一种荧光显像探针及其制备方法和应用与流程
[0001]
本发明属于荧光显像技术领域,具体涉及一种荧光显像探针及其制备方法和应用。
背景技术:
[0002]
如今,荧光成像已经成为一种有效的肿瘤成像技术。它的优点很多,例如:灵敏度高,成本低,非入侵性和无放射性。然而,较浅的组织穿透深度和较高的背景信号限制了荧光成像的进一步发展。在过去的十年中,近红外荧光(nirf)成像已经突破了上述两个障碍。近红外(nir)区域(700-900nm)是理想的成像区域。相对于可见光,生物组织吸收的近红外光非常少;因此,近红外光有更深的组织穿透深度。而且在nir区域内,生物体的自发荧光几乎不存在,所以nirf成像的背景信号很低。综上所述,nirf成像更适用于体内肿瘤荧光成像。mayeulcollot团队(chemical communications 2017,53,6117-6120)报告了一种nir荧光基团(本篇中称作g0,见图2),它是花菁染料ir780的衍生物。g0是具有良好的光稳定性的优异的荧光团,吸收峰在780nm,发射峰在808nm。因此,可以以g0为荧光基团设计新型分子探针。此外,在合成过程中使用5-氯-1-戊炔使得g0结构包含两个炔基,这方便通过铜(i)催化的炔-叠氮化物环化加成反应(cuaac)引入其他功能基团。
[0003]
尽管g0有许多优点,但仍然会在水溶液中聚集。通常,花菁染料是亲脂性的,因此它们在水溶液中的溶解度较低,会形成聚集体,这大大降低了探针对光的利用效率。许多研究表明,花菁染料在有机溶剂中具有较高的摩尔消光系数和光子量子产率,但是这些数据在水溶液中会大大降低。考虑到花菁染料的这一特性,为了获得更好的成像效果,新探针的设计必须降低其聚集趋势。为降低其聚集趋势,获得更好的成像效果,如中国专利文献cn1975418a中提供了一种叶酸-壳聚寡糖-花菁荧光染料生物探针及制备方法,这种新型探针的水溶性得到显著提高,并增加了染料与细胞的相容性,在与生物组织结合及成像时表现出荧光加合效应,探针分子与dna或细胞相互作用的光学灵敏性显著提高,且具有很好的癌细胞标记的靶向性。然而在上述文献中的探针仅做了体外的探针标记蛋白实验,未能进行体内实验,难以预期其是否适应于生物体内癌细胞的标记,而又因为生物体内的环境十分复杂,花菁染料类的荧光探针不仅面对在生物体内容易聚集的问题,还要面对肿瘤细胞靶向性,是否容易跨膜运输进入肿瘤细胞以及在肿瘤细胞内的滞留时间长等问题,然而,目前还未有相关报道提供一种同时满足上述条件的花菁染料类荧光探针,因此,本发明提供了一种兼具荧光强度高、靶向性强、跨膜运输强以及在肿瘤细胞内滞留时间长等优势的花菁染料荧光显像探针。
技术实现要素:
[0004]
为此,本发明要解决的技术问题是提供一种兼具荧光强度高、靶向性强、跨膜运输强以及在肿瘤细胞内的滞留时间长优势的花菁染料g0的荧光显像探针及其制备方法和应用。
[0005]
第一方面,本发明提供一种荧光显像探针,所述荧光显像探针为式g1或式g2,所述式g1或式g2结构式如下:
[0006][0007]
第二方面,本发明提供一种荧光显像探针的制备方法,式g0与biotin-peg5-azide通过叠氮-炔环加成反应,得到式g1或式g2;式g0的结构式为
[0008][0009]
进一步地,所述荧光显像探针的制备方法,包括如下步骤:
[0010]
1)将化合物g0、biotin-peg5-azide和催化剂溶解于有机溶液,常温搅拌,得到反应产物;优选的常温搅拌为25℃下搅拌1.5h以上;
[0011]
2)将反应产物中的液体蒸发后,得到的固体,经过分离,即得到探针。
[0012]
进一步地,所述催化剂为cuso4·
5h2o、l-抗坏血酸钠盐和thpta配体的混合物。
[0013]
进一步地,g0与biotin-peg5-azide的摩尔比为1:(0.1-0.7),且g0与cuso4·
5h2o、l-抗坏血酸钠盐和thpta配体的摩尔比为1:0.35:0.7:0.35时,得到g1。
[0014]
进一步地,g0与biotin-peg5-azide的摩尔比为1:2.2,且g0与cuso4·
5h2o、l-抗坏血酸钠盐和thpta配体的摩尔比为1:1:2:1时得到g2。
[0015]
进一步地,所述有机溶液为thf、ch2cl2、dmf或dmso溶液中的一种;所述分离为柱层析分离。
[0016]
一种肿瘤靶向荧光显像剂,包括上述的荧光显像探针或上述的荧光显像探针的制备方法得到的荧光显像探针。
[0017]
一种肿瘤靶向荧光显像剂的制备方法,将上述的荧光显像探针或上述的荧光显像探针的制备方法得到的荧光显像探针溶解于水或生理盐水,制备得到荧光显像剂。
[0018]
一种荧光显像探针使用方法,包括如下1)或2):
[0019]
1),将上述的荧光显像探针或上述的荧光显像探针的制备方法得到的荧光显像探针溶解于水或生理盐水,或上述的荧光显像剂的制备方法制备得到的荧光显像剂,加入细胞培养液中与肿瘤细胞共孵育;细胞培养液中探针的浓度为25μm,孵育时间为12h;优选的,所述肿瘤细胞选自生物素受体高表达的肿瘤细胞,优选为hela细胞或a549细胞。
[0020]
2),将权利要求1所述的荧光显像探针或上述的荧光显像探针的制备方法得到的荧光显像探针溶解于水或生理盐水,或所述的荧光显像剂的制备方法制备得到的荧光显像
剂,通过尾静脉注射给药;以20g动物体重为计,给药浓度为20-30μm;给药体积为150-250μl;优选的,给药浓度为25μm,给药体积为200μl;优选的,所述肿瘤为荷瘤裸鼠的肿瘤。
[0021]
所述方法为非疾病诊断治疗方法。
[0022]
上述的荧光显像探针或上述制备方法制备得到的荧光显像探针在制备肿瘤靶向的荧光显像剂中的应用。
[0023]
本发明具备以下优点:
[0024]
1、在本发明中,设计并合成了两种具有良好水溶性和生物靶向性的nirf探针g1和g2。探针的peg结构改善了探针的亲水性,体外光谱和脂质-水分配系数证实:g1和g2具有较低的聚集趋势。生物素结构增强了探针对br阳性肿瘤细胞的靶向性,g1和g2在体外细胞实验和体内荧光成像中均显示出良好的荧光成像效果。由于特别选择的亲水性基团—peg结构与靶向基团—生物素的之间的配合,使得探针g1和g2在亲水性和靶向性之间能够取得平衡,进而在荧光强度、肿瘤靶向性、跨膜运输性以及在肿瘤细胞内的滞留时间等几个方面获得平衡,使得制备的探针g1和g2兼具荧光强度高、靶向性强、跨膜运输强以及在肿瘤细胞内的滞留时间长优势。综上所述,探针g1和g2有发展前景的新型肿瘤靶向分子探针。
[0025]
2、本发明的探针的制备方法,通过调整投料比例,能够制备得到g1和g2。本发明提供的一种制备荧光显像探针的制备方法,通过控制反应物的投料比,能够最大程度的制备获得连接不同个数靶向基团的荧光探针g1和g2,降低了反应过程中副产物的生成,提高反应产率,降低后期反应产物的分离和纯化步骤。
附图说明
[0026]
为了更清楚地说明本发明具体实施方式或现有技术中的技术方案,下面将对具体实施方式或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图是本发明的一些实施方式,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
[0027]
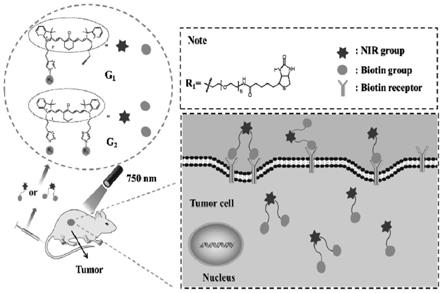
图1是实施例3中探针g1和g2在hela荷瘤小鼠体内的靶向机制图;
[0028]
图2是实施例1中化合物g1和g2的合成路线,反应条件:(a)cuso4·
5h2o,thpta,抗坏血酸钠,dmf与h2o的体积比=5/1,室温,0.5equivalent biotin-peg5-azide;(b)cuso4·
5h2o,thpta,抗坏血酸钠,dmf与h2o的体积比=5/1,室温,2.2equivalent biotin-peg5-azide;
[0029]
图3是实施例1中化合物g0的esi-ms分析图;纵坐标是离子的相对丰度(%);
[0030]
图4是实施例1中化合物g0的hplc分析图,检测波长为690nm;
[0031]
图5是实施例1中化合物g1的esi-ms分析图;纵坐标是离子的相对丰度(%);
[0032]
图6是实施例1中化合物g1的hplc分析图,检测波长为690nm;
[0033]
图7是实施例1中g1的核磁共振氢谱;
[0034]
图8是实施例1中g1的核磁共振碳谱;
[0035]
图9是实施例1中化合物g2的esi-ms分析图;纵坐标是离子的相对丰度(%)
[0036]
图10是实施例1中化合物g2的hplc分析图,检测波长为690nm;
[0037]
图11是实施例1中g2的核磁共振氢谱;
[0038]
图12是实施例1中g2的核磁共振碳谱;
[0039]
图13是实施例2中g0、g1和g2的hplc分析图,检测波长为690nm;
[0040]
图14是实施例2中在甲醇中(a、d和g)和水中(b、e和h),不同浓度的化合物g0、g1和g2的归一化吸收光谱;在甲醇中,吸光度与g0、g1和g2的浓度之间的拟合直线见c、f和i;a-c为g0,d-f为g1,g-i为g2;
[0041]
图15(a)是实施例2中使用甲醇作为溶剂,浓度为4μm的g0、g1和g2溶液的荧光光谱;纵坐标是吸光度(a.u.),横坐标为波长(nm);
[0042]
图15(b)是实施例2中使用水作为溶剂,浓度为4μm的g0、g1和g2溶液的荧光光谱;纵坐标是吸光度(a.u.),横坐标为波长(nm);
[0043]
图15(c)是实施例2中使用水作为溶剂,浓度为4μm的g0、g1和g2溶液的nir荧光图像;
[0044]
图15(d)是实施例2中平均荧光强度定量分析图,纵坐标为辐射效率;
[0045]
图16(a)是实施例3中g1和g2分别在3.125、6.25、12.5、25和50μm的浓度下,在hela细胞中孵育12h的毒性分析图;
[0046]
图16(b)是实施例3中g1和g2分别在3.125、6.25、12.5、25和50μm的浓度下,在hela细胞中孵育24h的毒性分析图;
[0047]
图16(c)是实施例3中g1和g2分别在3.125、6.25、12.5、25和50μm的浓度下,在lo2细胞中孵育12h的毒性分析图;
[0048]
图16(d)是实施例3中g1和g2分别在3.125、6.25、12.5、25和50μm的浓度下,在lo2细胞中孵育24h的毒性分析图;
[0049]
图17是实施例3中用25μm的g1和g2溶液分别孵育hela细胞和lo2细胞12h后的荧光图像;biotin(+)表示细胞用200μm生物素溶液预处理2小时,而生物biotin(-)表示细胞仅与探针一起孵育;每幅图中的左图为明场下细胞显像图,右图为细胞的荧光显像图;
[0050]
图18(a)是实施例4中探针g1和g2在正常小鼠体内,不同时间点的nir图像;
[0051]
图18(b)是实施例4中探针g1和g2在正常小鼠肾脏中,不同时间点荧光强度的定量;
[0052]
图18(c)是实施例4中探针g1和g2在正常小鼠12和192h肝脏代谢的nir图像;
[0053]
图19(a)是实施例4中探针g1和g2在hela荷瘤小鼠体内,不同时间点的nir图像;
[0054]
图19(b)为实施例4中探针g1在hela荷瘤小鼠体内,不同时间点肿瘤和肌肉荧光强度的定量;
[0055]
图19(c)为是实施例4中探针g2在hela荷瘤小鼠体内,不同时间点肿瘤和肌肉荧光强度的定量;
[0056]
图19(d)为是实施例4中探针g1和g2在hela荷瘤小鼠体内,不同时间点肿瘤和肌肉荧光强度比值(t/m)。
[0057]
图20(a)为实施例4中g1在hela荷瘤小鼠解剖后的肿瘤组织和主要器官的体外荧光图;
[0058]
图20(b)为实施例4中g2在hela荷瘤小鼠解剖后的肿瘤组织和主要器官的体外荧光图;
[0059]
图20(c)为g1和g2在hela荷瘤小鼠解剖后的肿瘤和主要器官荧光强度的定量分析。
具体实施方式
[0060]
5-氯-1-戊炔,2,3,3-三甲基吲哚啉和碘化钾购自安耐吉化工(中国上海);biotin-peg5-azide购自华腾公司(中国湖南);2-氯-1-甲酰基-3-(羟基亚甲基)-1-环己烯购自玉涵公司(中国上海)。除非另有说明,否则所有试剂均直接使用。
[0061]
探针的吸收光谱通过uv-vis光谱仪(lambda 25;perkin elmer)获得。发射光谱通过稳态和瞬态荧光光谱仪(qm/tm;pti)获得。高效液相色谱(hplc)用于分析化合物,主要由hplc泵(waters 1525;usa),双波长吸收uv/vis检测器(waters2487;usa)和c18色谱柱(10μm,6mm
×
250mm;elitehlc)组成。核磁共振谱记录在全数字核磁共振谱仪(avanceⅲ400mhz;bruker)上。通过四极串联质谱仪(sq-detect 2;waters),电喷雾电离质谱仪(esi-ms)验证化合物的质荷比。正常裸鼠和荷瘤小鼠的nir荧光图像是通过ivis荧光体内成像系统(caliper life sciences;perkinelmer)中的获得的。
[0062]
本发明中的室温范围为25
±
2℃。
[0063]
实施例1化合物g1和g2的合成
[0064]
1化合物g0的合成
[0065]
按照已报道的方法(chemical communications2017,53,6117-6120)获得了g0,具体方法:将ki(5.8g,34.93mmol,6eq)溶解在乙腈(25ml)中;然后加入5-氯-1-戊炔(1841μl,17.45mmol,3eq)。反应液在50℃下搅拌40分钟。向其中添加2,3,3-三甲基吲哚啉(933μl,5.82mmol,1eq)。将反应混合物在85℃下加热24小时。将反应液过滤,然后通过减压蒸馏去除乙腈。所得固体通过柱色谱法(流动相为:meoh与dcm体积比为2-5%)纯化得到化合物1,收率为17%(224mg,0.991mmol)。将化合物1(221mg,0.977mmol,1eq)和2-氯-1-甲酰基-3-(羟基亚甲基)-1-环己烯(84.1mg,0.489mmol,0.5eq)溶解于无水乙腈(3ml)。然后加入乙酸酐(123μl)和醋酸钠(40mg,0.489mmol,0.5eq)。将该溶液在85℃下搅拌5小时。反应后的混合物通过柱色谱法(流动相为1-5%甲醇的二氯甲烷溶液,硅胶200-300目)纯化,得到金黄色深绿色固体(g0),产率为80%(230mg,0.391mmol)。c
40 h
44
cln
2+
([m]
+
)的esi-ms计算值588.2,实测值588.32。
[0066]
esi-ms结果见图3,hplc分析结果见图4。
[0067]
2探针g1的合成
[0068]
将化合物g0(38.0mg,0.0648mmol,1eq)和biotin-peg5-azide(24.1mg,0.0454mmol,0.7eq)添加到cuso4·
5h2o(5.7mg,0.0227mmol,0.35eq)中。然后加入l-抗坏血酸钠盐(9.0mg,0.0454mmol,0.7eq)和thpta配体(9.85mg,0.0227mmol,0.35eq)。将所有化合物溶解在二甲基甲酰胺(dmf)和h2o的体积比为5∶1的5ml dmf水溶液中。将反应混合物在25℃下搅拌1.5h。之后,将溶剂蒸发至干。通过柱层析分离(流动相为:甲醇与二氯甲烷的体积比为1-20%,硅胶200-300目)纯化固体,具体方式,流动相从甲醇:二氯甲烷体积比为1%至20%依次增加,每次增加5%,到20%时洗脱出目标产物。以62.5%的产率获得30mg(0.0268mmol)g1,g1为深绿色固体。c
62
h
84
cln8o7s
+
([m
+
])的esi-ms计算值是1119.59,实测值是1119.46。
[0069]
测试纯化获得的g1的核磁共振和光谱特征:氢谱和碳谱的数据结果如下:
[0070]
氢谱解析:1h nmr(400mhz,chloroform-d)δ8.45(d,j=14.2hz,1h),8.31(d,j=13.7hz,1h),7.97(s,1h),7.38(dd,j=11.3,7.8hz,4h),7.33
–
7.28(m,2h),7.22(t,j=
7.4hz,2h),7.12(d,j=7.9hz,1h),6.31(d,j=14.2hz,1h),6.13(d,j=13.8hz,1h),4.60(s,4h),4.23(s,2h),4.14(t,j=7.1hz,2h),3.90(s,2h),3.63(d,j=6.4hz,18h),3.56(s,2h),3.44(s,2h),3.18(s,1h),2.96(s,2h),2.68
–
2.62(m,4h),2.39
–
2.34(m,2h),2.27(s,2h),2.06
–
2.01(m,2h),1.94(s,1h),1.72(s,18h),1.43(s,4h),1.33(t,j=7.2hz,2h)。
[0071]
碳谱解析:
13
c nmr(101mhz,cdcl3)δ174.22,170.98,151.16,146.54,143.26,142.26,141.67,140.64,129.15,128.74,128.15,127.27,126.21,124.80,122.28,122.22,111.58,109.96,102.62,99.59,82.49,70.48,70.27,70.11,69.86,69.06,55.31,50.85,49.84,48.94,46.16,44.08,42.60,39.41,35.25,28.27,28.05,27.77,27.67,26.75,26.37,26.22,25.72,25.38,21.94,20.59,16.04。
[0072]
3探针g2的合成
[0073]
将g0(37.1mg,0.0632mmol,1eq)和biotin-peg5-azide(73.9mg,0.139mmol,2.2eq)添加到cuso4·
5h2o(15.8mg,0.0632mmol,1eq)中。然后加入l-抗坏血酸钠盐(25mg,0.01264mmol,2eq)和thpta配体(27.4mg,0.0632mmol,1eq)。所有化合物都溶解在二甲基甲酰胺(dmf)和h2o的体积比为5∶1的5ml dmf溶液中。将反应混合物在25℃下搅拌1.5h。之后,将溶剂蒸发至干。通过柱层析分离(流动相为:1-30%(体积百分含量)甲醇的二氯甲烷溶液,硅胶200-300目)纯化固体,具体方式:流动相从甲醇比二氯甲烷为1%至30%依次增加,每次增加5%(v/v),到30%时洗脱出目标产物。以60%的产率获得了深绿色固体形式的g2(58mg,0.035mmol)。c
84 h
123
cln
14
o
14
s
2+
([m
+
])的esi-ms计算值1651.85,实测值是1652.36。
[0074]
测试纯化获得的g2的核磁共振和光谱特征:氢谱和碳谱的数据结果如下:
[0075]
氢谱解析:1h nmr(400mhz,chloroform-d)δ8.35(d,j=13.5hz,2h),7.75(s,2h),7.39(dd,j=14.8,7.3hz,4h),7.24(dd,j=15.7,8.0hz,6h),6.68(s,2h),6.27(d,j=13.6hz,2h),5.41
–
5.25(m,4h),4.57(s,4h),4.35(s,2h),4.24(s,2h),3.90(s,4h),3.62(d,j=8.4hz,36h),3.55(s,4h),3.41(s,4h),3.15(s,2h),2.92(s,4h),2.75(d,j=12.2hz,2h),2.66(s,2h),2.24(d,j=7.0hz,4h),2.01(dd,j=14.3,8.5hz,4h),1.72(s,12h),1.66(d,j=6.8hz,4h),1.43(s,2h),1.26(s,12h).
[0076]
碳谱解析:
13
c nmr(101mhz,cdcl3)δ173.81,172.42,150.82,144.71,142.10,141.02,129.90,128.95,127.73,125.36,123.10,122.17,111.02,101.45,70.44,70.39,70.07,69.83,69.48,62.04,60.48,55.57,50.28,49.32,43.96,40.50,39.20,35.69,31.89,29.77,29.68,29.61,29.51,29.31,29.23,28.18,28.11,27.99,27.21,26.35,25.56,22.67,22.49。
[0077]
如图2所示,通过cuaac反应将g0与biotin-peg5-azide连接,得到探针g1和g2。通过控制biotin-peg5-azide的当量分别获得g1和g2。探针g1和g2通过质谱,hplc,核磁共振氢谱和碳谱进行了表征(图5-12)。预期探针上的不同结构基团发挥不同的功能:(1)g0作为近红外荧光基团;(2)生物素残基用于增强探针对肿瘤的特异性;(3)peg结构改善探针的亲水性,降低探针的聚集趋势。此外,g1和g2的结构差异会影响探针的成像效果,因此本发明后续的实验中研究它们的亲水性和靶向效率的差异。
[0078]
实施例2化合物g1和g2光化学和光物理性质的测定
[0079]
1吸收光谱和荧光光谱的测定
[0080]
选择甲醇作为溶剂,制备g0,g1和g2的储备溶液(20mm),用于测定光谱。用甲醇和水
分别将g0、g1和g2的储备溶液稀释至不同浓度(4μm,3μm,2μm,1μm,0.5μm),以获得吸收光谱。获得了在λmax和浓度下的吸光度拟合直线。准备4μm的g0、g1和g2溶液以测量荧光光谱。本发明分别使用甲醇和水作为溶剂,以比较探针在不同溶剂中的荧光强度。
[0081]
2光子量子产率的测定
[0082]
分别用甲醇和水作溶剂准备g1、g2和吲哚菁绿(icg)溶液(1μm);将icg用作参考化合物(在甲醇中,φ=0.043;在水中,φ=0.005)。根据以下公式计算探针的量子产率(φ):
[0083][0084]
其中i代表测得的荧光光谱的积分面积;η是溶剂的折射率;a是溶液的吸光度;x表示探针;st是参考化合物。
[0085]
3脂质/水分配系数(log p)
[0086]
将g0(20μl,100μm)和1ml水依次添加到1ml正辛醇中。将溶液充分震荡后,将其高速离心3分钟。吸取150μl有机相以测量其吸光度。根据图14c,f,i计算其浓度(co)。g0的脂质/水分配系数(log p)可根据以下公式计算:
[0087]
log p=log(c
o
/c
w
)
ꢀꢀꢀ
(2)
[0088]
其中c
o
代表探针在正辛醇中的浓度,c
w
代表探针在水中的浓度。然后通过相同的方法测得g1和g2的log p。
[0089]
为了比较g0,g1和g2的亲水性,本发明测量了三者的log p,它是反映亲水性的重要参数。g0,g1和g2的log p分别为1.893
±
0.229、1.039
±
0.106和0.441
±
0.143(g0>g1>g2)。如图2所示,g2连接两个peg5基团,g1连接一个peg5基团,而g0不连接peg。这证明增加peg增强了探针的亲水性。此外,本发明通过hplc比较了三个探针的极性(图13),表明g0<g1<g2。此结果与亲水性顺序一致。
[0090]
接下来,本发明测量了g0、g1和g2的光物理和光化学性质(图14,表1)。在甲醇中,g1和g2均在784nm处有吸收峰;g0的吸收峰在780nm。g1和g2分别在809nm和807nm处有荧光发射峰,而g0的发射峰在808nm处。这些微小的波长变化表明,生物素和peg的引入不会改变nir荧光团的基本荧光性质。值得注意的是,在甲醇中,g0,g1和g2的最大吸收强度遵循比尔-朗伯定律,表明三者没有发生聚集(图14c,f,i)。但是,g0在水中的摩尔消光系数(ε)为94248m-1
cm-1
,仅为甲醇中的42%(表1)。这表明g0在水中发生了聚集,对光的吸收大大降低。该实验结果与其他花菁染料的特性一致。例如,吲哚菁绿(icg)是美国食品和药物管理局(fda)批准的用于临床的nir荧光成像剂。在甲醇中,icg的ε是230000m-1
cm-1
,而在水中,ε只有157000m-1
cm-1
(下降31%)。如表1所示,与g0相比,g1在水中的ε是141372m-1
cm-1
(g0的1.5倍);g2的ε是161568m-1
cm-1
(g0的1.71倍)。以上数据表明,peg结构降低了探针的聚集趋势,增强了探针对光的吸收强度。化合物在水中的吸收强度的差异也与log p的计算结果一致。
[0091]
表1.g0,g1和g2的光物理和光化学数据
[0092][0093]
a
λ
ex
=750nm;
b,d icg作为参比化合物(φ=4.3%in meoh;φ=0.5%in h2o);
c
λ
ex
=750nm.;em是发射波长;ex为激发波长
[0094]
如图15a,b所示,在甲醇中,g0、g1和g2的吸光度差别不大(g0:11280a.u.;g1:12429a.u;g2:12077a.u.)。但是,在水中,三者的吸光度差异很大。在水中,g1和g2的荧光强度分别是g0的1.67倍和2.04倍(图15)。类似的差异也反映在荧光量子产率上(表1)。g1和g2在水中的荧光量子产率(φ)分别是g0的1.98倍和2.76倍。ivis光谱成像系统也显示g1和g2在水中与g0相比具有较强的荧光强度(图15c)。在水中g1和g2的强度分别是g0的1.49倍和1.70倍(图15d)。以上实验结果证明,g1和g2是具有较强亲水性的新型花菁探针。
[0095]
实施例3体外肿瘤细胞特异性成像
[0096]
1细胞培养
[0097]
为了验证探针在肿瘤部位的特异性积累,选择hela细胞作为br阳性肿瘤细胞,选择lo2细胞作为br阴性正常细胞。将hela和lo2细胞置于含有10%(v/v)胎牛血清(fbs)的高葡萄糖dmem培养基中,并在5%(v/v)co2的潮湿稳定环境中培养。孵育温度为37℃。
[0098]
2细胞毒性测定
[0099]
进行mtt测定以评估探针g1和g2的细胞毒性。将hela和lo2细胞分别接种到96孔板中,将密度控制为每孔10000个细胞。将细胞在37℃下孵育24h,以使细胞附着。将g1和g2分别溶于dmf,得到20mm g1和g2储备溶液,用完全培养基(含有10%(v/v)胎牛血清(fbs)的高葡萄糖dmem培养基中)将g1和g2的储备溶液稀释至目标浓度。除去培养基后,将不同浓度(3.125至50μm)的探针溶液添加到96孔板中。然后将细胞在37℃下孵育12和24h。将mtt溶解在磷酸盐缓冲液(pbs,ph=7.4),浓度为5mg/ml,并向每个孔中添加20μl浓度为5mg/ml mtt溶液。然后将细胞孵育4h。用pbs洗涤细胞3次,除去培养基,并加入150μl dmso以溶解甲臜。用酶标仪读取每个孔在470nm处的平均吸光度。对照组是将上述步骤g1和g2探针溶液替换为相同体积的双蒸水。根据实验组和对照组的平均吸光度之比评价细胞毒性。
[0100]
3体外肿瘤细胞特异性成像
[0101]
选择n,n-二甲基甲酰胺(dmf)作为g1和g2溶剂,以制备用于细胞和动物实验的储备溶液(g1和g2的浓度均为20mm)。在6孔板中,分别接种hela和lo2细胞,将密度控制为每孔50000个细胞。将细胞在37℃下,5%co2培养箱中培养过夜。用培养基将g1和g2的储备溶液稀释至25μm,取体积3ml添加到6孔板中。将细胞在5%co2气氛下于37℃孵育。持续6h和12h。然后将细胞用pbs洗涤3次。除去培养基后,加入适量的pbs以维持细胞形态。最后,通过荧光显微镜观察细胞。
[0102]
在进行细胞荧光成像实验之前,应该评估两种探针的细胞毒性。如图16所示,探针g1和g2对hela和lo2细胞均显示出低毒性。例如,将细胞与25μm的g1溶液孵育24h后,hela和lo2的细胞活力分别为87.84%和88.46%。在将细胞与25μm的g2溶液孵育24h后,hela和lo2的细胞活力分别为80.46%和85.10%。因此,探针g1和g2可用于后续的细胞和动物实验。
[0103]
为了验证探针g1和g2对br阳性肿瘤细胞的靶向能力,选择hela和lo2细胞进行细胞荧光成像实验。将两种探针分别与细胞孵育12h后,在hela细胞中观察到明亮的荧光信号,而在lo2细胞中几乎没有荧光信号,这说明探针g1和g2可以特异性识别hela细胞(图17)。
[0104]
为了确认两种探针的特异性细胞成像是由br介导的,本发明还设计了一种竞争摄取实验。将hela细胞用过量的生物素预处理2h,以此占据大量的br位点。与仅施加探针g1和g2实验组相比,在用过量生物素预处理的对照组中,荧光信号明显减弱(图17)。该结果表明,生物素基团和br的相互作用使得探针可以特异性识别hela细胞。
[0105]
实施例4体内实验
[0106]
1正常裸鼠中探针g1和g2的动态生物分布
[0107]
选择正常裸鼠(4周大,雌性)以检测探针在体内的分布。将g1和g2分别用生理盐水稀释至25μm,并通过尾静脉将200μl探针溶液(浓度为25μm)注入正常裸鼠中,在不同的时间点(0、1、4、12、24、72、168、192h)通过成像系统监测正常裸鼠,结果见图18。注射探针后1h时,在小鼠肾脏中发现了明亮的荧光信号(图18a)。此外,在肝脏中也检测到了明亮的荧光信号(图18c)。像其他花菁探针一样,探针g1和g2主要由肝脏和肾脏代谢。还发现了g1和g2代谢速率的差异,如图18所示,在注射24h时,在注射g1的小鼠中仍检测到非常强的荧光信号,而注射g2的小鼠中,荧光信号相对较弱。168h时,探针g2几乎已从小鼠身上清除,而小鼠中仍存在少量探针g1。这表明探针g2的代谢速度更快。
[0108]
2荷瘤小鼠培养
[0109]
将hela细胞(1
×
107个)悬浮在pbs溶液中,皮下注射到裸鼠(4-6周大的雌性小鼠)的前肢腋窝中,以构建hela肿瘤异种移植模型。饲养小鼠至肿瘤体积达到100-120mm3。
[0110]
3荷瘤小鼠的体内荧光成像
[0111]
将探针g1和g2分别用生理盐水稀释至25μm,然后通过尾静脉将200μl探针溶液注射到hela肿瘤异种移植小鼠体内。使用成像系统在注射后的不同时间点对小鼠进行成像,见图19。从图19中可以看出,对于g1和g2,在注射后6h时,在肿瘤部位都观察到了明亮的荧光信号,并且持续了162h。连续的肿瘤成像对于诊断疾病和进行手术很有帮助。不同的是,与g1相比,g2在正常组织中的清除速度更快。
[0112]
为了进一步验证探针的肿瘤靶向能力,发明人定量分析了肿瘤和肌肉处的荧光强度,并计算了肿瘤和肌肉信号值之比(t/m)(图19)。对于g1,在注射后24h以内,肿瘤处的荧
光强度迅速增加,然后保持稳定,明显高于肌肉处的荧光强度。对于g2,注射12h之后,肿瘤部位的荧光强度便趋于稳定,并且明显强于肌肉的荧光强度。在荧光成像中,t/m是nirf探针的重要参数。当t/m超过2.5时,则认为探针在肿瘤中的富集是显著的。如图19所示,15.2h时,g1的t/m超过了2.5,在10.5h时,g2的t/m超过了2.5。具体数据如下:对于g1,t/m分别是3.06(24h),3.59(72h),4.01(96h),4.69(168h);对于g2,t/m分别是2.88(24h),3.94(72h),4.19(96h),4.27(168h)。以上数据表明,探针g1和g2可以特异性识别肿瘤组织。
[0113]
可以看出,肿瘤组织对g1的摄取高于g2。探针注射后72h时,肿瘤部位g1的荧光强度约为g2的2.39倍。本发明认为,g2的较强亲水性不利于探针跨膜运输。但是,这并不是唯一的原因。如图18和图19所示,探针g2在小鼠中的代谢比g1快。短时间内大量的g2会被排泄掉,所以肿瘤处g2的积累更少。
[0114]
4.生物分布
[0115]
为了进一步了解探针在肿瘤和其他器官中的分布情况,在注射探针后72h时,处死并解剖荷瘤小鼠。如图20所示,在肿瘤处检测到了强荧光信号,而肺,胃,脾,心脏和肠的荧光信号较弱,这验证了探针g1和g2在肿瘤处的选择性积累。在肝脏和肾脏中也检测到明亮的荧光信号,表明探针g1和g2主要由肝脏代谢,肾脏排泄,这与图18的结果一致。g1在肿瘤部位的荧光强度是g2的2.52倍(图20),此结果与图19一致。
[0116]
在本项研究中,本发明设计了两种具有良好水溶性的nirf探针(g1和g2)。探针的peg结构改善了探针的亲水性,生物素结构增强了探针对br阳性肿瘤细胞的靶向性。体外光谱和脂质-水分配系数证实g1和g2具有较低的聚集趋势。g1和g2在体外细胞实验和体内荧光成像中均显示出良好的荧光成像效果。综上所述,探针g1和g2是具有发展前景的新型肿瘤靶向分子探针。
[0117]
显然,上述实施例仅仅是为清楚地说明所作的举例,而并非对实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动。这里无需也无法对所有的实施方式予以穷举。而由此所引伸出的显而易见的变化或变动仍处于本发明创造的保护范围之中。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1