明提供一种手性1-叔丁基-3-甲基-6-甲基哌嗪-1,3-二甲酸酯的制备方法,包括如下步骤:
[0014]
(1)boc-d-丙氨酸和l-丝氨酸甲酯盐酸盐进行缩合反应,得到式i所示化合物,反应式如下:
[0015][0016]
(2)步骤(1)得到的式i所示化合物与脱保护剂进行脱保护反应,生成的产物进行关环反应,得到式ii所示化合物,反应式如下:
[0017][0018]
(3)步骤(2)得到的式ii所示化合物进行还原反应,生成的产物与盐酸进行反应,得到式iii所示化合物,反应式如下:
[0019][0020]
(4)步骤(3)得到的式iii所示化合物与缚酸剂进行反应,生成的产物与boc氨基保护剂进行氨基保护反应,得到式iv所示化合物,反应式如下:
[0021][0022]
(5)步骤(4)得到的式iv所示化合物进行氧化反应,得到式v所示化合物,反应式如下:
[0023][0024]
(6)步骤(5)得到的式v所示化合物进行关环反应,得到式vi所示化合物,反应式如下:
[0025][0026]
(7)步骤(6)得到的式vi所示化合物进行开环反应,得到所述手性1-叔丁基-3-甲基-6-甲基哌嗪-1,3-二甲酸酯;
[0027][0028]
本发明所述手性1-叔丁基-3-甲基-6-甲基哌嗪-1,3-二甲酸酯为(3r,6r)-1-叔丁基-3-甲基-6-甲基哌嗪-1,3-二甲酸酯,所述制备方法以手性boc-d-丙氨酸和手性l-丝氨
酸甲酯盐酸盐为起始原料,经过缩合反应、脱氨基保护反应、关环反应、还原反应、氨基保护反应、氧化反应、关环反应和开环反应,得到目标产物(3r,6r)-1-叔丁基-3-甲基-6-甲基哌嗪-1,3-二甲酸酯。本发明使用boc氨基保护剂对氨基进行保护,降低了生产成本,且原料易得,反应条件温和,能够应用于大规模的工业化生产。
[0029]
作为本发明的优选技术方案,步骤(1)所述缩合反应在缩合剂和缚酸剂存在下进行。
[0030]
优选地,所述boc-d-丙氨酸与l-丝氨酸甲酯盐酸盐的摩尔比为(0.8~1.2):1,例如可以是0.8:1、0.85:1、0.9:1、0.95:1、1:1、1.05:1、1.1:1、1.15:1或1.2:1等。
[0031]
优选地,所述缩合剂选自1-(3-二甲氨基丙基)-3-乙基碳二亚胺、二环己基碳二亚胺、o-苯并三氮唑-四甲基脲六氟磷酸盐、2-(7-氮杂苯并三氮唑)-n,n,n
′
,n
′-
四甲基脲六氟磷酸酯、苯并三氮唑-1-基氧基三(二甲基氨基)磷鎓六氟磷酸盐或六氟磷酸苯并三唑-1-基-氧基三吡咯烷基磷中的任意一种或至少两种的组合。
[0032]
优选地,所述缩合剂与boc-d-丙氨酸的摩尔比为1:(1~1.2),例如可以是1:1、1:1.02、1:1.04、1:1.06、1:1.08、1:1.1、1:1.12、1:1.14、1:1.16、1:1.18或1:1.2等。
[0033]
优选地,所述缚酸剂选自三乙胺、n-甲基吗啉或二异丙基乙胺中的任意一种或至少两种的组合。
[0034]
优选地,所述缚酸剂与boc-d-丙氨酸的摩尔比为1:(1.1~1.3),例如可以是1:1.1、1:1.12、1:1.14、1:1.16、1:1.18、1:1.2、1:1.22、1:1.24、1:1.26、1:1.28或1:1.3等。
[0035]
优选地,步骤(1)所述缩合反应的温度为23~28℃,例如可以是23℃、24℃、25℃、26℃、27℃或28℃等。
[0036]
优选地,步骤(1)所述缩合反应的时间为9~11h,例如可以是9h、9.2h、9.4h、9.6h、9.8h、10h、10.2h、10.4h、10.6h、10.8h、或11h等。
[0037]
优选地,步骤(1)所述缩合反应在有机溶剂存在下进行。
[0038]
优选地,所述有机溶剂选自二氯甲烷、二氯乙烷或三氯甲烷中的任意一种或至少两种的组合。
[0039]
优选地,步骤(1)所述缩合反应进行前还包括boc-d-丙氨酸、l-丝氨酸甲酯盐酸盐、缩合剂、缚酸剂与有机溶剂的混合。
[0040]
优选地,所述混合的温度为-5~5℃,例如可以是-5℃、-4℃、-3℃、-2℃、-1℃、0℃、1℃、2℃、3℃、4℃或5℃等。
[0041]
优选地,所述混合的时间为5~15min,例如可以是5min、6min、7min、8min、9min、10min、11min、12min、13min、14min或15min。
[0042]
优选地,步骤(1)所述缩合反应完成后还包括后处理的步骤。
[0043]
优选地,所述后处理的方法包括:萃取、洗涤、干燥、浓缩,得到式i所示化合物。
[0044]
作为本发明的优选技术方案,步骤(2)中所述脱保护剂选自三氟乙酸、hcl或h2so4中的任意一种或至少两种的组合。
[0045]
优选地,步骤(2)所述脱保护剂与式i所示化合物质量比为(8-10.5):1,例如可以是8:1、8.5:1、9:1、9.5:1、10:1或10.5:1等。
[0046]
优选地,步骤(2)所述脱保护剂的加入温度为-5~5℃,例如可以是-5℃、-4℃、-3℃、-2℃、-1℃、0℃、1℃、2℃、3℃、4℃或5℃等。
[0047]
优选地,所述脱保护反应的温度为23~28℃,例如可以是23℃、24℃、25℃、26℃、27℃或28℃等。
[0048]
优选地,所述脱保护反应的时间为3~5h;,例如可以是3h、3.2h、3.4h、3.6h、3.8h、4h、4.2h、4.4h、4.6h、4.8h或3h等。
[0049]
优选地,所述脱保护反应在有机溶剂存在下进行。
[0050]
优选地,所述有机溶剂选自二氯甲烷、二氯乙烷或三氯甲烷中的任意一种或至少两种的组合。
[0051]
优选地,所述脱保护反应后还包括去除有机溶剂的步骤。
[0052]
优选地,步骤(2)中所述关环反应在碱性物质存在下进行。
[0053]
优选地,所述碱性物质选自碳酸钾、碳酸钠或碳酸铯中的任意一种或至少两种的组合。
[0054]
优选地,所述碱性物质与式i所示化合物的质量比为(0.8-1.1):1,例如可以是0.8:1、0.83:1、0.85:1、0.88:1、0.92:1、0.95:1、0.98:1、1:1、1.02:1、1.05:1、1.07:1或1.1:1等。
[0055]
优选地,步骤(2)所述关环反应的温度为55~75℃,例如可以是55℃、57℃、59℃、61℃、63℃、65℃、67℃、69℃、71℃、73℃或75℃等。
[0056]
优选地,步骤(2)所述关环反应的时间为3~5h,例如可以是3h、3.2h、3.4h、3.6h、3.8h、4h、4.2h、4.4h、4.6h、4.8h或3h等。
[0057]
优选地,步骤(2)所述关环反应在溶剂存在下进行。
[0058]
优选地,所述溶剂选自甲醇、乙醇或乙腈中的任意一种或至少两种的组合。
[0059]
优选地,步骤(2)所述关环反应完成后还包括后处理的步骤。
[0060]
优选地,所述后处理的方法包括:浓缩、过滤,打浆、抽滤、干燥,得到式ii所示化合物。
[0061]
作为本发明的优选技术方案,步骤(3)所述还原反应在还原剂存在下进行反应。
[0062]
优选地,所述还原剂选自硼烷四氢呋喃、硼烷二甲硫醚、氢化铝锂或红铝中的任意一种或至少两种的组合。
[0063]
优选地,所述还原剂与式ii所示化合物的摩尔比为(5~7):1,例如可以是5:1、5.2:1、5.4:1、5.6:1、5.8:1、6:1、6.2:1、6.4:1、6.6:1、6.8:1或7:1等。
[0064]
优选地,所述还原剂的加入温度为-5~5℃,例如可以是-5℃、-4℃、-3℃、-2℃、-1℃、0℃、1℃、2℃、3℃、4℃或5℃等。
[0065]
优选地,所述还原反应的温度为35~45℃,例如可以是35℃、36℃、37℃、38℃、39℃、40℃、41℃、42℃、43℃、44℃或45℃等。
[0066]
优选地,所述还原反应的时间为3~5h,例如可以是3h、3.2h、3.4h、3.6h、3.8h、4h、4.2h、4.4h、4.6h、4.8h或3h等。
[0067]
优选地,所述还原反应在有机溶剂存在下进行。
[0068]
优选地,所述有机溶剂选自二氯甲烷、二氯乙烷或三氯甲烷中的任意一种或至少两种的组合。
[0069]
优选地,在所述还原反应前,步骤(3)还包括所述式ii所示化合物与还原剂混合。
[0070]
优选地,所述混合的温度为23~28℃,例如可以是23℃、24℃、25℃、26℃、27℃或
28℃等。
[0071]
优选地,所述混合的时间为4~6h,例如可以是4h、4.2h、4.4h、4.6h、4.8h、5h、5.2h、5.4h、5.6h、5.8h或6h等。
[0072]
优选地,步骤(3)所述盐酸与式ii所示化合物与的摩尔比为(9-11):1,例如可以是9:1、9.2:1、9.4:1、9.6:1、9.8:1、10:1、10.2:1、10.4:1、10.6:1、10.8:1或11:1等。
[0073]
优选地,步骤(3)所述盐酸的克当量浓度为4~6n,例如可以是4n、4.2n、4.4n、4.6n、4.8n、5n、5.2n、5.4n、5.6n、5.8n或6n等。
[0074]
优选地,步骤(3)所述盐酸的加入温度为-5~5℃,例如可以是-5℃、-4℃、-3℃、-2℃、-1℃、0℃、1℃、2℃、3℃、4℃或5℃等。
[0075]
优选地,步骤(3)所述产物与盐酸反应的温度为35~45℃,例如可以是35℃、36℃、37℃、38℃、39℃、40℃、41℃、42℃、43℃、44℃或45℃等。
[0076]
优选地,步骤(3)所述产物与盐酸反应的时间为1.5~2.5h,例如可以是1.5h、1.6h、1.7h、1.8h、1.9h、2h、2.1h、2.2h、2.3h、2.4h或2.5h等。
[0077]
优选地,步骤(3)所述产物与盐酸反应完成后还包括后处理的步骤。
[0078]
优选地,所述后处理的方法包括:将所述产物与盐酸反应完成后的体系降温、过滤、洗涤、干燥,得到式iii所示化合物。
[0079]
作为本发明的优选技术方案,步骤(4)所述缚酸剂选自三乙胺、二异丙基乙胺或n-甲基吗啉中的任意一种或至少两种的组合。
[0080]
优选地,步骤(4)所述缚酸剂与式iii所示化合物的摩尔比为1:(0.3~0.5),例如可以是1:0.3、1:0.32、1:0.34、1:0.36、1:0.38、1:0.4、1:0.42、1:0.44、1:0.46、1:0.48或1:0.5等。
[0081]
优选地,步骤(4)所述缚酸剂与式iii所示化合物反应的温度为-5~5℃,例如可以是-5℃、-4℃、-3℃、-2℃、-1℃、0℃、1℃、2℃、3℃、4℃或5℃等。
[0082]
优选地,步骤(4)所述缚酸剂与式iii所示化合物反应的时间为20~40min,例如可以是20min、22min、24min、26min、28min、30min、32min、34min、36min、38min或40min等。
[0083]
优选地,步骤(4)所述缚酸剂与式iii所示化合物的反应在有机溶剂存在下进行。
[0084]
优选地,所述有机溶剂选自二氯甲烷、二氯乙烷或三氯甲烷中的任意一种或至少两种的组合。
[0085]
优选地,步骤(4)所述boc氨基保护剂与式iii所示化合物的摩尔比为(2.1~3.1):1,例如可以是2.1:1、2.2:1、2.3:1、2.4:1、2.5:1、2.6:1、2.7:1、2.8:1、2.9:1、3:1或3.1:1等。
[0086]
优选地,所述氨基保护反应的温度为23~28℃,例如可以是23℃、24℃、25℃、26℃、27℃或28℃等。
[0087]
优选地,所述氨基保护反应的时间为11~13h,例如可以是11h、11.2h、11.4h、11.6h、11.8h、12h、12.2h、12.4h、12.6h、12.8h或13h等。
[0088]
优选地,所述氨基保护反应完成后还包括后处理的步骤。
[0089]
优选地,所述后处理的方法包括:洗涤、浓缩、打浆、过滤、干燥,得到式iv所示化合物。
[0090]
作为本发明的优选技术方案,步骤(5)中所述氧化反应的氧化剂为tempo共氧化体
系,所述tempo共氧化体系包括tempo、碱金属溴盐和次氯酸钠的组合。
[0091]
优选地,所述氧化剂为氧化剂溶液。
[0092]
优选地,所述氧化剂溶液的溶剂为去离子水。
[0093]
优选地,所述tempo、碱金属溴盐和次氯酸钠的摩尔比为1:(10~40):(190~300),例如可以是1:10:190、1:12:200、1:15:210、1:18:220、1:20:230、1:22:240、1:25:190、1:28:250、1:30:260、1:32:270、1:35:280、1:37:290或1:40:300等。
[0094]
优选地,所述氧化剂与式iv所示化合物的摩尔比为(2.1~3.4):1,例如可以是2.1:1、2.2:1、2.3:1、2.4:1、2.5:1、2.6:1、2.7:1、2.8:1、2.9:1、3:1、3.1:1、3.2:1、3.3:1或3.4:1等。
[0095]
优选地,所述氧化反应在碱性物质存在下进行反应。
[0096]
优选地,所述碱性物质选自碳酸氢钾和/或碳酸氢钠。
[0097]
优选地,所述碱性物质与式iv所示化合物的摩尔比为(1.5~2.5):1,例如可以是1.5:1、1.6:1、1.7:1、1.8:1、1.9:1、2:1、2.1:1、2.2:1、2.3:1、2.4:1或2.5:1等。
[0098]
优选地,所述氧化反应在有机溶剂存在下进行。
[0099]
优选地,所述有机溶剂选自二氯甲烷、二氯乙烷或三氯甲烷中的任意一种或至少两种的组合。
[0100]
作为本发明的优选技术方案,步骤(5)所述氧化反应包括第一反应阶段和第二反应阶段。
[0101]
优选地,所述第一反应阶段的温度为-5~5℃,例如可以是-5℃、-4℃、-3℃、-2℃、-1℃、0℃、1℃、2℃、3℃、4℃或5℃等。
[0102]
优选地,所述第一反应阶段的时间为1.5~2.5h,例如可以是1.5:1、1.6:1、1.7:1、1.8:1、1.9:1、2:1、2.1:1、2.2:1、2.3:1、2.4:1或2.5:1等。
[0103]
优选地,所述第二反应阶段的温度为23~28℃,例如可以是23℃、24℃、25℃、26℃、27℃或28℃等。
[0104]
优选地,所述第二反应阶段的温度为11~13h,例如可以是11h、11.2h、11.4h、11.6h、11.8h、12h、12.2h、12.4h、12.6h、12.8h或13h等。
[0105]
优选地,步骤(5)所述氧化反应完成后还包括后处理的步骤。
[0106]
优选地,所述后处理的方法包括:向氧化反应后的体系中加入具有还原性的溶液混合、分液,保留有机相,然后浓缩、干燥,得到式v所示化合物。
[0107]
作为本发明的优选技术方案,步骤(6)所述关环反应在氯化试剂存在下进行。
[0108]
优选地,所述氯化试剂选自草酰氯、三氯化磷、氯化亚砜或五氯化磷中的任意一种或至少两种的组合。
[0109]
优选地,所述氯化试剂与式v所示化合物的摩尔比为(1.1~1.3):1,例如可以是1.1:1、1.12:1、1.14:1、1.16:1、1.18:1、1.2:1、1.22:1、1.24:1、1.26:1、1.28:1或1.3:1等。
[0110]
优选地,所述氯化试剂的加入温度为-5~5℃,例如可以是-5℃、-4℃、-3℃、-2℃、-1℃、0℃、1℃、2℃、3℃、4℃或5℃等。
[0111]
优选地,步骤(6)所述关环反应的温度为23~28℃,例如可以是23℃、24℃、25℃、26℃、27℃或28℃等。
[0112]
优选地,步骤(6)所述关环反应的时间为1.5~2.5h,例如可以是1.5h、1.6h、1.7h、
1.8h、1.9h、2h、2.1h、2.2h、2.3h、2.4h或2.5h等。
[0113]
优选地,步骤(6)所述关环反应在有机溶剂存在下进行。
[0114]
优选地,所述有机溶剂选自二氯甲烷、二氯乙烷或三氯甲烷中的任意一种或至少两种的组合。
[0115]
优选地,步骤(6)所述关环反应完成后还包括后处理的步骤。
[0116]
优选地,所述后处理的方法包括:浓缩、打浆、抽滤、干燥,得到式vi所示化合物。
[0117]
作为本发明的优选技术方案,步骤(7)所述开环反应在甲醇存在下进行。
[0118]
优选地,所述甲醇与式vi所示化合物的质量比为(9~11):1,例如可以是9:1、9.2:1、9.4:1、9.6:1、9.8:1、10:1、10.2:1、10.4:1、10.6:1、10.8:1或11:1等。
[0119]
优选地,所述开环反应的温度为55~75℃,例如可以是55℃、57℃、59℃、61℃、63℃、65℃、67℃、69℃、71℃、73℃或75℃等。
[0120]
优选地,所述开环反应的时间为4~6h,例如可以是4h、4.2h、4.4h、4.6h、4.8h、5h、5.2h、5.4h、5.6h、5.8h或6h等。
[0121]
优选地,所述开环反应完成后还包括后处理的步骤。
[0122]
优选地,所述后处理的方法包括:浓缩、洗涤、干燥,得到所述手性1-叔丁基-3-甲基-6-甲基哌嗪-1,3-二甲酸酯。
[0123]
作为本发明的优选技术方案,所述制备方法包括具体如下步骤:
[0124]
(1)将boc-d-丙氨酸、l-丝氨酸甲酯盐酸盐、缩合剂、缚酸剂和有机溶剂在-5~5℃下混合5~15min后,升温至23~28℃反应9~11h,得到化合物所述boc-d-丙氨酸与l-丝氨酸甲酯盐酸盐的摩尔比为(0.8~1.2):1,所述缩合剂与boc-d-丙氨酸的摩尔比为1:(0.8~1.2),所述缚酸剂与boc-d-丙氨酸的摩尔比为1:(0.8~1.2);
[0125]
(2)将步骤(1)得到的化合物和有机溶剂混合,在-5~5℃下向其中滴加脱保护剂,滴加完成后升温至23~28℃反应3~5h,脱溶后,加入溶剂和碱性物质,在温度为55~75℃条件下,反应3~5h,得到化合物所述脱保护剂与化合物的质量比为(7.5~11):1,所述碱性物质与化合物的质量比为1:(1~1.2);
[0126]
(3)将步骤(2)得到的化合物和有机溶剂混合,在-5~5℃下向其中滴
加还原剂,滴加完成后升温至23~28℃混合4~6h,然后升温至35℃~45℃反应3~5h后,降温至-5~5℃,向其中滴加盐酸,滴加完成后,升温至35~45℃反应1.5~2.5h,得到化合物;所述还原剂与化合物的摩尔比为(5-7):1,所述盐酸与化合物的摩尔比为(9-11):1;
[0127]
(4)将步骤(3)得到的化合物、缚酸剂和有机溶剂混合,在-5~5℃的条件下反应20~40min后,向其中滴加boc氨基保护剂,滴加完成后,升温至23~28℃反应11~13h,得到化合物;所述缚酸剂与化合物的摩尔比为1:(0.3~0.5),所述boc氨基保护剂与化合物的摩尔比为(2.1~3.1):1;
[0128]
(5)将步骤(4)得到的化合物、氧化剂、碱性物质和有机溶剂混合后,在-5~5℃下,反应1.5~2.5h后,升温至23~28℃反应11~13h,得到化合物;所述氧化剂与化合物的摩尔比为(2.1~3.4):1,所述碱性物质与化合物的摩尔比为(1.5-2.5):1;
[0129]
(6)将步骤(5)得到的化合物和有机溶剂混合,在-5~5℃下向其中滴加氯化试剂,滴加完成后升温至23~28℃反应1.5~2.5h,得到化合物;所述氯化试剂与化合物的摩尔比为(1.1-1.3):1;
[0130]
(7)将步骤(6)得到的化合物和甲醇混合,在55~75℃的条件下反应4~6h,得到所述手性1-叔丁基-3-甲基-6-甲基哌嗪-1,3-二甲酸酯;所述甲醇与化合物质量比为(9~11):1;反应的工艺路线如下:
[0131][0132]
与现有技术相比,本发明至少具有以下有益效果:
[0133]
本发明提供的制备方法以手性boc-d-丙氨酸和手性l-丝氨酸甲酯盐酸盐为起始原料,经过缩合反应、脱氨基保护反应、关环反应、还原反应、氨基保护反应、氧化反应、关环反应和开环反应,得到目标产物(3r,6r)-1-叔丁基-3-甲基-6-甲基哌嗪-1,3-二甲酸酯,且制备目标产物的过程中,每个步骤的产率都较高,均≥88%。本发明中使用boc氨基保护剂对氨基进行保护,降低了生产成本,且原料易得,反应条件温和,能够应用于大规模的工业化生产。
具体实施方式
[0134]
下面通过具体实施方式来进一步说明本发明的技术方案。为本领域技术人员应该明了,所述实施例仅仅是帮助理解本发明,不应视为对本发明的具体限制。
[0135]
实施例1
[0136]
本实施例提供一种(3r,6r)-1-叔丁基-3-甲基-6-甲基哌嗪-1,3-二甲酸酯的制备方法,具体包括如下步骤:
[0137]
(1)
[0138]
将boc-d-丙氨酸(183.5g,0.97mol)加入到2.5l二氯甲烷中,冰浴下滴加1-(3-二
甲氨基丙基)-3-乙基碳二亚胺(205g,1.07mol),搅拌10min,再滴加l-丝氨酸甲酯盐酸盐(158.7g,1.02mol)和三乙胺(161ml,1.16mol),自然升温至室温,反应10h;向反应体系中加入3l纯水,静置、分液,得到有机相和水相,水相用1l二氯甲烷萃取1次,合并有机相,并依次经克当量浓度为1n的盐酸、饱和碳酸氢钠、纯水、饱和食盐水洗涤,无水硫酸钠干燥,浓缩,得到253g产物收率为90%。
[0139]
对产物进行检测:1h-nmr(cdcl3,400mhz):δ7.17(d,1h),5.18(br,1h),4.64-4.61(m,1h),4.18-4.15(m,1h),3.96(br,2h),3.76(s,3h),1.44(s,9h),1.39(d,3h)。
[0140]
(2)
[0141]
将步骤(1)得到的产物(250g,0.86mol)溶于1.5l二氯甲烷中,冰浴下滴加1.5l三氟乙酸,自然升温至室温,反应4h;薄层色谱法(tlc)检测显示原料消失后,减压脱溶,加入2l甲醇和碳酸钾(237g,1.72mol),室温下搅拌30min,然后加热回流4h;浓缩,搅拌下加入3l纯水,过滤,滤饼用石油醚打浆,抽滤,烘干,得到129g产物收率为95%。
[0142]
对产物进行检测:1h-nmr(cdcl3,400m):δ1.25(d,3h).3.54(m,1h),3.70(m,1h),3.76(m,1h),3.94(m,1h),5.11(t,1h),7.90(s,1h),8.10(s,1h)。
[0143]
(3)
[0144]
将步骤(2)得到的产物(120g,0.76mol)加入到2l二氯甲烷中,冰浴下滴加物质的量浓度为1m的硼烷四氢呋喃4.56l,自然升温至室温,搅拌5h,加热回流4h;降温至0℃,向反应体系中滴加克当量浓度为5n的盐酸1.5l,加热回流2h;冷却至0℃,搅拌2h,过滤,滤饼用四氢呋喃洗涤,烘干,得到139g产物收率为90%。
[0145]
对产物进行检测:1h-nmr(dmso-d6,400m):δ1.33(d,3h),3.12(m,2h),3.39-3.78(m,6h),5.62(s,1h),10.03(brs,4h)。
[0146]
(4
[0147]
将步骤(3)得到的产物(130g,0.64mol)加入到2.5l二氯甲烷中,冰浴下加入三乙胺(222ml,1.6mol),反应30min;向反应体系中滴加boc酸酐(293g,1.34mol),室温下反应
12h;经300ml纯水洗涤2次,克当量浓度为1n的盐酸400ml洗涤2次,200ml纯水洗涤2次,饱和食盐水洗涤1次,浓缩至干,用600ml石油醚打浆、过滤、烘干,得到211g产物收率为96%。
[0148]
对产物进行检测:1h-nmr(cdcl3,400m):δ1.22(d,3h),1.50(s,18h),3.10-3.30(m,2h),3.42-3.73(m,3h),3.80-4.02(m,1h),4.03-4.24(m,1h),4.25-4.43(m,1h)。
[0149]
(5)
[0150]
将步骤(4)得到的产物(209g,0.63mol)加入到2l二氯甲烷中,在冰浴下,加入2.5l纯水,将溴化钾(15g,0.126mol)、tempo(2g,0.0126mol)、碳酸氢钾(126g,1.26mol)溶于1l水中并滴加至反应体系中,搅拌20min,加入质量浓度为10%的次氯酸钠溶液880ml,滴加结束后,保持冰浴2h,自然升温至室温,过夜反应;滴加质量浓度为10%的硫代硫酸钠溶液1l,搅拌30min,静置,分液,水相用克当量浓度为1n的盐酸调节ph至1-2,用800ml二氯甲烷萃取两次,合并有机相、干燥、浓缩,得到184g产物,收率为90%。
[0151]
对产物进行检测:1h-nmr(cdcl3,400m):δ1.23(d,3h),1.50(s,18h),3.16-3.37(m,2h),3.45-3.84(m,2h),4.09-4.28(m,1h),4.78-4.89(m,1h)。
[0152]
(6)
[0153]
将步骤(5)得到的产物(180g,0.52mol)加入到2l二氯甲烷中,0℃下滴加草酰氯(55ml,0.65mol),自然升温至室温,反应2h;减压浓缩,残渣用正己烷打浆,抽滤,真空干燥,得到126g产物,收率为90%。
[0154]
(7)
[0155]
将步骤(6)得到的产物(120g,0.44mol)加入到1.5l甲醇中,加热回流5h;浓缩,残渣溶于二氯甲烷中,饱和食盐水洗涤,无水硫酸钠干燥,浓缩,真空干燥,得到106g目标产物(3r,6r)-1-叔丁基-3-甲基-6-甲基哌嗪-1,3-二甲酸酯,收率为93%。
[0156]
对目标产物进行检测:1h nmr(dmso-d6,400mhz):δ1.14(d,3h),1.39(s,9h),2.46(dd,1h),2.68(s,1h),2.95(dd,1h),3.17(dd,1h),3.58(d,1h),3.66(s,3h),3.98-4.10(m,
1h),4.11-4.18(m,1h)。
[0157]
实施例2
[0158]
本实施例提供一种(3r,6r)-1-叔丁基-3-甲基-6-甲基哌嗪-1,3-二甲酸酯的制备方法,具体包括如下步骤:
[0159]
(1)
[0160]
将boc-d-丙氨酸(189g,1mol)加入到2.5l二氯乙烷中,冰浴下滴加二环己基碳二亚胺(206g,1mol),搅拌5min,再滴加l-丝氨酸甲酯盐酸盐(176.8g,1.14mol)和n-甲基吗啉(148ml,1.7mol),自然升温至室温,反应9.5h;向反应体系中加入3l纯水,静置、分液,得到有机相和水相,水相用1l二氯乙烷萃取1次,合并有机相并依次经克当量浓度为1n的盐酸、饱和碳酸氢钠、纯水、饱和食盐水洗涤,无水硫酸钠干燥,浓缩,得到258g产物,收率为89%。
[0161]
(2)
[0162]
将步骤(1)得到的产物(255g,0.88mol)加入到1.5l二氯乙烷中,冰浴下滴加物质的量浓度为5m的盐酸溶液11.5l,自然升温至室温,反应4.5h;薄层色谱法(tlc)检测显示原料消失后,减压脱溶,加入2l乙醇和碳酸钠(128.26g,1.21mol),室温下搅拌30min,然后加热回流3h;浓缩,搅拌下加入3l纯水,过滤,滤饼用石油醚打浆,抽滤,烘干,得到130.8g产物收率为94%。
[0163]
(3)
[0164]
将步骤(2)得到的产物(125g,0.79mol)加入到2l二氯乙烷中,冰浴下滴加物质的量浓度为1m的硼烷二甲硫醚4.34l,自然升温至室温,搅拌4h,加热回流4.5h;降温至0℃,向反应体系中滴加克当量浓度为4n的盐酸1.5l,加热回流1.8h;冷却至0℃,搅拌2h,过滤,滤饼用四氢呋喃洗涤,烘干,得到147.5g产物,收率为92%。
[0165]
(4)
[0166]
将步骤(3)得到的产物(124g,0.61mol)加入到2.5l二氯乙烷中,冰浴下加入n-甲基吗啉(187.3ml,1.35mol),反应20min;向反应体系中滴加boc酸酐(373.3g,1.708mol),室温下反应11h;经300ml纯水洗涤2次,克当量浓度为1n的盐酸400ml洗涤2次,200ml纯水洗涤2次,饱和食盐水洗涤1次,浓缩至干,用600ml石油醚打浆、过滤、烘干,得到194.4g产物,收率为95%。
[0167]
(5)
[0168]
将步骤(4)得到的产物(210g,0.64mol)加入到2l二氯乙烷中,在冰浴下,加入2.5l纯水,将溴化钾(15g,0.126mol)、tempo(2g,0.0126mol)、碳酸氢钠(91.6g,1.09mol)溶于1l水中并滴加至反应体系中,搅拌20min,加入质量浓度为10%的次氯酸钠溶液880ml,滴加结束后,保持冰浴2h,自然升温至室温,过夜反应;滴加质量浓度为10%的硫代硫酸钠溶液1l,搅拌30min,静置,分液,水相用克当量浓度为1n的盐酸调节ph至1-2,用800ml二氯乙烷萃取两次,合并有机相、干燥、浓缩,得到194g产物,收率为88%。
[0169]
(6)
[0170]
将步骤(5)得到的产物(178g,0.517mol)加入到2l二氯乙烷中,0℃下滴加三氯化磷(60ml,0.57mol),自然升温至室温,反应1.5h;减压浓缩,残渣用正己烷打浆,抽滤,真空干燥,得到127g产物收率为91%。
[0171]
(7)
[0172]
将步骤(6)得到的产物(117g,0.43mol)加入到1.33l甲醇中,加热回流4h;浓缩,残渣溶于二氯乙烷中,饱和食盐水洗涤,无水硫酸钠干燥,浓缩,真空干燥,得到102g目标产物(3r,6r)-1-叔丁基-3-甲基-6-甲基哌嗪-1,3-二甲酸酯,收率为92%。
[0173]
实施例3
[0174]
本实施例提供一种(3r,6r)-1-叔丁基-3-甲基-6-甲基哌嗪-1,3-二甲酸酯的制备方法,具体包括如下步骤:
[0175]
(1)
[0176]
将boc-d-丙氨酸(200g,1.06mol)加入到2.5l三氯甲烷中,冰浴下滴加2-(7-氮杂苯并三氮唑)-n,n,n
′
,n
′-
四甲基脲六氟磷酸酯(334.6g,0.88mol),搅拌15min,再滴加l-丝氨酸甲酯盐酸盐(206g,1.33mol)和二异丙基乙胺(162.4ml,1.17mol),自然升温至室温,反应10.5h;向反应体系中加入3l纯水,静置、分液,得到有机相和水相,水相用1l三氯甲烷萃取1次,合并有机相并依次经克当量浓度为1n的盐酸、饱和碳酸氢钠、纯水、饱和食盐水洗涤,无水硫酸钠干燥,浓缩,得到280g产物,收率为91%。
[0177]
(2)
[0178]
将步骤(1)得到的产物(245g,0.84mol)加入到1.5l三氯甲烷中,冰浴下滴加物质的量浓度为3m的硫酸溶液7.2l,自然升温至室温,反应5h;薄层色谱法(tlc)检测显示原料消失后,减压脱溶,加入2l乙腈和碳酸铯(606g,1.86mol),室温下搅拌30min,然后加热回流3.5h;浓缩,搅拌下加入3l纯水,过滤,滤饼用石油醚打浆,抽滤,烘干,得到127.5g产物,收率为96%。
[0179]
(3)
[0180]
将步骤(2)得到的产物(128g,0.81mol)加入到2l三氯甲烷中,冰浴下滴加物质的量浓度为1m的氢化铝锂4.05l,自然升温至室温,搅拌4.5h,加热回流3h;降温至0℃,向反应体系中滴加克当量浓度为4.5n的盐酸1.5l,加热回流1.5h;冷却至0℃,搅拌2h,过滤,滤饼用四氢呋喃洗涤,烘干,得到146.3g产物,收率为89%。
[0181]
(4)
[0182]
将步骤(3)得到的产物(128g,0.63mol)加入到2.5l三氯甲烷中,冰浴下加入二异丙基乙胺(174ml,1.26mol),反应25min;向反应体系中滴加boc酸酐(426.9g,1.95mol),室温下反应11.5h;经300ml纯水洗涤2次,克当量浓度为1n的盐酸400ml洗涤2次,200ml纯水洗涤2次,饱和食盐水洗涤1次,浓缩至干,用600ml石油醚打浆、过滤、烘干,得到202g产物,收率为97%。
[0183]
(5)
[0184]
将步骤(4)得到的产物(200g,0.61mol)加入到2l三氯甲烷中,在冰浴下,加入2.5l纯水,将溴化钾(15g,0.126mol)、tempo(2g,0.0126mol)、碳酸氢钾(133g,1.33mol)溶于1l水中并滴加至反应体系中,搅拌20min,加入质量浓度为10%的次氯酸钠溶液880ml,滴加结束后,保持冰浴2h,自然升温至室温,过夜反应;滴加质量浓度为10%的硫代硫酸钠溶液1l,搅拌30min,静置,分液,水相用克当量浓度为1n的盐酸调节ph至1-2,用800ml三氯甲烷萃取两次,合并有机相、干燥、浓缩,得到186.8g产物,收率为89%。
[0185]
(6)
[0186]
将步骤(5)得到的产物(185g,0.54mol)加入到2l三氯甲烷中,0℃下滴加氯化亚砜(58ml,0.62mol),自然升温至室温,反应1.8h;减压浓缩,残渣用正己烷打浆,抽滤,真空干燥,得到129.8g产物,收率为89%。
[0187]
(7)
[0188]
将步骤(6)得到的产物(124g,0.46mol)加入到1.57l甲醇中,加热回流6h;浓缩,残渣溶于二氯甲烷中,饱和食盐水洗涤,无水硫酸钠干燥,浓缩,真空干燥,得到111.6g目标产物(3r,6r)-1-叔丁基-3-甲基-6-甲基哌嗪-1,3-二甲酸酯收率为94%。
[0189]
实施例4
[0190]
本实施例提供一种(3r,6r)-1-叔丁基-3-甲基-6-甲基哌嗪-1,3-二甲酸酯的制备方法,具体包括如下步骤:
[0191]
(1)
[0192]
将boc-d-丙氨酸(180g,0.95mol)加入到2.5l二氯甲烷中,冰浴下滴加六氟磷酸苯并三唑-1-基-氧基三吡咯烷基磷(468.3g,0.9mol),搅拌12min,再滴加l-丝氨酸甲酯盐酸盐(133.8g,0.86mol)和三乙胺(176.3ml,1.27mol),自然升温至室温,反应9h;向反应体系中加入3l纯水,静置、分液,得到有机相和水相,水相用1l二氯甲烷萃取1次,合并有机相并依次经克当量浓度为1n的盐酸、饱和碳酸氢钠、纯水、饱和食盐水洗涤,无水硫酸钠干燥,浓缩,得到242.7g产物,收率为88%。
[0193]
(2)
[0194]
将步骤(1)得到的产物(240g,0.83mol)加入到1.5l二氯甲烷中,冰浴下滴加1.57l三氟乙酸,自然升温至室温,反应3.5h;薄层色谱法(tlc)检测显示原料消失后,减压脱溶,加入2l甲醇和碳酸钾(192g,1.39mol),室温下搅拌30min,然后加热回流4.5h;浓缩,搅拌下加入3l纯水,过滤,滤饼用石油醚打浆,抽滤,烘干,得到122g产物,收率为93%。
[0195]
(3)
[0196]
将步骤(2)得到的产物(122g,0.77mol)加入到2l二氯甲烷中,冰浴下滴加物质的量浓度为3.46m的红铝的甲苯溶液1.44l,自然升温至室温,搅拌5h,加热回流3.5h;降温至0℃,向反应体系中滴加克当量浓度为6n的盐酸1.5l,加热回流2.3h;冷却至0℃,搅拌2h,过滤,滤饼用四氢呋喃洗涤,烘干,得到142.2g产物收率为91%。
[0197]
(4)
[0198]
将步骤(3)得到的产物(132g,0.65mol)加入到2.5l二氯甲烷中,冰浴下加入三乙胺(258ml,1.86mol),反应35min;向反应体系中滴加boc酸酐(354g,1.63mol),室温下反应
13h;经300ml纯水洗涤2次,克当量浓度为1n的盐酸400ml洗涤2次,200ml纯水洗涤2次,饱和食盐水洗涤1次,浓缩至干,用600ml石油醚打浆、过滤、烘干,得到201.6g产物,收率为94%。
[0199]
(5)
[0200]
将步骤(4)得到的产物(205g,0.62mol)加入到2l二氯甲烷中,在冰浴下,加入2.5l纯水,将溴化钾(15g,0.126mol)、tempo(2g,0.0126mol)、碳酸氢钾(155g,1.55mol)溶于1l水中并滴加至反应体系中,搅拌20min,加入质量浓度为10%的次氯酸钠溶液880ml,滴加结束后,保持冰浴2h,自然升温至室温,过夜反应;滴加质量浓度为10%的硫代硫酸钠溶液1l,搅拌30min,静置,分液,水相用克当量浓度为1n的盐酸调节ph至1-2,用800ml二氯甲烷萃取两次,合并有机相、干燥、浓缩,得到194.3g产物,收率为91%。
[0201]
(6)
[0202]
将步骤(5)得到的产物(174g,0.51mol)加入到2l二氯甲烷中,0℃下滴加五氯化磷(53ml,0.66mol),自然升温至室温,反应2.5h;减压浓缩,残渣用正己烷打浆,抽滤,真空干燥,得到126.7g产物,收率为92%。
[0203]
(7)
[0204]
将步骤(6)得到的产物(115g,0.426mol)加入到1.52l甲醇中,加热回流6h;浓缩,残渣溶于二氯甲烷中,饱和食盐水洗涤,无水硫酸钠干燥,浓缩,真空干燥,得到100g目标产物(3r,6r)-1-叔丁基-3-甲基-6-甲基哌嗪-1,3-二甲酸酯,收率为91%。
[0205]
实施例5
[0206]
本实施例提供一种(3r,6r)-1-叔丁基-3-甲基-6-甲基哌嗪-1,3-二甲酸酯的制备方法,具体包括如下步骤:
[0207]
(1)
[0208]
将boc-d-丙氨酸(185g,0.98mol)加入到2.5l二氯甲烷中,冰浴下滴加1-(3-二甲氨基丙基)-3-乙基碳二亚胺(162.8g,0.85mol),搅拌10min,再滴加l-丝氨酸甲酯盐酸盐(127.6g,0.82mol)和三乙胺(169.3ml,1.22mol),自然升温至室温,反应11h;向反应体系中加入3l纯水,静置、分液,得到有机相和水相,水相用1l二氯甲烷萃取1次,合并有机相并依次经克当量浓度为1n的盐酸、饱和碳酸氢钠、纯水、饱和食盐水洗涤,无水硫酸钠干燥,浓缩,得到261.7g产物,收率为92%。
[0209]
(2)
[0210]
将步骤(1)得到的产物(260g,0.9mol)加入到1.5l二氯甲烷中,冰浴下滴加1.78l三氟乙酸,自然升温至室温,反应3h;薄层色谱法(tlc)检测显示原料消失后,减压脱溶,加入2l甲醇和碳酸钾(286g,2.07mol),室温下搅拌30min,然后加热回流5h;浓缩,搅拌下加入3l纯水,过滤,滤饼用石油醚打浆,抽滤,烘干,得到135.2g产物,收率为95%。
[0211]
(3)
[0212]
将步骤(2)得到的产物(118g,0.75mol)加入到2l二氯甲烷中,冰浴下滴加物质的量浓度为1m的硼烷四氢呋喃4.05l,自然升温至室温,搅拌5h,加热回流5.5h;降温至0℃,向反应体系中滴加克当量浓度为5n的盐酸1.5l,加热回流2.5h;冷却至0℃,搅拌2.5h,过滤,滤饼用四氢呋喃洗涤,烘干,得到134g产物,收率为88%。
[0213]
(4)
[0214]
将步骤(3)得到的产物(136g,0.67mol)加入到2.5l二氯甲烷中,冰浴下加入三乙胺(291ml,2.1mol),反应40min;向反应体系中滴加boc酸酐(321g,1.47mol),室温下反应
13h;经300ml纯水洗涤2次,克当量浓度为1n的盐酸400ml洗涤2次,200ml纯水洗涤2次,饱和食盐水洗涤1次,浓缩至干,用600ml石油醚打浆、过滤、烘干,得到210g产物,收率为95%。
[0215]
(5)
[0216]
将步骤(4)得到的产物(208g,0.63mol)加入到2l二氯甲烷中,在冰浴下,加入2.5l纯水,将溴化钾(15g,0.126mol)、tempo(2g,0.0126mol)、碳酸氢钾(97.5g,0.975mol)溶于1l水中并滴加至反应体系中,搅拌20min,加入质量浓度为10%的次氯酸钠溶液880ml,滴加结束后,保持冰浴2h,自然升温至室温,过夜反应;滴加质量浓度为10%的硫代硫酸钠溶液1l,搅拌30min,静置,分液,水相用克当量浓度为1n的盐酸调节ph至1-2,用800ml二氯甲烷萃取两次,合并有机相、干燥、浓缩,得到199.6g产物,收率为92%。
[0217]
(6)
[0218]
将步骤(5)得到的产物(183g,0.53mol)加入到2l二氯甲烷中,0℃下滴加草酰氯(52ml,0.61mol),自然升温至室温,反应2.5h;减压浓缩,残渣用正己烷打浆,抽滤,真空干燥,得到126g产物收率为88%。
[0219]
(7)
[0220]
将步骤(6)得到的产物(110g,0.41mol)加入到1.53l甲醇中,加热回流6h;浓缩,残渣溶于二氯甲烷中,饱和食盐水洗涤,无水硫酸钠干燥,浓缩,真空干燥,得到100.5g目标产物(3r,6r)-1-叔丁基-3-甲基-6-甲基哌嗪-1,3-二甲酸酯,收率为95%。
[0221]
综上所述,本发明以手性boc-d-丙氨酸和手性l-丝氨酸甲酯盐酸盐为起始原料,经过缩合反应、脱氨基保护反应、关环反应、还原反应、氨基保护反应、氧化反应、关环反应和开环反应,制备得到了目标产物(3r,6r)-1-叔丁基-3-甲基-6-甲基哌嗪-1,3-二甲酸酯,且制备目标产物的过程中,每个步骤的产率均≥88%,原料易得,反应条件温和,能够应用于大规模的工业化生产。
[0222]
申请人声明,本发明通过上述实施例来说明本发明的详细工艺设备和工艺流程,
但本发明并不局限于上述详细工艺设备和工艺流程,即不意味着本发明必须依赖上述详细工艺设备和工艺流程才能实施。所属技术领域的技术人员应该明了,对本发明的任何改进,对本发明产品各原料的等效替换及辅助成分的添加、具体方式的选择等,均落在本发明的保护范围和公开范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1