苯并哌啶衍生物可药用盐及其制备方法与流程
本公开属于医药
技术领域:
,具体涉及化合物(e)-4-((2-(3,5-二氟-4-((1s,3r)-2-(2-氟-2-甲基丙基)-3-甲基-6-(1-甲基-1h-吡唑-4-基)-1,2,3,4-四氢异喹啉-1-基)苯氧基)乙基)氨基)-1-吗啉基丁-2-烯-1-酮的可药用盐及其制备方法。
背景技术:
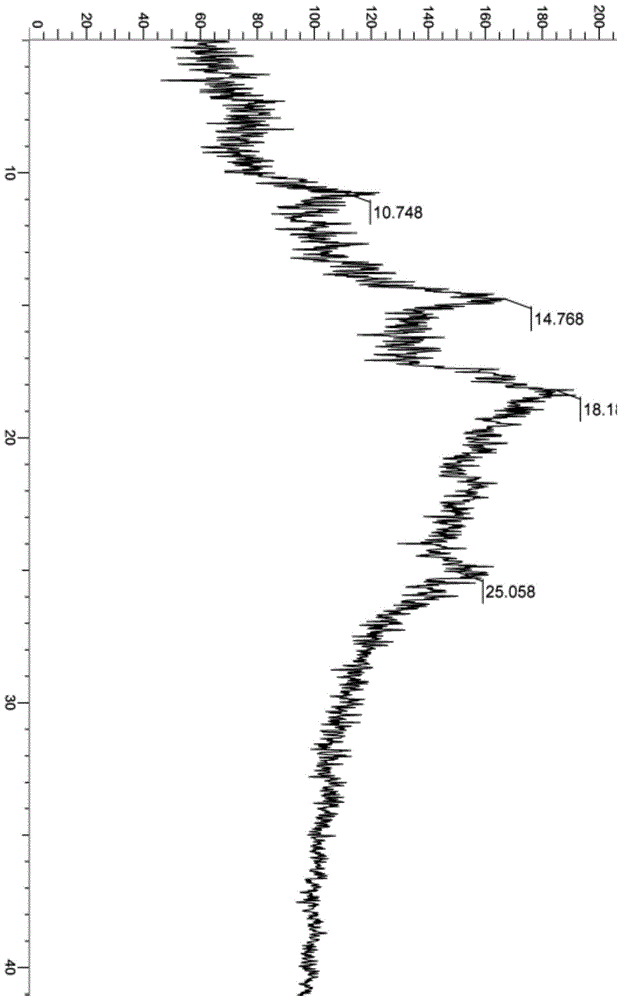
:pct/cn2019/087944(申请日2019年5月22日)描述了一种化合物(e)-4-((2-(3,5-二氟-4-((1s,3r)-2-(2-氟-2-甲基丙基)-3-甲基-6-(1-甲基-1h-吡唑-4-基)-1,2,3,4-四氢异喹啉-1-基)苯氧基)乙基)氨基)-1-吗啉基丁-2-烯-1-酮,作为一种esr1基因突变的雌激素受体拮抗剂,可用于经过芳香化酶抑制剂(aromataseinhibitor,ai)治疗而进展的转移性雌激素受体阳性乳腺癌病人,近一半药物分子都是以盐的形式存在,成盐可改善药物某一些不理想的物理化学或生物学性质。开发出相对于(e)-4-((2-(3,5-二氟-4-((1s,3r)-2-(2-氟-2-甲基丙基)-3-甲基-6-(1-甲基-1h-吡唑-4-基)-1,2,3,4-四氢异喹啉-1-基)苯氧基)乙基)氨基)-1-吗啉基丁-2-烯-1-酮,在理化性质或药学性质方面具有更优异的性质的盐是具有重要意义的。同时,鉴于固体药物晶型及其稳定性对其在临床治疗中的重要性,深入研究化合物(e)-4-((2-(3,5-二氟-4-((1s,3r)-2-(2-氟-2-甲基丙基)-3-甲基-6-(1-甲基-1h-吡唑-4-基)-1,2,3,4-四氢异喹啉-1-基)苯氧基)乙基)氨基)-1-吗啉基丁-2-烯-1-酮可药用盐的多晶型,对开发适合工业生产且生物活性良好的药物也是具有重要意义。技术实现要素:本公开(thedisclosure)中提供了化合物(e)-4-((2-(3,5-二氟-4-((1s,3r)-2-(2-氟-2-甲基丙基)-3-甲基-6-(1-甲基-1h-吡唑-4-基)-1,2,3,4-四氢异喹啉-1-基)苯氧基)乙基)氨基)-1-吗啉基丁-2-烯-1-酮的可药用盐,其中,所述可药用盐选自盐酸盐,硫酸盐、氢溴酸盐、甲磺酸盐、磷酸盐、柠檬酸盐、乙酸盐、马来酸盐、酒石酸盐、琥珀酸盐、苹果酸盐、草酸盐、苯甲酸盐、5-磺基水杨酸盐或富马酸盐,优选甲磺酸盐、硫酸盐、草酸盐和5-磺基水杨酸盐,更优选草酸盐。一些实施方案提供的可药用盐中所述化合物与酸分子的化学配比为所述化合物与酸分子的化学配比为2:1~1:2,优选为2:1、1:1、1:1.5、1:2。在可选实施方案中,所述化合物与硫酸(根)的化学配比为1:1或2:1。在可选实施方案中,所述化合物与甲磺酸(根)的化学配比为1:1。在可选实施方案中,所述化合物与草酸(根)的化学配比为1:1或1:1.5。在可选实施方案中,所述化合物与5-硝基水杨酸(根)的化学配比为1:1。本公开中还提供了制备前述可药用盐的方法,包括:化合物(e)-4-((2-(3,5-二氟-4-((1s,3r)-2-(2-氟-2-甲基丙基)-3-甲基-6-(1-甲基-1h-吡唑-4-基)-1,2,3,4-四氢异喹啉-1-基)苯氧基)乙基)氨基)-1-吗啉基丁-2-烯-1-酮与酸成盐的步骤,所述酸选自盐酸(或氯化氢溶液),硫酸、氢溴酸、甲磺酸、磷酸、柠檬酸、乙酸、马来酸、酒石酸、琥珀酸、草酸、苯甲酸、5-磺基水杨酸或富马酸,优选甲磺酸、硫酸、草酸和5-磺基水杨酸,更优选草酸。本公开中成盐所用溶剂选自环己烷、丙酮、甲醇、乙醇、异丙醇、二氯甲烷、异丙醚、四氢呋喃、乙酸异丙酯、丙酮、甲基叔丁基醚、乙腈、1,4-二氧六环、乙酸乙酯中的至少一种。进一步地,在可选实施方案中,制备前述可药用盐的方法还包括挥发溶剂或搅拌析晶,过滤、干燥等步骤。本公开中还提供了一种药物组合物,其含有前述化合物的可药用盐和任选自药学上可接受的载体、稀释剂或赋形剂中的至少一种的药用辅料。本公开中还提供了前述可药用盐或药物组合物在制备用于治疗乳腺癌的药物中的用途,所述乳腺癌为雌激素受体阳性,进一步地,所述乳腺癌为经过芳香化酶抑制剂(aromataseinhibitor,ai)治疗而进展的。另一方面,本公开中还提供了化合物(e)-4-((2-(3,5-二氟-4-((1s,3r)-2-(2-氟-2-甲基丙基)-3-甲基-6-(1-甲基-1h-吡唑-4-基)-1,2,3,4-四氢异喹啉-1-基)苯氧基)乙基)氨基)-1-吗啉基丁-2-烯-1-酮草酸盐的晶型a,以衍射角2θ角度表示的x-射线粉末衍射图谱,在14.768处有特征峰。在可选实施方案中,所述晶型a,以衍射角2θ角度表示的x-射线粉末衍射图谱,在14.768,18.110处有特征峰。在可选实施方案中,所述晶型a,以衍射角2θ角度表示的x-射线粉末衍射图谱,在10.478,14.768,18.110,25.058处有特征峰。一些优选实施方案中提供所述晶型a,以衍射角2θ角度表示的x-射线粉末衍射图谱如图1所示。制备化合物(e)-4-((2-(3,5-二氟-4-((1s,3r)-2-(2-氟-2-甲基丙基)-3-甲基-6-(1-甲基-1h-吡唑-4-基)-1,2,3,4-四氢异喹啉-1-基)苯氧基)乙基)氨基)-1-吗啉基丁-2-烯-1-酮草酸盐的晶型a的方法,包括:(a)将化合物(e)-4-((2-(3,5-二氟-4-((1s,3r)-2-(2-氟-2-甲基丙基)-3-甲基-6-(1-甲基-1h-吡唑-4-基)-1,2,3,4-四氢异喹啉-1-基)苯氧基)乙基)氨基)-1-吗啉基丁-2-烯-1-酮加入溶剂(i)中,搅拌溶解或加热溶解,(b)加入草酸,搅拌析晶。本法所述溶剂(i)所用体积(ml)为化合物重量(g)的1~50倍,可以为1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44、45、46、47、48、49、50倍;所述溶剂(i)优选乙腈。本公开所述晶型制备方法还包括过滤、洗涤或干燥等步骤。本公开还提供了一种药物组合物,其含有前述晶型a或由前述方法制备的晶型a和任选自药学上可接受的载体、稀释剂或赋形剂中的药用辅料。本公开还提供了一种药物组合物,由前述晶型a和任选自药学上可接受的载体、稀释剂或赋形剂制备而成。本公开还提供了一种药物组合物的制备方法,包括将前述晶型a或由前述方法制备的晶型a,与药学上可接受的载体、稀释剂或赋形剂混合的步骤。本公开还提供了前述的晶型a或由前述方法制备的晶型a或由前述组合物在制备用于治疗乳腺癌的药物的用途,所述乳腺癌优选雌激素受体阳性,所述乳腺癌更优选经过芳香化酶抑制剂治疗而进展的。本公开“药物组合物”表示含有一种或多种本文所述化合物或其生理学上/可药用的盐或前体药物与其他化学组分的混合物,以及其他组分例如生理学/可药用的载体和赋形剂。药物组合物的目的是促进对生物体的给药,利于活性成分的吸收进而发挥生物活性。本公开中所述的“x-射线粉末衍射图谱”为使用cu-kα辐射测量得到。本公开中所述的“x-射线粉末衍射图谱或xrpd”是指根据布拉格公式2dsinθ=nλ(式中,λ为x射线的波长,衍射的级数n为任何正整数,一般取一级衍射峰,n=1),当x射线以掠角θ(入射角的余角,又称为布拉格角)入射到晶体或部分晶体样品的某一具有d点阵平面间距的原子面上时,就能满足布拉格方程,从而测得了这组x射线粉末衍射图。本公开所述的“2θ或2θ角度”是指衍射角,θ为布拉格角,单位为°或度;每个特征峰2θ的误差范围为±0.20,可以为-0.20、-0.19、-0.18、-0.17、-0.16、-0.15、-0.14、-0.13、-0.12、-0.11、-0.10、-0.09、-0.08、-0.07、-0.06、-0.05、-0.04、-0.03、-0.02、-0.01、0.00、0.01、0.02、0.03、0.04、0.05、0.06、0.07、0.08、0.09、0.10、0.11、0.12、0.13、0.14、0.15、0.16、0.17、0.18、0.19、0.20。本公开中化合物与酸分子的化学配比测定存在一定程度的误差,一般而言,正负10%均属于合理误差范围内。随其所用之处的上下文而有一定程度的误差变化,该误差变化不超过正负10%,可以为正负9%、正负8%、正负7%、正负6%、正负5%、正负4%、正负3%、正负2%或正负1%,优选正负5%”本公开中所述的“差示扫描量热分析或dsc”是指在样品升温或恒温过程中,测量样品与参考物之间的温度差、热流差,以表征所有与热效应有关的物理变化和化学变化,得到样品的相变信息。本公开中所述的“差示扫描量热分析或dsc”是指在样品升温或恒温过程中,测量样品与参考物之间的温度差、热流差,以表征所有与热效应有关的物理变化和化学变化,得到样品的相变信息。本公开中所述干燥温度一般为25℃~100℃,优选40℃~70℃,可以常压干燥,也可以减压干燥。优选干燥在减压下干燥。本公开中所用试剂可通过商业途径获得。本公开中实验所用仪器的测试条件:1、差示扫描量热仪(differentialscanningcalorimeter,dsc)仪器型号:dsc3+吹扫气:氮气升温速率:10.0℃/min温度范围:25-300℃2、x-射线粉末衍射谱(x-raypowderdiffraction,xrpd)(1)仪器型号:brukerd8discovera25x-射线粉末衍射仪射线:单色cu-kα射线(λ=1.5406)扫描方式:θ/2θ,扫描范围:5-48°电压:40kv,电流:40ma3、热重分析仪(thermogravimetricanalysis,tga)仪器型号:tga2吹扫气:氮气升温速率:10.0℃/min温度范围:25-300℃化合物的结构是通过核磁共振(nmr)或/和质谱(ms)来确定的。nmr位移(δ)以10-6(ppm)的单位给出。nmr的测定是用brukeravance-400核磁仪,测定溶剂为氘代二甲基亚砜(dmso-d6)、氘代氯仿(cdcl3)、氘代甲醇(cd3od),内标为四甲基硅烷(tms)。ms的测定用finniganlcqad(esi)质谱仪(生产商:thermo,型号:finniganlcqadvantagemax)。高效液相色谱法(hplc)分析使用agilenthplc1200dad、agilenthplc1200vwd和watershplce2695-2489高压液相色谱仪。手性hplc分析测定使用agilent1260dad高效液相色谱仪。高效液相制备使用waters2767、waters2767-sqdetecor2、shimadzulc-20ap和gilson-281制备型色谱仪。手性制备使用shimadzulc-20ap制备型色谱仪。实施例中的反应进程的监测采用薄层色谱法(tlc),反应所使用的展开剂,纯化化合物采用的柱层析的洗脱剂的体系和薄层色谱法的展开剂体系包括:a:二氯甲烷/甲醇体系,b:正己烷/乙酸乙酯体系,c:石油醚/乙酸乙酯体系,溶剂的体积比根据化合物的极性不同而进行调节,也可以加入少量的三乙胺和醋酸等碱性或酸性试剂进行调节。附图说明图1:化合物(e)-4-((2-(3,5-二氟-4-((1s,3r)-2-(2-氟-2-甲基丙基)-3-甲基-6-(1-甲基-1h-吡唑-4-基)-1,2,3,4-四氢异喹啉-1-基)苯氧基)乙基)氨基)-1-吗啉基丁-2-烯-1-酮草酸盐晶型a的xrpd谱图。具体实施方式以下将结合实施例或实验例更详细地解释本公开,本公开中的实施例或实验例仅用于说明本公开中的技术方案,并非限定本公开中的实质和范围。实施例1:(e)-4-((2-(3,5-二氟-4-((1s,3r)-2-(2-氟-2-甲基丙基)-3-甲基-6-(1-甲基-1h-吡唑-4-基)-1,2,3,4-四氢异喹啉-1-基)苯氧基)乙基)氨基)-1-吗啉基丁-2-烯-1-酮(参照pct/cn2019/087944中方法制备)第一步(e)-4-溴-1-吗啉基丁-2-烯-1-酮c将化合物a(500mg,3.0mmol)溶于二氯甲烷(30ml),冰水浴冷却下加入2-(7-氧化苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸盐(856mg,3.6mmol)和三乙胺(368mg,3.6mmol)。缓慢滴加入吗啡啉(314mg,3.6mmol),滴加完毕后继续保持冰水浴冷却状态1小时,然后室温继续搅拌6小时。将反应液倒入冰水中,用二氯甲烷萃取(100ml×3),合并有机相,无水硫酸钠干燥,过滤,滤液减压浓缩。用柱色谱法以展开剂体系b纯化所得残余物,得到标题产物c(271mg),产率:38%。第二步(e)-4-((2-(3,5-二氟-4-((1s,3r)-2-(2-氟-2-甲基丙基)-3-甲基-6-(1-甲基-1h-吡唑-4-基)-1,2,3,4-四氢异喹啉-1-基)苯氧基)乙基)氨基)-1-吗啉基丁-2-烯-1-酮1将化合物c(303mg,1.3mmol)溶于n,n-二甲基甲酰胺(8ml),室温下加入二异丙基乙胺(393mg,3.0mmol),然后加入化合物j(720mg,1.5mmol),室温搅拌1.5小时。加入水(15ml),乙酸乙酯萃取(20ml×3),有机层用饱和氯化钠溶液洗涤(20ml×3),无水硫酸钠干燥,过滤浓缩得粗品。用薄层色谱法以展开剂体系a纯化所得残余物,得到标题产物1(230mg),产率:24%。msm/z(esi):626.1[m+1];1hnmr(400mhz,cd3od)7.88(s,1h),7.76(s,1h),7.28(s,1h),7.18(d,1h),6.90-6.74(dt,1h),6.70-6.59(m,2h),6.56(d,2h),5.10(s,1h),4.11(t,2h),3.90(s,3h),3.74-3.60(m,9h),3.55(d,2h),3.40-3.30(m,1h),3.06(t,2h),2.94(t,1h),2.61(dd,1h),2.30(dd,1h),1.14(d,3h),1.09(d,3h),1.00(d,3h).中间体j的制备(参照pct/cn2019/087944中方法制备):第一步(r)-(1-(3-(苄氧基)苯基)丙基-2-基)氨基甲酸叔丁酯1c将苄氧基-3-溴苯1a(3.65g,13.91mmol)溶于四氢呋喃(40ml),氩气保护,搅拌均匀后加入正丁基锂(13.9mmol,5.8ml),-78℃搅拌30分钟。30分钟后将(r)-4-甲基-1,2,3-氧杂噻唑烷-3-甲酸叔丁酯2,2-二氧化物1b(3.3g,13.90mmol,采用专利申请“wo2017182493”公开的方法制备而得)溶于20ml四氢呋喃中,将上述溶液加入至反应液中,-78℃搅拌反应0.5小时。缓慢升温到0℃时,冰浴下搅拌30分钟后,停止反应。用10%柠檬酸溶液淬灭反应(20ml),再加入水(100ml),搅拌10分钟。用乙酸乙酯萃取(100ml×2),合并有机相,无水硫酸钠干燥,过滤,滤液减压浓缩,用薄层色谱法以展开剂体系b纯化所得残余物,得到标题产物1c(2.37g),产率:49.5%。msm/z(esi):286.2[m-56+1]第二步(r)-1-(3-(苄氧基)苯基)丙基-2-胺1d将化合物1c(2.66g,7.79mmol)溶于二氯甲烷(15ml),加入三氟乙酸(3ml),室温搅拌反应5小时,停止反应。减压浓缩反应液,用饱和碳酸氢钠溶液(100ml)将反应液调至ph8左右,无水硫酸钠干燥,过滤,滤液减压浓缩,得到粗品标题产物1d(2.28g),产物不经纯化直接进行下一步反应。msm/z(esi):242.2[m+1]第三步(r)-n-(1-(3-(苄氧基)苯基)丙基-2-基)-2-氟-2-甲基丙基-1-胺1f将化合物1d粗品(0.24g,998.64umol)溶于二氧六环(10ml),加入二异丙基乙胺(0.38g,2.99mmol),(2-氟-2-甲基-丙基)三氟甲烷磺酸酯1e(0.34g,1.49mmol,采用专利申请“wo2017182493”公开的方法制备而得),氩气保护。油浴90℃搅拌反应3小时。原料消失,停止反应。冷却反应液,浓缩反应液,加入饱和碳酸氢钠溶液(15ml),用乙酸乙酯萃取(50ml×2),合并有机相,无水硫酸钠干燥,过滤,滤液减压浓缩,用薄层色谱法以展开剂体系b纯化所得残余物,得到标题产物1f(170mg),产率:54%。msm/z(esi):316.3[m+1]第四步(r)-3-(2-((2-氟-2-甲基丙基)氨基)丙基)苯酚1g将化合物1f(0.70g,2.21mmol)溶于甲醇(10ml),加入50%湿钯碳(140mg),氢气置换3次后搅拌反应3小时停止反应。过滤,滤液减压浓缩,得到粗品标题产物1g(499mg),产物不经纯化直接进行下一步反应。msm/z(esi):226.2[m+1]第五步(1s,3r)-1-(4-溴-2,6-二氟苯基)-2-(2-氟-2-甲基丙基)-3-甲基-1,2,3,4-四氢异喹啉-6-酚1i将化合物1g(1.00g,4.44mmol)溶于甲苯(10ml)中,加入2,6-二氟-4-溴苯甲醛1h(1.18g,5.34mmol),乙酸(2.13g,35.45mmol)。加完后反应在85℃油浴中搅拌16小时,停止反应。减压浓缩,用柱色谱法以展开剂体系b纯化所得残余物,得到标题产物1i(1.35g),产率:71%。msm/z(esi):428.1[m+1];第六步(1s,3r)-6-(苄氧基)-1-(4-溴-2,6-二氟苯基)-2-(2-氟-2-甲基丙基)-3-甲基-1,2,3,4-四氢异喹啉1k将化合物1i(1.3g,3.0mmol)溶于丙酮(20ml),加入苄基溴1j(0.8mg,4.5mmol),碳酸钾(1.3g,9.0mmol)。反应在70℃油浴回流3小时,停止反应。过滤,用丙酮(20ml)洗,滤液减压浓缩得粗品,用柱色谱法以展开剂体系b纯化所得残余物,得到标题产物1k(1.0g),产率:64%。msm/z(esi):518.1[m+1];第七步(2-(4-((1s,3r)-6-(苄氧基)-2-(2-氟-2-甲基丙基)-3-甲基-1,2,3,4-四氢异喹啉-1-基)-3,5-二氟苯氧基)乙基)氨基甲酸叔丁酯1m将化合物1k(500mg,1.0mmol),化合物1l(310mg,1.9mmol)和甲苯(15ml)依次加入反应瓶中,氩气保护,然后加入[(2-二-叔丁基膦基-3-甲氧基-6-甲基-2′,4′,6′-三异丙基-1,1′-联苯基)-2-(2-氨基联苯基)]钯(ii)甲磺酸甲酯(80mg,0.09mmol),碳酸铯(785mg,2.4mmol),抽换氩气3次,升温至90℃搅拌3小时。减压浓缩,用柱色谱法以展开剂体系b纯化所得残余物,得到标题产物1m(480mg),产率:83%。msm/z(esi):599.3[m+1];第八步(2-(3,5-二氟-4-((1s,3r)-2-(2-氟-2-甲基丙基)-6-羟基-3-甲基-1,2,3,4-四氢异喹啉-1-基)苯氧基)乙基)氨基甲酸叔丁酯1n将化合物1m(500mg,0.8mmol)溶于甲醇(15ml),加入20%氢氧化钯碳(58mg,0.08umol),氢气置换3次,30℃下氢化反应4小时,停止反应。过滤旋干得到化合物1n(400mg),产率:94%。直接用于下一步反应。msm/z(esi):509.3[m+1];第九步(1s,3r)-1-(4-(2-((叔丁氧羰基)氨基)乙氧基)-2,6-二氟苯基)-2-(2-氟-2-甲基丙基)-3-甲基-1,2,3,4-四氢异喹啉-6-基三氟甲磺酸酯1o将化合物1n(450mg,0.9mmol)溶于二氯甲烷(20ml),冰浴下加入4-二甲氨基吡啶(10mg,0.08umol),三乙胺(180mg,1.8mmol),然后加入n-苯基双(三氟甲烷磺酸亚胺)(506mg,1.4mmol),室温反应过夜,减压浓缩得粗品。用柱色谱法以展开剂体系b纯化所得残余物,得到标题产物1o(566mg),产率:100%。第十步(2-(3,5-二氟-4-((1s,3r)-2-(2-氟-2-甲基丙基)-3-甲基-6-(1-甲基-1h-吡唑-4-基)-1,2,3,4-四氢异喹啉-1-基)苯氧基)乙基)氨基甲酸叔丁酯1q将化合物1o(140mg,0.2mmol),1-甲基吡唑-4-硼酸频哪醇酯1p(90mg,0.4mmol,采用公知的方法“organicprocessresearch&development,2010,14(4),849-858”制备而得)和1,4-二氧六环(6ml),水(1.5ml)加入反应瓶中,然后加入1,1'-双二苯基膦二茂铁二氯化钯(15mg,0.02mmol),无水碳酸钠(54mg,0.5mmol),氩气置换三次保护,升温至90℃搅拌4小时。反应降至室温,加20ml水,用乙酸乙酯萃取(15ml×3),有机层用饱和食盐水15ml洗涤,无水硫酸钠干燥,过滤浓缩。用柱色谱法以展开剂体系b纯化所得残余物,得到标题产物1q(108mg),产率:86%。msm/z(esi):573.3[m+1];第十一步2-(3,5-二氟-4-((1s,3r)-2-(2-氟-2-甲基丙基)-3-甲基-6-(1-甲基-1h-吡唑-4-基)-1,2,3,4-四氢异喹啉-1-基)苯氧基)乙烷-1-胺j将化合物1q(105mg,0.2mmol)溶于二氯甲烷(4ml),室温下滴加氯化氢的二氧六环溶液(5m,0.4ml),室温搅拌2小时,减压浓缩得化合物j,产率:99%。测试例1本发明化合物对雌激素受体报告基因活性的抑制作用1、实验目的本实验的目的是测试本发明化合物对雌激素受体报告基因活性的抑制作用,根据ic50大小评价化合物的体外活性。2、实验方法表达雌激素受体反应元件控制的荧光素酶报告基因ere-luc(金唯智生物科技有限公司合成)的mcf7细胞(atcc,htb-22)mcf7/ere-luc使用含有10%胎牛血清和500μg/mlg418的mem(gehealthcare,sh30024.01)培养基进行培养。实验第一天,使用含有10%活性炭处理的胎牛血清(biosun,bs-0004-500)的mem不完全培养基将mcf7/ere-luc细胞以30,000个/孔的密度种于96孔板,每孔100μl细胞悬液,放置37℃,5%co2的细胞培养箱培养过夜。第二天,每孔加入10μl用不完全培养基配制的β-雌二醇和不同浓度的待测化合物,β-雌二醇的终浓度是0.1nm,化合物的终浓度是从10μm开始进行10倍梯度稀释的9个浓度点,设置含有0.5%dmso的空白对照,放置37℃,5%co2的细胞培养箱培养20小时。第三天,取出96孔板,每孔加入100μlone-glotmluciferaseassaysystem(promega,e6110)检测荧光素酶的活性,室温放置3分钟至细胞充分裂解后,使用多标记微孔板酶标仪(perkinelmer,victor3)读取发光信号值,用graphpadprism软件根据化合物的浓度和发光信号值计算化合物抑制活性的ic50值。3、测试结果本发明中化合物对雌激素受体报告基因活性的抑制作用通过以上的试验进行测定,经graphpadprism计算得到,ic50=2nm。测试例2本发明化合物对mcf7细胞增殖的抑制效应1、实验目的本实验的目的是测定本发明化合物对mcf7细胞增殖的抑制活性,根据ic50大小评价化合物的体外活性。2、实验方法mcf7细胞(atcc,htb-22)用含有10%胎牛血清的mem(gehealthcare,sh30024.01)完全培养基进行培养。实验第一天,使用完全培养基将mcf7细胞以3,000个/孔的密度种于96孔板,每孔100μl细胞悬液,放置37℃,5%co2的细胞培养箱培养过夜。第二天吸掉培养基,每孔更换为135μl含有2%胎牛血清的mem不完全培养基,同时每孔加入15μl用不完全培养基配制的不同浓度的待测化合物,化合物的终浓度是从100nm开始进行4倍梯度稀释的9个浓度点,设置含有0.5%dmso的空白对照,放置37℃,5%co2的细胞培养箱培养144小时。第八天,取出96孔细胞培养板,每孔加入150μlluminescentcellviabilityassay(promega,g7573),室温放置10分钟后,使用多标记微孔板酶标仪(perkinelmer,victor3)读取发光信号值,用graphpadprism软件根据化合物的浓度和发光信号值计算化合物抑制活性的ic50值。3、数据分析用graghpadprism对化学发光信号值与化合物的对数浓度作图,得出化合物的ic50值。编号ic50(nm)maxinhibition(%)10.1897测试例3表达erα突变体mcf7细胞增殖抑制实验生物学评价1、实验目的本实验的目的是测定本发明化合物对表达erα突变体mcf7细胞增殖的抑制活性。2、实验方法定点突变和细胞系构建人雌激素受体α(estrogenreceptorα,erα)蛋白的突变体erαy537s与erαd538g使用双引物pcr的方式以野生型esr1基因的cdna(accessionno.nm000125)为模板进行定点突变获得。突变所使用的引物序列如下(下划线标出的核苷酸为突变的位点):y537s:f-aagaacgtggtgcccctctctgacctgctgctggagatg;r-catctccagcagcaggtcagagaggggcaccacgttctt;d538g:f-aacgtggtgcccctctatggcctgctgctggagatgctg;r-cagcatctccagcagcaggccatagaggggcaccacgtt。将突变体esr1的cdna克隆至目标慢病毒载体pcdh-cmv-mcs-ef1-puro上。然后将带有突变体esr1基因序列的慢病毒质粒以及慢病毒包装质粒通过lipofectamine3000transfectionreagent(thermofisherscientific,cat#l3000075)转染到hek-293t细胞(atcc,crl-3216)中。转染后48小时,将带有病毒的培养基上清过滤、超速离心获得病毒沉淀,用适量的培养基重悬溶解后,加入到mcf7细胞(atcc,htb-22)中,并加入终浓度为8μg/ml的polybrene孵育过夜。转染两天后,在细胞培养液中加入1μg/ml的嘌呤霉素进行抗性筛选,约两周后得到能够稳定表达erαy537s与erαd538g突变体的mcf7细胞系。细胞增殖抑制实验将表达erα突变体的mcf7细胞用含有10%胎牛血清的mem(gehealthcare,sh30024.01)完全培养基进行培养。实验第一天,使用完全培养基将细胞以3,000个/孔的密度种于96孔板,每孔100μl细胞悬液,放置37℃,5%co2的细胞培养箱培养过夜。第二天吸掉培养基,每孔更换为135μl含有2%胎牛血清的mem不完全培养基,同时每孔加入15μl用不完全培养基配制的不同浓度的待测化合物,化合物的终浓度是从100nm开始进行4倍梯度稀释的9个浓度点,设置含有0.5%dmso的空白对照,放置37℃,5%co2的细胞培养箱培养144小时。第八天,取出96孔细胞培养板,每孔加入150μlluminescentcellviabilityassay(promega,g7573),室温放置10分钟后,使用多标记微孔板酶标仪(perkinelmer,victor3)读取发光信号值,用graphpadprism软件根据化合物的浓度和发光信号值计算化合物抑制活性的ic50值。实施例2:草酸盐将100mg化合物(e)-4-((2-(3,5-二氟-4-((1s,3r)-2-(2-氟-2-甲基丙基)-3-甲基-6-(1-甲基-1h-吡唑-4-基)-1,2,3,4-四氢异喹啉-1-基)苯氧基)乙基)氨基)-1-吗啉基丁-2-烯-1-酮加入0.67ml丙酮搅拌溶解,然后加入二水合草酸的丙酮的溶液200μl(100mg/ml),室温搅拌,过滤,真空干燥,得到产物草酸盐。实施例3:草酸盐将800mg化合物(e)-4-((2-(3,5-二氟-4-((1s,3r)-2-(2-氟-2-甲基丙基)-3-甲基-6-(1-甲基-1h-吡唑-4-基)-1,2,3,4-四氢异喹啉-1-基)苯氧基)乙基)氨基)-1-吗啉基丁-2-烯-1-酮溶于乙酸乙酯(4ml)中,滴加无水草酸(231mg)的乙酸乙酯(4ml)溶液,析出固体,室温搅拌过夜,过滤,收集滤饼,真空干燥,得到产物(920mg,黄色固体)。所得产物经xrpd检测,无特征峰,为无定型;dsc谱在63.39℃、121.85℃、211.31℃有吸热峰峰值;干燥失重(tga)试验显示,25℃-145℃失重2.69%。实施例4:草酸盐将30mg化合物(e)-4-((2-(3,5-二氟-4-((1s,3r)-2-(2-氟-2-甲基丙基)-3-甲基-6-(1-甲基-1h-吡唑-4-基)-1,2,3,4-四氢异喹啉-1-基)苯氧基)乙基)氨基)-1-吗啉基丁-2-烯-1-酮溶于异丙醇(0.2ml)中,滴加草酸(4.3mg)的0.2ml异丙醇溶液,升温至50℃,搅拌溶清,降至室温,滴加0.25ml异丙醚,浑浊,升温至50℃溶清,缓慢降至室温,搅拌析晶,过滤,干燥得到产物(4.3mg,黄色固体)。经x-射线粉末衍射检测,无特征峰,为无定型。实施例5:草酸盐将30mg化合物(e)-4-((2-(3,5-二氟-4-((1s,3r)-2-(2-氟-2-甲基丙基)-3-甲基-6-(1-甲基-1h-吡唑-4-基)-1,2,3,4-四氢异喹啉-1-基)苯氧基)乙基)氨基)-1-吗啉基丁-2-烯-1-酮溶于乙腈(0.2ml)中,滴加草酸(4.3mg)的0.2ml乙腈溶液,升温至50℃,搅拌溶清,缓慢降至室温,搅拌析晶,过滤,干燥得到产物(4.3mg,黄色固体)经x-射线粉末衍射检测,其xrpd图谱见图1并定义该晶型为晶型a,其特征峰位置如下表1所示:表1实施例6:草酸盐将30mg化合物(e)-4-((2-(3,5-二氟-4-((1s,3r)-2-(2-氟-2-甲基丙基)-3-甲基-6-(1-甲基-1h-吡唑-4-基)-1,2,3,4-四氢异喹啉-1-基)苯氧基)乙基)氨基)-1-吗啉基丁-2-烯-1-酮的草酸盐(无定型)溶于1,4-二氧六环(0.3ml)中,加入乙酸乙酯(1.0ml),升温至70℃溶清,缓慢降至室温,搅拌析晶,过滤,干燥得到产物(8mg,黄色固体)。经x-射线粉末衍射检测,无特征峰,为无定型。实施例7:甲磺酸盐将100mg化合物(e)-4-((2-(3,5-二氟-4-((1s,3r)-2-(2-氟-2-甲基丙基)-3-甲基-6-(1-甲基-1h-吡唑-4-基)-1,2,3,4-四氢异喹啉-1-基)苯氧基)乙基)氨基)-1-吗啉基丁-2-烯-1-酮加入环己烷搅拌溶解,然后加入甲磺酸(15.40mg),室温搅拌,过滤,真空干燥,得到产物。所得产物经xrpd检测,无特征峰,为无定型。所得产物的硫元素检测结果:4.23%,表明该盐中化合物与甲磺酸的摩尔比约为1:1。实施例8:硫酸盐将800mg化合物(e)-4-((2-(3,5-二氟-4-((1s,3r)-2-(2-氟-2-甲基丙基)-3-甲基-6-(1-甲基-1h-吡唑-4-基)-1,2,3,4-四氢异喹啉-1-基)苯氧基)乙基)氨基)-1-吗啉基丁-2-烯-1-酮加入乙酸异丙酯(4ml)中,滴加硫酸(251mg)的乙酸异丙酯(4ml)溶液,搅拌析晶,过滤,收集滤饼,真空干燥,得到产物(950mg,黄色固体)。所得产物经xrpd检测,无特征峰,为无定型。实施例9:硫酸盐将40mg化合物(e)-4-((2-(3,5-二氟-4-((1s,3r)-2-(2-氟-2-甲基丙基)-3-甲基-6-(1-甲基-1h-吡唑-4-基)-1,2,3,4-四氢异喹啉-1-基)苯氧基)乙基)氨基)-1-吗啉基丁-2-烯-1-酮(63.93μmol)加入1,4-二氧六环(0.2ml)中,滴加硫酸(13.2mg)的1,4-二氧六环(0.3ml)溶液,升温至70℃,搅拌0.5小时,析出固体,继续搅拌3小时,缓慢降至室温,室温搅拌48小时,过滤,收集滤饼,真空干燥,得到产物(15mg,黄色固体)。所得产物经xrpd检测,无特征峰,为无定型。实施例10:5-磺基水杨酸盐将1.0g化合物(e)-4-((2-(3,5-二氟-4-((1s,3r)-2-(2-氟-2-甲基丙基)-3-甲基-6-(1-甲基-1h-吡唑-4-基)-1,2,3,4-四氢异喹啉-1-基)苯氧基)乙基)氨基)-1-吗啉基丁-2-烯-1-酮溶于乙酸异丙酯(5ml)中,室温下滴加5-磺基水杨酸(406.3mg)的醋酸异丙酯(15ml)溶液,搅拌析晶16小时,过滤,收集滤饼,干燥,得到产物(1.1g,黄色固体)。经x-射线粉末衍射检测,无特征峰,为无定型;dsc谱在57.41℃有吸热峰峰值;干燥失重(tga)试验显示,25℃-90℃失重4.0%,90-225℃失重8.76%。经nmr检测,5-磺基水杨酸与化合物1的成盐比例为:1:1。1hnmr(400mhz,cd3od)δppm8.31(d,1h),7.92(s,1h),7.86(dd,1h),7.78(s,1h),7.37(s,1h),7.28(d,1h),6.79-6.96(m,3h),6.62-6.77(m,3h),5.58(br.s.,1h),4.95(m,1h),4.30(br.s.,2h),3.82-3.96(m,6h),3.62(br.s.,9h),3.49(br.s.,2h),3.33-3.38(m,1h),2.86(dd,1h),2.64-2.80(m,1h),1.32-1.38(m,3h),1.28-1.32(m,3h),1.23(d,3h)。实施例11:5-磺基水杨酸盐将30mg化合物(e)-4-((2-(3,5-二氟-4-((1s,3r)-2-(2-氟-2-甲基丙基)-3-甲基-6-(1-甲基-1h-吡唑-4-基)-1,2,3,4-四氢异喹啉-1-基)苯氧基)乙基)氨基)-1-吗啉基丁-2-烯-1-酮的5-磺基水杨酸盐加入异丙醇(1.1ml)中,升温至70℃,溶清,缓慢降至室温,搅拌析晶,过滤,收集滤饼,干燥,得到产物(11mg,黄色固体)。经x-射线粉末衍射检测,无特征峰,为无定型。实施例12:5-磺基水杨酸盐将100mg化合物(e)-4-((2-(3,5-二氟-4-((1s,3r)-2-(2-氟-2-甲基丙基)-3-甲基-6-(1-甲基-1h-吡唑-4-基)-1,2,3,4-四氢异喹啉-1-基)苯氧基)乙基)氨基)-1-吗啉基丁-2-烯-1-酮加入醋酸异丙酯(1ml)中,室温下滴加5-磺基水杨酸(40.6mg)的醋酸异丙酯(1ml)溶液,搅拌过夜,过滤,收集滤饼,真空干燥,得到标题产物(115mg,黄色固体)。经x-射线粉末衍射检测,无特征峰,为无定型。实施例13:稳定性研究将无定型游离碱和无定型甲磺酸盐分别置于25℃,rh60%条件下和40℃,rh75%条件下,进行稳定性考察,数据如下:表2实验结果:在25℃,rh60%或40℃,rh75%条件下,无定型游离碱呈现明显降解,温度越高,降解越明显,40℃,rh75%条件下放置26天,主峰峰纯度降至78.01%。相应地,化合物成盐后,其稳定性有所提高。实施例14:稳定性研究将无定型硫酸盐(实施例8)、无定型草酸盐(实施例3)样品敞口平摊放置,考察在加热(40℃、60℃)、光照(4500lux)、高湿(rh75%、rh90%)条件下样品的稳定性,取样考察期为30天,数据如下:表3实验结果:在光照、高温40℃、高温60℃、高湿75%、高湿90%条件下,放置30天,硫酸盐或草酸盐样品在各条件下纯度均略有下降,高湿条件下吸潮,建议样品避光、干燥、低温保存。实施例15:将20mg化合物(e)-4-((2-(3,5-二氟-4-((1s,3r)-2-(2-氟-2-甲基丙基)-3-甲基-6-(1-甲基-1h-吡唑-4-基)-1,2,3,4-四氢异喹啉-1-基)苯氧基)乙基)氨基)-1-吗啉基丁-2-烯-1-酮草酸盐无定型样品加入环己烷(0.3ml)中,室温搅拌96小时,过滤,收集滤饼,真空干燥,得到产物(12mg,黄色固体)。经x-射线粉末衍射检测,无特征峰,为无定型。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1