制备哌嗪的方法与流程
1.本发明涉及哌嗪的制备领域,具体地,涉及一种由n-羟乙基哌嗪或/和n-氨乙基哌嗪制备哌嗪的方法。
背景技术:
2.哌嗪是一种重要的乙撑胺类产品,英文名为piperazine,简称为pip,分子式为c4h10n2,哌嗪及其衍生物是非常重要的精细化工产品,多用于医药、农药、染料中间体,在医药领域应用极为广泛,是多种医药产品的原材料,随着医药需求不断扩大,尤其是喹诺酮类药物需求量不断提升,哌嗪及其衍生物的市场需求不断攀升。
3.一乙胺又名乙胺,氨基乙烷,乙烷胺,英文名为monoethylamine,分子式为c2h7n,是一种具有多用途的精细化工产品,可广泛用于医药、农药、选矿药剂、涂料、光化学品、纺织、高聚物、食品化学品、干洗剂等。
4.us10266541中记述以乙醇胺为原料,使用环胺催化剂,生成哌嗪和三乙烯二胺,从实施例1和实施例2可以看出,其产物中存在n-羟乙基哌嗪和n-氨乙基哌嗪,且n-氨乙基哌嗪副产较高。
5.国内的哌嗪等乙撑胺产品基本依赖进口,且现有的乙醇胺法制备乙二胺及哌嗪路线,会产生大量的重组分,其组成主要是n-羟乙基乙二胺、n-羟乙基哌嗪和n-氨乙基哌嗪,这几种重组分在分离上需要大量能耗,同时会严重影响目标产物的收率,而且将n-羟乙基哌嗪和n-氨乙基哌嗪等重组分返回反应器前进行循环的话,易发生结焦等问题,影响催化剂使用寿命。
6.因此需要开发一种制备哌嗪的新路线,以提高哌嗪的收率以及选择性。
技术实现要素:
7.本发明的目的是为了克服现有技术存在的制备哌嗪时,收率以及选择性低,使得分离能耗高的技术缺陷,提供一种由n-羟乙基哌嗪或/和n-氨乙基哌嗪制备哌嗪的方法。
8.本发明的发明人发现,将n-羟乙基哌嗪或n-氨乙基哌嗪氨化制备哌嗪不仅可以提高乙醇胺法制备乙撑胺装置的经济效益,也提供了一条制备哌嗪的新路线。因此,本发明提供了一种制备哌嗪的方法,该方法包括:在氢气和催化剂的存在下,使n-羟乙基哌嗪或/和n-氨乙基哌嗪与氨源接触进行反应,其中,所述n-羟乙基哌嗪或/和n-氨乙基哌嗪与氨源的摩尔比为1:5-30,所述催化剂包括载体和负载在所述载体上的活性组分和任选的助剂,所述活性组分包括钴和/或镍,所述助剂为vib族金属中的至少一种、ib族金属中的至少一种和iib族金属中的至少一种的组合。
9.与现有技术相比,本发明在制备哌嗪时表现出较高的选择性和转化率,具有较大的现实意义,不仅可以提高乙醇胺法制备哌嗪装置的经济效益,还提供了一条制备哌嗪的新路线,同时可以较高收率联产一乙胺。
10.进一步地,本发明的发明人研究发现,配合特定的助剂能够进一步改善催化剂的
催化活性。而且,改变催化剂的碱性并不是提高催化剂活性和选择性的最佳方案,相反,改变催化剂的氨吸附量和孔结构更有利于提高由n-羟乙基哌嗪或/和n-氨乙基哌嗪制备哌嗪的选择性和转化率,从而以克服技术偏见的方式进一步提高了哌嗪的收率。
具体实施方式
11.在本文中所披露的范围的端点和任何值都不限于该精确的范围或值,这些范围或值应当理解为包含接近这些范围或值的值。对于数值范围来说,各个范围的端点值之间、各个范围的端点值和单独的点值之间,以及单独的点值之间可以彼此组合而得到一个或多个新的数值范围,这些数值范围应被视为在本文中具体公开。
12.本发明提供了一种制备哌嗪的方法,其特征在于,该方法包括:在氢气和催化剂的存在下,使n-羟乙基哌嗪或/和n-氨乙基哌嗪与氨源接触进行反应,其中,所述n-羟乙基哌嗪或/和n-氨乙基哌嗪与氨源的摩尔比为1:5-30,所述催化剂包括载体和负载在所述载体上的活性组分和任选的助剂,所述活性组分包括钴和/或镍,所述助剂为vib族金属中的至少一种、ib族金属中的至少一种和iib族金属中的至少一种的组合。
13.根据本发明,所述接触的条件可以包括:温度为150-240℃,优选为160-230℃。
14.根据本发明,所述接触的条件还可以包括:压力为6-19mpa,优选为7-18mpa。
15.根据本发明,所述接触的条件还可以包括:n-羟乙基哌嗪或/和n-氨乙基哌嗪的液相体积空速为0.05-0.5m3/(m3·
h),优选为0.1-0.4m3/(m3·
h)。
16.根据本发明,所述接触的条件还可以包括:氢气、氨源与n-羟乙基哌嗪或/和n-氨乙基哌嗪的摩尔比为1-4:5-30:1。优选为1-3:6-28:1。
17.根据本发明,所述n-羟乙基哌嗪或/和n-氨乙基哌嗪可以以纯物质的形式进行反应,也可以配制成溶液进行反应,为了进一步提高转化率,减少重组分的生成,提高哌嗪的选择性,防止由于温度变化导致部分高凝固点产物析出堵塞管道,优选地,所述n-羟乙基哌嗪或/和n-氨乙基哌嗪以溶液的方式进行反应。对所述溶液中n-羟乙基哌嗪或/和n-氨乙基哌嗪的浓度没有特别的要求,可以为10-95重量%。所述溶液中的溶剂可以为各种能够溶解n-羟乙基哌嗪或/和n-氨乙基哌嗪且不与其他反应原料反应的溶剂,可以为水,也可以为有机溶剂,优选地,所述溶液中的溶剂选自水和/或1,4-二氧六环。
18.根据本发明,所述氨源为能够提供氨基和/或胺基的反应物,可以选自氨、c1-12的伯胺和c2-12的仲胺中的至少一种,优选为氨、一甲胺、二甲胺、甲基乙基胺、一乙胺和二乙胺中的至少一种。“c1-12”指代碳原子数为1、2、3、4、5、6、7、8、9、10、11、12的伯胺或仲胺。
19.根据本发明,所述方法还可以包括对氨化反应的产物进行分离纯化以分别得到哌嗪和一乙胺的步骤。分离纯化的方式可以为精馏。
20.根据本发明,所述氨化反应可以是连续的,也可以是间歇的,可以选择使用固定床管式反应器、高压搅拌釜、鼓泡床反应器、流化床反应器、微通道反应器等反应设备中的一种。
21.本发明中,所述载体可以为本领域常见的载体。根据本发明的优选实施方式,所述载体包括氧化铝载体、掺杂元素和其它载体,所述其它载体选自氧化硅和/或分子筛;所述载体的氨吸附量为0.2-0.6mmol/g;所述载体中孔径在7-27nm范围内的孔容占所述载体孔容的百分比大于65%。
22.根据本发明,所述载体使用的氧化铝前驱体在制备时掺加了氧化硅前驱体和/或分子筛前驱体等,在制备成载体后可进一步大幅改善催化剂的扩散性和孔结构稳定性能。因此,根据本发明一种优选的实施方式,所述载体中氧化铝载体的含量占氧化铝载体与其它载体的总量的70重量%以上,优选为80-97重量%。
23.根据本发明的优选实施方式,所述掺杂元素的含量占载体的含量的0.05-5重量%,优选为0.08-3重量%。
24.根据本发明的优选实施方式,所述载体的氧化铝前驱体中掺杂元素以硼酸根离子、氟离子、磷酸根离子、硫酸根离子和硒酸根离子中的至少一种的方式掺杂。所述掺杂元素优选选自硼、氟、磷、硫和硒中的至少一种。在制备载体的前驱体过程就掺杂进非金属元素,使得掺杂元素主要存在于载体的体相中,而不是附于表面。
25.根据本发明的优选实施方式,所述载体的氨吸附量为0.3-0.6mmol/g。
26.根据本发明的优选实施方式,所述载体中孔径在7-27nm范围内的孔容占所述载体孔容的百分比为70-90%。根据本发明的优选实施方式,孔径小于7nm的孔容占所述载体孔容的百分比为0-8%,优选为0-5%。根据本发明的优选实施方式,所述载体的比表面积为120-210m2/g。
27.根据本发明的优选实施方式,所述载体的孔容为0.45-1.1ml/g。
28.本发明中,所述载体的比表面积、孔容和不同孔径的孔的占比通过氮吸附-脱附方法测得,具体参见gb/t6609.35-2009。
29.根据本发明,相对于每100g的载体,所述活性组分的含量可以为10-46g,优选为18-38g。
30.根据本发明,相对于每100克的载体,所述助剂的含量可以为0.1-10g,优选为0.5-6g。
31.根据本发明,为了更好发挥出本发明的催化剂的性能、调优反应产物比例、减少不需要的副反应,所述催化剂还含有如前所述的助剂。所述助剂中vib族金属、ib族金属和iib族金属的重量比优选为0.1-10:0.1-10:1,更优选为0.2-8:0.2-8:1。优选地,所述vib族金属选自钼和/或钨。优选地,所述ib族金属选自铜、银和金中的至少一种。优选地,所述iib族金属选自锌。
32.根据本发明,所述载体可以采用现有的能够得到氨吸附量和孔结构等满足上述范围的方法制备得到,且获取氨吸附量和孔结构满足上述范围的载体是本领域技术人员能够实施的。但根据本发明的一种优选实施方式,所述载体通过包括以下步骤的方法制备得到:将含有氧化铝前驱体、掺杂元素和其它载体前驱体的混合物依次进行成型、干燥和焙烧,所述其它载体前驱体选自氧化硅前驱体(如硅溶胶)和/或分子筛前驱体(如zsm-5)。成型的方法可以使用捏合、滚球或打片等。
33.在以上载体的制备方法中,本领域技术人员能够理解的是:如果提供载体前驱体的原料中已经含有所需量的掺杂元素,那么只需使用这种原料进行成型即可,如果提供载体前驱体的原料中不含掺杂元素或掺杂元素的含量较低(不足),那么可以额外引入掺杂元素。
34.在以上载体的制备方法中,本领域技术人员能够根据最终载体中某成分(如掺杂元素)的量确定某成分原料(如载体改性剂)的用量,因此,本文中部分原料用量未示出。
35.在以上载体的制备方法中,所述掺杂元素优选由硼酸、氢氟酸、磷酸、硫酸和硒酸中的至少一种提供。
36.在以上载体的制备方法中,所述氧化铝前驱体优选为拟薄水铝石。所述拟薄水铝石的比表面积优选为260-400m2/g。所述拟薄水铝石的孔容优选为0.8-1.2。所述拟薄水铝石可以由碳化法、有机铝水解法、硫酸铝法和硝酸法中的至少一种方法制备,优选使用硫酸铝法制备的拟薄水铝石。选用具有特定孔结构的拟薄水铝石能够获得性能更优的催化剂。
37.在以上载体的制备方法中,所述干燥的条件可以包括:温度为80-150℃(例如可以为80℃、85℃、90℃、95℃、100℃、105℃、110℃、115℃、120℃、130℃、140℃、150℃,或者上述任意两个数值之间的任意值),时间6-20h(例如可以为6h、7h、7.5h、8h、8.5h、9h、9.5h、10h、10.5h、10h、11h、11.5h、12h、12.5h、13h、14h、14.5h、15h、15.5h、16h、17h、18h、19h、20h,或者上述任意两个数值之间的任意值)。
38.在以上载体的制备方法中,所述焙烧的条件可以包括:温度为500-1100℃(例如可以为500℃、550℃、600℃、650℃、680℃、700℃、750℃、800℃、850℃、900℃、950℃、990℃、1000℃、1050℃、1100℃,或者上述任意两个数值之间的任意值),时间为2-20h(例如可以为2h、2.5h、3h、3.5h、4h、4.5h、5h、5.5h、6h、7h、8h、9h、9.5h、10h、10.5h、11h、12h、15h、18h、20h,或者上述任意两个数值之间的任意值)。
39.根据本发明,所述催化剂可以还原后使用。可以用含有氢气的气体在350-500℃下还原,优选在350-450℃下还原。氢气可以是纯氢气,也可以是惰性气体稀释的氢气,例如氮气与氢气的混合气。在还原时逐渐升高还原温度,升温不宜太快,例如不超过20℃/小时。可以通过监测还原体系中h2o的产生确定还原的时间,也即当还原体系不再产生新的h2o时,结束还原,本领域技术人员能够据此对还原的时间进行选择,对此不再详述,例如,在最高温度下还原时间可以为2-5h。还原可以在反应器中直接进行,然后进行催化反应。也可以在单独的反应器中还原,也称为器外还原,还原后从反应器中卸出前可以用含有氧气的混合气进行钝化,钝化温度例如为10-60℃,特别是20-40℃。器外还原且钝化的催化剂装填到反应器中在使用前可以使用氢气或氢气与氮气的混合物活化,活化温度例如为150-250℃,优选170-200℃。可以通过监测活化体系中h2o的产生确定活化的时间,也即当活化体系不再产生新的h2o时,结束活化,本领域技术人员能够据此对活化的时间进行选择,对此不再详述,例如,在最高温度下活化时间例如为1-5h,优选为2-3h,也可以不活化直接使用,取决于催化剂中的活性组分和助剂被氧化的程度。
40.本发明还提供了一种制备如前所述的催化剂的方法,该方法包括:将活性组分和助剂负载于载体上。
41.可以理解的是,制备所述催化剂的方法中还可以包括:按照前述方法制备所述载体的步骤。
42.本发明中,将活性组分和助剂负载于载体上的方法可以为浸渍法,也即,使用含活性组分前驱体和助剂前驱体的溶液浸渍载体,之后进行干燥和焙烧。浸渍法是将载体放在合适的含有所述活性组分和助剂的前驱体的溶液中浸泡,前驱体吸附负载在载体上。浸渍方法细分,包括干浸法、湿浸法、多次浸渍法、混合浸渍法和喷浸法等。干、湿浸渍法是指载体在浸渍活性组分前驱体之前的状态,是干燥的还是预先用水浸湿的。多次浸渍法是指一种或几种组分的前驱体混合溶液分多次浸渍,或者不同前驱体分组分批次浸渍上去,多次
浸渍法在每次浸渍后都需要干燥和焙烧以“锚定”浸渍上去的组分。混合浸渍法是在活性组分和助剂所使用的前驱体之间不发生沉淀反应时一起浸渍上去。喷浸法是用喷枪把浸渍溶液喷到连续转动的载体上,使浸渍液正好将载体的孔体积填充饱和。本发明的催化剂可以根据加工厂的情况合理地选择这些浸渍方法。
43.浸渍载体的金属(活性组分或助剂)优选以金属盐的溶液形式使用,例如硝酸盐、甲酸盐、草酸盐、乳酸盐等。溶剂优选使用水,一些有机溶剂也可以使用,例如乙醇。金属盐溶液浸渍载体可以以任何需要的顺序进行,也可以是用多种含有一种或多种金属盐的溶液连续进行浸渍。所有或单一浸渍步骤可以分几次进行,还可以改变浸渍顺序。选择溶液的浓度使需要量的金属负载在载体上。浸渍后的载体优选在80-150℃下干燥,更优选80-120℃下干燥。根据干燥温度、干燥物料多少和干燥设备等情况,合理地选择干燥时间,例如8小时,以干燥后的含水量不影响后续的焙烧为准则。干燥后在150-500℃下焙烧将盐中的结晶水除去或将盐分解为氧化物,优选在300-500℃下焙烧1-6h。多次浸渍的情况下,最好在每次浸渍后都进行干燥和焙烧。
44.本发明中,活性组分或助剂负载操作对催化剂的微观结构影响不大,因此,所得催化剂与载体具有类似的孔结构。
45.根据本发明,所述方法包括先对催化剂进行筛选,筛选出组成或结构参数满足上述要求的催化剂,再按照上述方式进行氨化反应。
46.以下将通过实施例对本发明进行详细描述。以下实施例中,拟薄水铝石粉体的干基(al2o3)含量为72重量%;硅溶胶购自青岛海洋化工有限公司。
47.制备例1
48.将硫酸铝法制备的拟薄水铝石粉体(比表面积380m2/g,孔容1.02ml/g,拟薄水铝石粉体中含掺杂元素s,相对100g的以al2o3计的拟薄水铝石粉体,含有s元素2.15g;拟薄水铝石粉体制备过程中,在开始时掺加有sio2前驱体水玻璃(硅酸钠水溶液),使得焙烧后载体中源于sio2前驱体的sio2的质量占载体总质量的4%)用含5vol%硝酸的稀酸水捏合,捏合后挤成直径5mm条状,切成4mm长短,在120℃下干燥8h,然后在650℃下焙烧5h,制成所需的载体。
49.将186.5g六水合硝酸钴(工业级,纯度98%)、6.83g六水合硝酸锌(分析纯)和5.65g三水合硝酸铜(分析纯)用水溶解为148ml溶液,分两次用喷浸法将该溶液负载在获得的100g载体上,每次喷浸后都在120℃下干燥4小时,然后在400℃下焙烧4小时。然后将1.8g四水合钼酸铵(分析纯)用水溶解为74ml溶液,喷浸在上面获得的半成品上,在120℃下干燥4小时,然后在400℃下焙烧4小时。然后用氢气逐渐升温还原,升温还原速率为20℃/小时,最后在430℃下还原3小时,获得催化剂a-1。
50.制备例2
51.将硫酸铝法制备的拟薄水铝石粉体(比表面积375m2/g,孔容0.98ml/g,拟薄水铝石粉体中含掺杂元素b,相对100g的以al2o3计的拟薄水铝石粉体,含有b元素0.53g;拟薄水铝石粉体制备过程中,在开始时掺加有sio2前驱体水玻璃(硅酸钠水溶液),使得焙烧后载体中源于sio2前驱体的sio2的质量占载体总质量的11%))用含5vol%硝酸的稀酸水捏合,捏合后挤成3mm粗的三叶草形状,在100℃下干燥12h,然后在590℃下焙烧8h,制成所需的载体。
52.将151.7g六水合硝酸镍(工业级,纯度98%)、6.83g六水合硝酸锌(分析纯)和5.65g三水合硝酸铜(分析纯)用水溶解为156ml溶液,分两次用喷浸法将该溶液负载在获得的100g载体上,每次喷浸后都在120℃下干燥4小时,然后在400℃下焙烧4小时。然后将3.7g四水合钼酸铵用水溶解为78ml溶液,喷浸在上面获得的半成品上,在120℃下干燥4小时,然后在400℃下焙烧4小时。然后用氢气逐渐升温还原,升温还原速率为20℃/小时,最后在430℃下还原3小时,获得催化剂a-2。
53.制备例3
54.将硫酸铝法制备的拟薄水铝石粉体(比表面积380m2/g,孔容1.02ml/g,拟薄水铝石粉体中含掺杂元素s,相对100g的以al2o3计的拟薄水铝石粉体,含有s元素2.15g;拟薄水铝石粉体制备过程中,在开始时掺加有sio2前驱体水玻璃(硅酸钠水溶液),使得焙烧后载体中源于sio2前驱体的sio2的质量占载体总质量的4%)用含5vol%硝酸的稀酸水捏合,捏合后挤成直径5mm条状,切成4mm长短,在120℃下干燥8h,然后在650℃下焙烧5h,制成所需的载体。
55.将55.4g六水合硝酸钴(工业级,纯度98%)、6.83g六水合硝酸锌(分析纯)和5.65g三水合硝酸铜(分析纯)用水溶解为144ml溶液,分两次用喷浸法将该溶液负载在获得的100g载体上,每次喷浸后都在120℃下干燥4小时,然后在400℃下焙烧4小时。然后将1.8g四水合钼酸铵用水溶解为72ml溶液,喷浸在上面获得的半成品上,在120℃下干燥4小时,然后在400℃下焙烧4小时。然后用氢气逐渐升温还原,升温还原速率为20℃/小时,最后在430℃下还原3小时,获得催化剂a-3。
56.制备例4
57.将硫酸铝法制备的拟薄水铝石粉体(比表面积340m2/g,孔容1.13ml/g,拟薄水铝石粉体中含掺杂元素p,相对100g的以al2o3计的拟薄水铝石粉体,含有p元素0.18g。在拟薄水铝石粉体制备过程中,在开始时掺加有zsm-5分子筛前驱体(zsm-5粉体,南开大学催化剂厂,sio2/al2o3=45(摩尔比),下同),使得焙烧后载体中源自拟薄水铝石粉体al2o3占载体总质量的85%)用含5vol%硝酸的稀酸水捏合,捏合后挤成直径4mm齿状球,在80℃下干燥20h,然后在530℃下焙烧6h,制成所需的载体。
58.将126g六水合硝酸钴(工业级,纯度98%)、4.55g硝酸锌和0.79g硝酸银(分析纯)用水溶解为210ml溶液,分两次用喷浸法将该溶液负载在获得的100g载体上,每次喷浸后都在120℃下干燥4小时,然后在400℃下焙烧4小时。然后将3.7g四水合钼酸铵(分析纯)用水溶解为105ml溶液,喷浸在上面获得的半成品上,在120℃下干燥4小时,然后在400℃下焙烧4小时。然后用氢气逐渐升温还原,升温还原速率为20℃/小时,最后在430℃下还原3小时,获得催化剂a-4。
59.制备例5
60.按照制备例4的方法制备载体,不同的是,在拟薄水铝石粉体制备过程中,在开始时掺加有zsm-5分子筛前驱体,使得焙烧后载体中源自拟薄水铝石粉体al2o3占载体总质量的85%。
61.将126g六水合硝酸钴(工业级,纯度98%)、9.1g六水合硝酸锌(分析纯)和1.57g硝酸银(分析纯)用水溶解为208ml溶液,分两次用喷浸法将该溶液负载在获得的100g载体上,每次喷浸后都在120℃下干燥4小时,然后在400℃下焙烧4小时。然后将7.4g四水合钼酸铵
(分析纯)用水溶解为104ml溶液,喷浸在上面获得的半成品上,在120℃下干燥4小时,然后在400℃下焙烧4小时。然后用氢气逐渐升温还原,升温还原速率为20℃/小时,最后在430℃下还原3小时,获得催化剂a-5。
62.制备例6
63.将硫酸铝法制备的拟薄水铝石粉体(比表面积341m2/g,孔容1.11ml/g,拟薄水铝石粉体中含掺杂元素p,相对100g的以al2o3计的拟薄水铝石粉体,含有p元素4.2g。在拟薄水铝石粉体制备过程中,在开始时掺加有zsm-5分子筛前驱体,使得焙烧后载体中源自拟薄水铝石粉体al2o3占载体总质量的85%)用含5vol%硝酸的稀酸水捏合,捏合后挤成直径4mm齿状球,在80℃下干燥20h,然后在530℃下焙烧6h,制成所需的载体。
64.其余步骤与制备例4相同,获得催化剂a-6。
65.制备例7
66.将硫酸铝法制备的拟薄水铝石粉体(比表面积288m2/g,孔容0.93ml/g,拟薄水铝石粉体中含掺杂元素s,相对100g的以al2o3计的拟薄水铝石粉体,,含有s元素0.88g。拟薄水铝石粉体制备过程中,在开始时掺加有sio2前驱体水玻璃(硅酸钠水溶液),使得焙烧后载体中源于sio2前驱体的sio2的质量占载体总质量的8%)用含5vol%硝酸的稀酸水捏合,捏合后挤成直径4mm三叶草,在100℃下干燥8h,然后在850℃下焙烧4h,制成所需的载体。
67.将201.6g六水合硝酸钴(工业级,纯度98%)、2.28g六水合硝酸锌(分析纯)和8.48g三水合硝酸铜(分析纯)用水溶解为146ml溶液,分两次用喷浸法将该溶液负载在获得的100g载体上,每次喷浸后都在120℃下干燥4小时,然后在400℃下焙烧4小时。然后将2.7g偏钨酸铵(分析纯)用水溶解为73ml溶液,喷浸在上面获得的半成品上,在120℃下干燥4小时,然后在400℃下焙烧4小时。然后用氢气逐渐升温还原,升温还原速率为20℃/小时,最后在430℃下还原3小时,获得催化剂a-7。
68.制备例8
69.采用制备例7中制备的载体。
70.将100.8g六水合硝酸钴(工业级,纯度98%)、2.28g六水合硝酸锌(分析纯)和8.48g三水合硝酸铜(分析纯)用水溶解为150ml溶液,分两次用喷浸法将该溶液负载在获得的100g载体上,每次喷浸后都在120℃下干燥4小时,然后在400℃下焙烧4小时。然后将2.7g偏钨酸铵(分析纯)用水溶解为75ml溶液,喷浸在上面获得的半成品上,在120℃下干燥4小时,然后在400℃下焙烧4小时。然后用氢气逐渐升温还原,升温还原速率为20℃/小时,最后在430℃下还原3小时,获得催化剂a-8。
71.制备例9
72.将硫酸铝法制备的拟薄水铝石粉体(比表面积281m2/g,孔容0.87ml/g,拟薄水铝石粉体中含掺杂元素f,相对100g的以al2o3计的拟薄水铝石粉体,含有f元素0.82g;拟薄水铝石粉体制备过程中,在开始时掺加有sio2前驱体水玻璃(硅酸钠水溶液),使得焙烧后载体中源于sio2前驱体的sio2的质量占载体总质量的21%)用含5vol%硝酸的稀酸水捏合,捏合后挤成4mm粗的三叶草形状,在90℃下干燥18h,然后在770℃下焙烧9h,制成所需的载体。
73.将176.4g六水合硝酸钴(工业级,纯度98%)、2.28g六水合硝酸锌(分析纯)和11.3g三水合硝酸铜(分析纯)用水溶解为130ml溶液,分两次用喷浸法将该溶液负载在获得的100g载体上,每次喷浸后都在120℃下干燥4小时,然后在400℃下焙烧4小时。然后将2.0g
偏钨酸铵(分析纯)用水溶解为65ml溶液,喷浸在上面获得的半成品上,在120℃下干燥4小时,然后在400℃下焙烧4小时。然后用氢气逐渐升温还原,升温还原速率为20℃/小时,最后在430℃下还原3小时,获得催化剂a-9。
74.制备例10
75.将硫酸铝法制备的拟薄水铝石粉体(比表面积274m2/g,孔容0.85ml/g,拟薄水铝石粉体中含掺杂元素s,相对100g的以al2o3计的拟薄水铝石粉体,含有s元素0.95g;拟薄水铝石粉体制备过程中,在开始时掺加有sio2前驱体水玻璃(硅酸钠水溶液),使得焙烧后载体中源于sio2前驱体的sio2的质量占载体总质量的25%)用含5vol%硝酸的稀酸水捏合,捏合后挤成3.5mm粗的三叶草形状,在150℃下干燥6h,然后在930℃下焙烧6h,制成所需的载体。
76.将227.5g六水合硝酸镍(工业级,纯度98%)、2.28g六水合硝酸锌(分析纯)和1.10g硝酸银(分析纯)用水溶解为156ml溶液,分3次用喷浸法将该溶液负载在获得的100g载体上,每次喷浸后都在120℃下干燥4小时,然后在400℃下焙烧4小时。然后将5.4g偏钨酸铵(分析纯)用水溶解为52ml溶液,喷浸在上面获得的半成品上,在120℃下干燥4小时,然后在400℃下焙烧4小时。然后用氢气逐渐升温还原,升温还原速率为20℃/小时,最后在430℃下还原3小时,获得催化剂a-10。
77.制备例11
78.将硫酸铝法制备的拟薄水铝石粉体(比表面积265m2/g,孔容0.81ml/g,拟薄水铝石粉体中含掺杂元素p,相对100g的以al2o3计的拟薄水铝石粉体,含有p元素3.1g;拟薄水铝石粉体制备过程中,在开始时掺加有sio2前驱体水玻璃(硅酸钠水溶液),使得焙烧后载体中源于sio2前驱体的sio2的质量占载体总质量的28%)用含5vol%硝酸的稀酸水捏合,捏合后挤成3mm粗的三叶草形状,在100℃下干燥8h,然后在1020℃下焙烧5h,制成所需的载体。
79.将141.1g六水合硝酸钴(工业级,纯度98%)、2.28g六水合硝酸锌(分析纯)和2.20g硝酸银(分析纯)用水溶解为144ml溶液,分3次用喷浸法将该溶液负载在获得的100g载体上,每次喷浸后都在120℃下干燥4小时,然后在400℃下焙烧4小时。然后将5.4g偏钨酸铵(分析纯)用水溶解为48ml溶液,喷浸在上面获得的半成品上,在120℃下干燥4小时,然后在400℃下焙烧4小时。然后用氢气逐渐升温还原,升温还原速率为20℃/小时,最后在430℃下还原3小时,获得催化剂a-11。
80.制备例12
81.按照制备例1的方法制备催化剂,不同的是,使用的拟薄水铝石粉体不含掺杂元素,且比表面积为380m2/g,孔容为1.02ml/g,获得催化剂a-12。
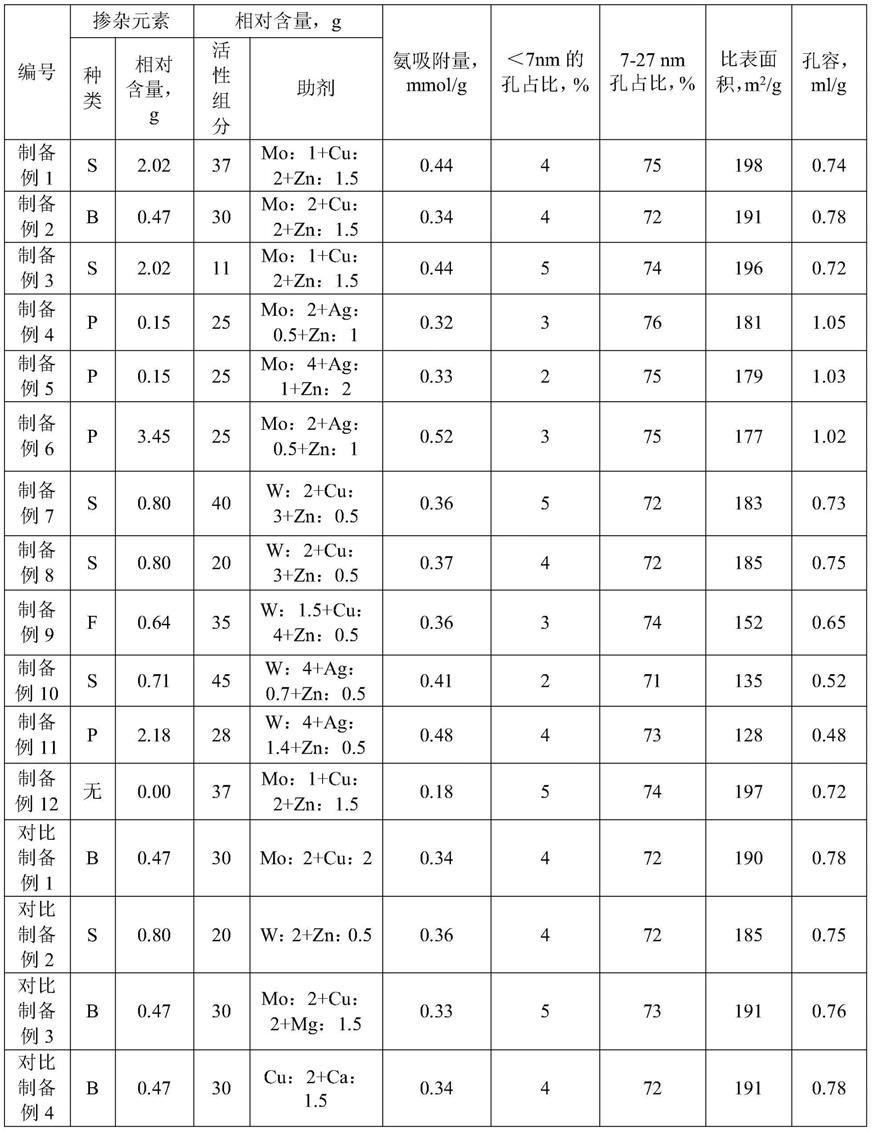
82.对比制备例1
83.按照制备例2的方法制备催化剂,不同之处在于,喷浸法采用的溶液中不含六水合硝酸锌,获得催化剂d-1。
84.对比制备例2
85.按照制备例8的方法制备催化剂,不同之处在于,喷浸法采用的溶液中不含三水合硝酸铜,获得催化剂d-2。
86.对比制备例3
87.按照制备例2的方法制备催化剂,不同的是,将六水合硝酸锌替换为16.03g的六水
合硝酸镁(分析纯)。获得催化剂d-3。
88.对比制备例4
89.按照制备例2的方法制备催化剂,不同的是,不加入钼酸铵,同时将六水合硝酸锌替换为8.84g的四水合硝酸钙。获得催化剂d-4。
90.测试例1
91.通过等离子体发射光谱仪分析载体和催化剂的元素组成,掺杂元素以相对100g的载体的含量表示,活性组分和助剂的含量均以相对100g的载体的含量表示;通过nh
3-tpd、bet氮吸附脱附法方法对以上制备的载体进行表征,具体步骤如下,结果如表1所示。
92.nh
3-tpd测试
93.测试仪器:全自动化学吸附仪(automated catalyst characterization system)仪器型号:autochem 2920,美国micromeritics公司产品
94.测试条件:准确称取约0.1g样品放入样品管中,在he气吹扫条件下以10℃/min升至600℃,停留1h,降至120℃,再改变气体为10%nh
3-he混合气,吸附60min,然后再改变为he气吹扫1h,基线稳定后开始计数,以10℃/min升至600℃,保持30min,停止记录,完成实验。对峰面积进行积分计算,计算得到nh3脱附量。
95.bet测试
96.仪器名称:全自动物化吸附分析仪(automatic micropore&chemisorption analyzer);仪器型号:asap2420,美国micromeritics(麦克仪器公司)
97.测试条件:实验气体:n2(纯度99.999%);脱气条件:以10℃/min升到350℃,抽真空4h;分析条件:介孔等温线全分析。得到比表面积和孔容。
98.表1
[0099][0100]
实施例1
[0101]
将制备例制备的催化剂a-1至d-3分别量取100毫升装在固定床反应器中,使用氢气在220℃下活化2小时,然后降温至175℃,用氢气将系统压力升高至12mpa,然后用计量泵将氨计量后送入反应系统,经预热至170℃进入反应器上端,用计量泵将30wt%的n-羟乙基哌嗪和70wt%的1,6-二氧六环的混合溶液送入反应器上端,氢气经气体质量流量计稳定送入,氢气:氨:n-羟乙基哌嗪的摩尔比为3:25:1,n-羟乙基哌嗪的液相体积空速为0.3h-1
,在反应器进行催化氨化反应,反应稳定后(也即反应20h时),反应液取样分析,分析结果列于表2中。
[0102]
取样分析方法为气相色谱分析,通过配制标准样品的校正因子进行校准;
[0103]
以反应液中各组分的摩尔含量计算转化率和选择性。
[0104][0105]nx
表示转化为一摩尔产物x所需要的n-羟乙基哌嗪的摩尔数;
[0106]
产物x包括哌嗪、乙二胺、羟乙基乙二胺、n-氨乙基哌嗪、一乙胺、微量其他二聚产物;
[0107][0108]nx
表示转化为一摩尔产物x所需要的n-羟乙基哌嗪的摩尔数;
[0109]n哌嗪
表示转化为一摩尔哌嗪所需要的n-羟乙基哌嗪的摩尔数;
[0110]
产物x包括哌嗪、乙二胺、羟乙基乙二胺、n-氨乙基哌嗪、一乙胺、微量其他二聚产物;其他产物的选择性只需要将公式中分子的n
哌嗪
及相应摩尔含量换成对应值便可。
[0111]
表2
[0112][0113][0114]
实施例2
[0115]
将催化剂a-1至d-3分别量取100毫升装在固定床反应器中,使用氢气在220℃下活化2小时,然后降温至200℃,用氢气将系统压力升高至9.3mpa,然后用计量泵将氨计量后送入反应系统,经预热至180℃进入反应器上端,用计量泵将20wt%的n-氨乙基哌嗪和80wt%的1,6-二氧六环的混合溶液送入反应器上端,氢气经气体质量流量计稳定送入,氢气:氨:
n-氨乙基哌嗪的摩尔比为1:10:1,n-氨乙基哌嗪的液相体积空速为0.1h-1
,在反应器进行催化氨化反应,反应稳定后(也即反应20h时),反应液取样分析,以反应液中各组分的摩尔含量计算转化率和选择性。
[0116]
分析结果列于表3中。
[0117][0118]nx
表示转化为一摩尔产物x所需要的n-氨乙基哌嗪的摩尔数;
[0119]
产物x包括哌嗪、乙二胺、二乙烯三胺、一乙胺、微量其他二聚产物;
[0120][0121]nx
表示转化为一摩尔产物x所需要的n-氨乙基哌嗪的摩尔数;
[0122]n哌嗪
表示转化为一摩尔哌嗪所需要的n-氨乙基哌嗪的摩尔数;
[0123]
产物x包括哌嗪、乙二胺、二乙烯三胺、一乙胺、微量其他二聚产物;
[0124]
其他产物的选择性只需要将公式中分子的n
哌嗪
及相应摩尔含量换成对应值便可。
[0125]
表3
[0126][0127]
实施例3
[0128]
将催化剂a-1至d-3分别量取100毫升装在固定床反应器中,使用氢气在220℃下活化2小时,然后降温至160℃,用氢气将系统压力升高至16mpa,然后用计量泵将氨计量后送入反应系统,经预热至150℃进入反应器上端,用计量泵将20wt%的n-氨乙基哌嗪、20wt%
的n-羟乙基哌嗪、60wt%的1,6-二氧六环的混合溶液送入反应器上端,氢气经气体质量流量计稳定送入,氢气:氨:n-氨乙基哌嗪和n-羟乙基哌嗪的总的摩尔比为2:18:1,n-羟乙基哌嗪和n-氨乙基哌嗪的总的液相体积空速为0.4h-1
,在反应器进行催化氨化反应,反应稳定后(也即反应20h时),反应液取样分析,以反应液中各组分的摩尔含量计算转化率和选择性。分析结果列于表4中。
[0129][0130][0131]nx
表示转化为一摩尔产物x所需要的n-氨乙基哌嗪(或n-羟乙基哌嗪)的摩尔数;
[0132]n哌嗪
表示转化为一摩尔哌嗪所需要的n-氨乙基哌嗪(或n-羟乙基哌嗪)的摩尔数;
[0133]
产物x包括哌嗪、羟乙基乙二胺、二乙烯三胺、乙二胺、一乙胺、微量其他二聚产物;
[0134]
其他产物的选择性只需要将公式中分子的n
哌嗪
及相应摩尔含量换成对应值便可。
[0135]
表4
[0136][0137][0138]
实施例4
[0139]
按照实施例3的方法制备哌嗪,不同的是,催化剂为a-1,温度为155℃,压力为18.5mpa,氢气:氨:n-氨乙基哌嗪和n-羟乙基哌嗪的总的摩尔比为4:5:1,n-羟乙基哌嗪和n-氨乙基哌嗪的总的液相体积空速为0.3h-1
,反应液取样分析结果列于表5中。
[0140]
表5
[0141][0142]
以上详细描述了本发明的优选实施方式,但是,本发明并不限于此。在本发明的技术构思范围内,可以对本发明的技术方案进行多种简单变型,包括各个技术特征以任何其它的合适方式进行组合,这些简单变型和组合同样应当视为本发明所公开的内容,均属于本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1