一种环氧氯丙烷副产物制备甲基缩水甘油醚的方法与流程
[0001]
本发明涉及精细有机化学技术领域,尤其涉及一种环氧氯丙烷副产物制备甲基缩水甘油醚的方法。
背景技术:
[0002]
环氧氯丙烷主要用于生产环氧树脂、表面活性剂、医药、农药、离子交换树脂等,国内年用量在70万吨左右。双氧水直接氧化法环氧氯丙烷生产工艺因其原子利用率高、废水量少、能耗低而成为环氧氯丙烷绿色生产的发展方向。采用钛硅分子筛催化双氧水直接氧化法生产环氧氯丙烷过程中会产生副产物3-氯-1-甲氧基-2-丙醇,由于没有合适的应用,通常作为废液处理,处理难度大、成本高且造成资源浪费。
[0003]
高纯甲基缩水甘油醚可作为低卤环氧树脂的活性稀释剂降低体系粘度,还可用于生物医药、有机合成等领域,前景广阔。
[0004]
leal-duaso等人报道了甲醇与环氧氯丙烷在氢氧化钾的催化下65℃反应1.5h,得到甲基缩水甘油醚,收率为61%(参见“glycerol-derived solvents:synthesis and properties of symmetric glyceryl diethers”,leal-duaso等,sustainable chemistry&engineering,第7卷第15期,第13004-13014页)
[0005]
isono等人报道了甲醇与(s)-(+)-环氧氯丙烷在三氟化硼乙醚的催化下在0℃下反应2h,后用naoh闭环,(r)-(-)-环氧丙基甲基醚收率为16%(参见“design and synthesis of thermoresponsive aliphatic polyethers with a tunable phase transition temperature”,isono等,polym.chem.,第8卷,第5698-5707页)
[0006]
2009年李春钢等人报道了乙醇与环氧氯丙烷在三氟化硼乙醚的催化下进行开环反应,后用30%naoh闭环,用乙醚萃取得到乙基缩水甘油醚,收率为78%(参见“甲磺司特的合成”,李春钢等,广东药学院学报,第25卷第3期,第278-280页)。
[0007]
上述传统的甲基缩水甘油醚的合成方法均存在选择性差、副产物多、收率低,同时过多的副产物增加了甲基缩水甘油醚在分离提纯时的难度,导致产品质量差、分离成本高等缺点。
[0008]
因此,需要开发一种甲基缩水甘油醚的制备方法,克服现有技术中选择性差及副产物多的问题。
技术实现要素:
[0009]
为解决上述技术问题,本发明提供一种环氧氯丙烷副产物制备甲基缩水甘油醚的方法,所述方法包括利用环氧氯丙烷副产物3-氯-1-甲氧基-2-丙醇皂化得到皂化液并经提纯,制得高纯甲基缩水甘油醚,解决了环氧氯丙烷副产物3-氯-1-甲氧基-2-丙醇处理难题,实现了资源化利用,而且制得的甲基缩水甘油醚总氯低、纯度高,应用前景广阔。
[0010]
为达此目的,本发明采用以下技术方案:
[0011]
第一方面,本发明提供一种环氧氯丙烷副产物制备甲基缩水甘油醚的方法,所述
方法包括:将环氧氯丙烷副产物皂化得到皂化液,所述皂化液经提纯,得到甲基缩水甘油醚;所述环氧氯丙烷副产物中含有3-氯-1-甲氧基-2-丙醇。
[0012]
本发明所述方法利用环氧氯丙烷副产物3-氯-1-甲氧基-2-丙醇皂化反应,生成甲基缩水甘油醚,反应的选择性高,副产物少,产品中总氯含量低,且解决了环氧氯丙烷副产物3-氯-1-甲氧基-2-丙醇处理难题,实现了资源化利用。
[0013]
优选地,所述环氧氯丙烷副产物为双氧水直接氧化法产生的环氧氯丙烷副产物。
[0014]
优选地,所述环氧氯丙烷副产物中3-氯-1-甲氧基-2-丙醇的质量浓度为5~60%。
[0015]
优选地,所述环氧氯丙烷副产物中还含有水。
[0016]
优选地,所述环氧氯丙烷副产物中水的质量浓度为40~95%,例如可以是40%、47%、53%、59%、65%、71%、77%、83%、89%或95%等,但不限于所列举的数值,该范围内其他未列举的数值同样适用。
[0017]
优选地,所述皂化包括:向环氧氯丙烷副产物中滴加碱溶液,进行皂化反应。
[0018]
本发明采用上述滴加方式,更有利于提高产品的选择性和纯度,减少副产物的产生。
[0019]
优选地,所述碱溶液为氢氧化钠溶液。
[0020]
优选地,所述碱溶液中碱按物质的量计为3-氯-1-甲氧基-2-丙醇的0.8~1.3倍,例如可以是0.8倍、0.86倍、0.92倍、0.97倍、1.03倍、1.08倍、1.14倍、1.19倍、1.25倍或1.3倍等,但不限于所列举的数值,该范围内其他未列举的数值同样适用,优选为1.0~1.1倍。
[0021]
优选地,所述碱溶液的滴加时间为0.5~4h,例如可以是0.5h、0.9h、1.3h、1.7h、2.1h、2.5h、2.9h、3.3h、3.7h或4h等,但不限于所列举的数值,该范围内其他未列举的数值同样适用,优选1~3h。
[0022]
优选地,所述碱溶液滴加过程中,皂化反应的温度为0~80℃,例如可以是0℃、9℃、18℃、27℃、36℃、45℃、54℃、63℃、72℃或80℃等,但不限于所列举的数值,该范围内其他未列举的数值同样适用,优选30~50℃。
[0023]
本发明通过控制反应温度,更有利于控制反应的选择性,减少后续提纯的步骤。
[0024]
优选地,所述碱溶液滴加后,继续进行皂化反应。
[0025]
优选地,所述碱溶液滴加后,皂化反应的温度为10~100℃,例如可以是10℃、20℃、30℃、40℃、50℃、60℃、70℃、80℃、90℃或100℃等,但不限于所列举的数值,该范围内其他未列举的数值同样适用,优选30~70℃。
[0026]
优选地,所述碱溶液滴加后,皂化反应的时间为0.5~3h,例如可以是0.5h、0.8h、1.1h、1.4h、1.7h、1.9h、2.2h、2.5h、2.8h或3h等,但不限于所列举的数值,该范围内其他未列举的数值同样适用,优选0.5~2h。
[0027]
优选地,所述提纯依次包括:萃取、脱水和精馏,得到甲基缩水甘油醚。
[0028]
优选地,所述萃取包括:利用萃取剂对所述皂化液进行萃取,得到萃取油相。
[0029]
优选地,所述脱水包括:所述萃取油相经脱水,得到脱水后产物。
[0030]
优选地,所述精馏包括:所述脱水后产物经精馏分离,得到甲基缩水甘油醚。
[0031]
优选地,所述萃取剂不溶于水。
[0032]
优选地,所述萃取剂包括酯类、酮类、醚类或烷基醇类中的任意一种或至少两种的组合,其中典型非限制性的组合为酯类和酮类的组合,酯类和醚类的组合,烷基醇类和酯类
的组合,醚类和酮类的组合,酮类和烷基醇类的组合,烷基醇类和醚类的组合,优选为醋酸正丁酯、环己酮、苯甲醚或异辛醇中的任意一种或至少两种的组合,其中典型非限制性的组合为醋酸正丁酯和环己酮的组合,醋酸正丁酯和苯甲醚的组合,醋酸正丁酯和异辛醇的组合,环己酮和苯甲醚的组合,环己酮和异辛醇的组合,苯甲醚和异辛醇的组合。
[0033]
本发明选用上述溶剂进行萃取,其与甲基缩水甘油醚具有较佳的相似相溶性且不溶于水,能够提高萃取效率。
[0034]
优选地,所述萃取剂与皂化液的质量比为0.1~2:1,例如可以是0.1:1、0.4:1、0.6:1、0.8:1、1:1、1.2:1、1.4:1、1.6:1、1.8:1或2:1等,但不限于所列举的数值,该范围内其他未列举的数值同样适用,优选0.3~1.0:1。
[0035]
优选地,所述萃取采用萃取塔进行。
[0036]
优选地,所述萃取的空速为0.1~10h-1
,例如可以是0.1h-1
、1.2h-1
、2.3h-1
、3.4h-1
、4.5h-1
、5.6h-1
、6.7h-1
、7.8h-1
、8.9h-1
或10h-1
等,但不限于所列举的数值,该范围内其他未列举的数值同样适用,优选0.5~2.0h-1
。
[0037]
本发明将萃取的空速控制在0.1~10h-1
,既有利于提高萃取效率,又有利于节约萃取剂的用量,减少成本。
[0038]
优选地,所述脱水采用脱水塔进行。
[0039]
优选地,所述脱水的塔釜温度为90~120℃,例如可以是90℃、94℃、97℃、100℃、104℃、107℃、110℃、114℃、117℃或120℃等,但不限于所列举的数值,该范围内其他未列举的数值同样适用,优选90~110℃。
[0040]
优选地,所述脱水还得到第一甲基缩水甘油醚水溶液。
[0041]
优选地,所述第一甲基缩水甘油醚水溶液循环至萃取过程中。
[0042]
优选地,所述精馏分离的真空度为-0.01~-0.1mpa,例如可以是-0.01mpa、-0.02mpa、-0.03mpa、-0.04mpa、-0.05mpa、-0.06mpa、-0.07mpa、-0.08mpa、-0.09mpa或-0.1mpa等,优选为-0.05~-0.08mpa。
[0043]
优选地,所述精馏分离的塔顶温度为40~110℃,例如可以是40℃、47.8℃、55.6℃、63.4℃、71.2℃、78.9℃、86.7℃、94.5℃、102.3℃或110℃等,但不限于所列举的数值,该范围内其他未列举的数值同样适用,优选为50~90℃。
[0044]
优选地,所述精馏分离的回流比为0.5~5:1,例如可以是0.5:1、1:1、1.5:1、2:1、2.5:1、3:1、3.5:1、4:1、4.5:1或5:1等,但不限于所列举的数值,该范围内其他未列举的数值同样适用,优选为1~3:1。
[0045]
优选地,所述萃取还得到萃取水相,所述萃取水相经共沸处理,回收第二甲基缩水甘油醚水溶液。
[0046]
优选地,所述第二甲基缩水甘油醚水溶液循环至萃取中。
[0047]
优选地,所述提纯之后还包括溶剂回收。
[0048]
优选地,所述溶剂回收后循环至萃取过程中作萃取剂。
[0049]
作为本发明优选的技术方案,所述方法包括如下步骤:
[0050]
(1)在0~80℃条件下,用0.5~4h向环氧氯丙烷副产物中滴加碱溶液,进行皂化反应,所述碱溶液滴加后,继续在10~100℃下进行皂化反应0.5h~3h,得到皂化液,所述皂化液经提纯,得到甲基缩水甘油醚;
[0051]
其中,所述碱溶液中碱按物质的量计为3-氯-1-甲氧基-2-丙醇的0.8~1.3倍;
[0052]
(2)在萃取塔中利用萃取剂对所述皂化液进行萃取,得到萃取油相和萃取水相;萃取剂与皂化液的质量比为0.1~2:1,空速为0.1~10h-1
;
[0053]
所述萃取剂包括酯类、酮类、醚类或烷基醇类中的任意一种或至少两种的组合;
[0054]
所述萃取水相经共沸处理,回收第二甲基缩水甘油醚水溶液,所述第二甲基缩水甘油醚水溶液循环至萃取中;
[0055]
(3)采用脱水塔对所述萃取油相进行脱水,得到脱水后产物和第一甲基缩水甘油醚水溶液;所述脱水的塔釜温度为90~120℃。所述第一甲基缩水甘油醚水溶液循环至萃取过程中;
[0056]
(4)所述脱水后产物经精馏分离,得到甲基缩水甘油醚;所述精馏分离的真空度为-0.01~-0.1mpa,塔顶温度为40~110℃,回流比为0.5~5:1,回收溶剂循环至步骤(2)中作萃取剂。
[0057]
第二方面,本发明提供一种双氧水直接氧化法生产环氧氯丙烷联产甲基缩水甘油醚的方法,所述方法包括第一方面所述的环氧氯丙烷副产物制备甲基缩水甘油醚的方法。
[0058]
本发明第二方面提供的双氧水直接氧化法生产环氧氯丙烷联产甲基缩水甘油醚的方法中通过采用第一方面所述的方法处理副产物,制得的甲基缩水甘油醚纯度和附加值高,大大降低了双氧水直接氧化法生产环氧氯丙烷工艺的成本,提高了经济价值。
[0059]
本发明所述甲基缩水甘油醚可用于环氧树脂稀释剂、生物医药和有机合成等领域。
[0060]
与现有技术相比,本发明至少具有以下有益效果:
[0061]
(1)本发明提供的环氧氯丙烷副产物制备甲基缩水甘油醚的方法实现解决了环氧氯丙烷副产物3-氯-1-甲氧基-2-丙醇处理难题,3-氯-1-甲氧基-2-丙醇转化率≥88wt%,实现了资源化利用,降低双氧水直接氧化法环氧氯丙烷生产成本,提高经济性;
[0062]
(2)本发明提供的环氧氯丙烷副产物制备甲基缩水甘油醚的方法制得的甲基缩水甘油醚总氯低、纯度高,甲基缩水甘油醚纯度≥99wt%,且甲基缩水甘油醚收率≥88wt%,满足高端市场需求;
[0063]
(3)本发明提供的环氧氯丙烷副产物制备甲基缩水甘油醚的方法流程短、收率高,反应条件温和、操作简便,易于产业化,应用前景广阔。
具体实施方式
[0064]
为便于理解本发明,本发明列举实施例如下。本领域技术人员应该明了,所述实施例仅仅是帮助理解本发明,不应视为对本发明的具体限制。
[0065]
实施例1
[0066]
本实施例提供一种环氧氯丙烷副产物制备甲基缩水甘油醚的方法,所述方法包括如下步骤:
[0067]
(1)向反应釜内泵入环氧氯丙烷的副产物(3-氯-1-甲氧基-2-丙醇的质量分数为33.56%),开启搅拌器,升温至30℃,滴加浓度为30wt%的氢氧化钠溶液,氢氧化钠用量为3-氯-1-甲氧基-2-丙醇的1.0倍,控制反应温度30℃,1h滴加完毕,保温反应1h;皂化反应结束,皂化液中剩余3-氯-1-甲氧基-2-丙醇的浓度为2.56wt%,3-氯-1-甲氧基-2-丙醇的转
化率为89.65wt%;皂化液中甲基缩水甘油醚的质量浓度为15.50wt%,甲基缩水甘油醚收率为88.75wt%;
[0068]
(2)在30℃下萃取塔上部泵入所述皂化液,萃取塔下部泵入异辛醇,异辛醇与皂化液质量流量比为0.3:1,空速0.5h-1
下,萃取塔顶出萃取油相中甲基缩水甘油醚的质量浓度为32.16wt%;萃取塔底部出萃余水相中甲基缩水甘油醚的质量浓度为1.73wt%,萃取效率为90.53wt%;
[0069]
萃取水相进入共沸塔,控制负压-0.02~-0.03mpa,塔釜温度90~105℃,共沸塔顶为甲基缩水甘油醚水溶液去萃取工序,塔釜甲基缩水甘油醚≤0.1wt%去废水处理工序;
[0070]
(3)萃取油相进入脱水塔,收集塔釜温度90℃前馏份,回用于下批次萃取,得到塔釜萃取油相含水≤200ppm;
[0071]
(4)步骤(3)中所述塔釜萃取油相转入精馏塔在-0.06~-0.08mpa回流比3:1下,收集塔顶温度50~80℃馏份,得到总氯含量为30ppm、含水130ppm、纯度为99.9wt%甲基缩水甘油醚,精馏釜液99.5wt%的异辛醇为萃取剂回用于下批次萃取。
[0072]
实施例2-3
[0073]
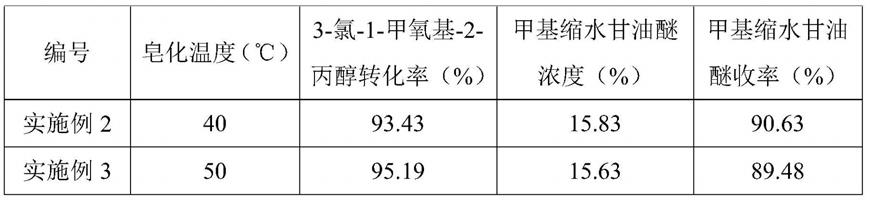
在实施例1基础上调整皂化温度,皂化温度分别调整至40℃和50℃,其他条件不变,3-氯-1-甲氧基-2-丙醇转化率及甲基缩水甘油醚收率见表1。
[0074]
表1
[0075][0076]
实施例4-5
[0077]
在实施例2基础上调整氢氧化钠用量,分别调整至3-氯-1-甲氧基-2-丙醇的1.05倍和1.1倍,其他条件不变,3-氯-1-甲氧基-2-丙醇转化率及甲基缩水甘油醚收率见表2。
[0078]
表2
[0079][0080]
实施例6-7
[0081]
在实施例4基础上调整皂化滴加时间,皂化滴加时间分别调整至2h和3h,其他条件不变,3-氯-1-甲氧基-2-丙醇转化率及甲基缩水甘油醚收率见表3。
[0082]
表3
[0083][0084]
实施例8-10
[0085]
在实施例6基础上调整皂化保温时间,皂化保温时间分别调整至0.5h、1.5h和2h,其他条件不变,3-氯-1-甲氧基-2-丙醇转化率及甲基缩水甘油醚收率见表4。
[0086]
表4
[0087][0088]
实施例11-12
[0089]
在实施例6基础上调整萃取温度,萃取温度分别调整至50℃、70℃,其他条件不变,产物甲基缩水甘油醚萃取效率见表5。
[0090]
表5
[0091][0092]
实施例13-14
[0093]
在实施例11基础上调整空速,空速分别调整为1h-1
和2h-1
,其他条件不变,产物甲基缩水甘油醚萃取效率见表6。
[0094]
表6
[0095]
[0096]
实施例15-17
[0097]
在实施例11基础上调整萃取剂,分布使用醋酸正丁酯、环己酮、苯甲醚为萃取剂,其他条件不变,产物甲基缩水甘油醚萃取效率见表7。
[0098]
表7
[0099][0100]
实施例18-19
[0101]
在实施例11基础上调整萃取剂用量,萃取剂用量分别调整至0.6:1和1:1(萃取剂与皂化反应液质量流量比),其他条件不变,产物甲基缩水甘油醚萃取效率见表8。
[0102]
表8
[0103][0104]
实施例20
[0105]
本实施例提供一种环氧氯丙烷副产物制备甲基缩水甘油醚的方法,所述方法包括如下步骤:
[0106]
(1)向反应釜内泵入环氧氯丙烷的副产物(3-氯-1-甲氧基-2-丙醇的质量分数为33.56%),开启搅拌器,控温至5℃,滴加浓度为25wt%的氢氧化钠溶液,氢氧化钠用量为3-氯-1-甲氧基-2-丙醇的1.3倍,控制反应温度10℃,0.5h滴加完毕,保温反应3h;皂化反应结束,皂化液中未检出3-氯-1-甲氧基-2-丙醇,3-氯-1-甲氧基-2-丙醇的转化率为100wt%;皂化液中甲基缩水甘油醚的质量浓度为13.16wt%,甲基缩水甘油醚收率为81.30wt%;
[0107]
(2)在60℃下萃取塔上部泵入所述皂化液,萃取塔下部泵入异辛醇,异辛醇与皂化液质量流量比为0.1:1,空速0.1h-1
下,萃取塔顶出萃取油相中甲基缩水甘油醚的质量浓度为27.71wt%;萃取塔底部出萃余水相中甲基缩水甘油醚的质量浓度为9.70wt%,萃取效率为29.13wt%;
[0108]
萃取水相进入共沸塔,控制负压-0.02mpa,塔釜温度95℃,共沸塔顶为甲基缩水甘油醚水溶液去萃取工序,塔釜甲基缩水甘油醚≤0.1wt%去废水处理工序;
[0109]
(3)萃取油相进入脱水塔,收集塔釜温度90℃前馏份,回用于下批次萃取,得到塔釜萃取油相含水≤200ppm;
[0110]
(4)步骤(3)中所述塔釜萃取油相转入精馏塔在-0.06mpa回流比5:1下,收集塔顶温度90℃~110℃范围的馏份,得到总氯含量为70ppm、含水110ppm、纯度为99.87wt%甲基缩水甘油醚,精馏釜液99.4wt%的异辛醇为萃取剂回用于下批次萃取。
[0111]
实施例21
[0112]
本实施例提供一种环氧氯丙烷副产物制备甲基缩水甘油醚的方法,所述方法包括如下步骤:
[0113]
(1)向反应釜内泵入环氧氯丙烷的副产物(3-氯-1-甲氧基-2-丙醇的质量分数为33.56%),开启搅拌器,升温至80℃,滴加浓度为40wt%的氢氧化钠溶液,氢氧化钠用量为3-氯-1-甲氧基-2-丙醇的0.8倍,控制反应温度100℃,1h滴加完毕,保温反应0.5h;皂化反应结束,皂化液中剩余3-氯-1-甲氧基-2-丙醇的浓度为5.92wt%,3-氯-1-甲氧基-2-丙醇的转化率为77.3wt%;皂化液中甲基缩水甘油醚的质量浓度为13.76wt%,甲基缩水甘油醚收率为74.59wt%;
[0114]
(2)在15℃下萃取塔上部泵入所述皂化液,萃取塔下部泵入异辛醇,异辛醇与皂化液质量流量比为2:1,空速10h-1
下,萃取塔顶出萃取油相中甲基缩水甘油醚的质量浓度为5.31wt%;萃取塔底部出萃余水相中甲基缩水甘油醚的质量浓度为2.86wt%,萃取效率为81.57wt%;
[0115]
萃取水相进入共沸塔,控制负压-0.02mpa,塔釜温度105℃,共沸塔顶为甲基缩水甘油醚水溶液去萃取工序,塔釜甲基缩水甘油醚≤0.1wt%去废水处理工序;
[0116]
(3)萃取油相进入脱水塔,收集塔釜温度110℃前馏份,回用于下批次萃取,得到塔釜萃取油相含水≤200ppm;
[0117]
(4)步骤(3)中所述塔釜萃取油相转入精馏塔在-0.08mpa回流比0.5:1下,收集塔顶温度40℃~60℃范围的馏份,得到总氯含量为100ppm、含水210ppm、纯度为89.81wt%甲基缩水甘油醚,精馏釜液99.2wt%的异辛醇为萃取剂回用于下批次萃取。
[0118]
上述实施例中各物质纯度的测试方法采用气相色谱定量分析得到。
[0119]
上述实施例中由于操作过程中存在温度波动和压降,故某些工艺参数为数值范围,实际为运行过程中参数波动的范围或全塔/釜中塔顶和塔釜的温度和压力范围。
[0120]
本发明提供一种环氧氯丙烷副产物制备甲基缩水甘油醚的方法通过将环氧氯丙烷副产物皂化后提纯,综合皂化温度、混合方式、萃取温度和空速等工艺参数的选择,制得的甲基缩水甘油醚总氯低、纯度高,甲基缩水甘油醚纯度≥99wt%,且甲基缩水甘油醚收率≥88wt%,满足高端市场需求,副产物中3-氯-1-甲氧基-2-丙醇转化率≥88wt%,反应条件温和、操作简便,易于产业化,应用前景广阔。
[0121]
申请人声明,本发明通过上述实施例来说明本发明的详细工艺设备和工艺流程,但本发明并不局限于上述详细工艺设备和工艺流程,即不意味着本发明必须依赖上述详细工艺设备和工艺流程才能实施。所属技术领域的技术人员应该明了,对本发明的任何改进,对本发明产品各原料的等效替换及辅助成分的添加、具体方式的选择等,均落在本发明的保护范围和公开范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1