[0001]
本发明涉及上转换材料技术领域,尤其涉及一种化合物及其制备方法和三重态-三重态湮灭上转换体系。
背景技术:[0002]
上转换是将低能量长波长光转换为短波长高能量光的过程,即反斯托克斯发光。目前,实现上转换的方法一般有双光子吸收上转换,稀土离子能量转移上转换和三重态-三重态湮灭上转换。与双光子吸收和稀土离子能量转移上转换相比,三重态-三重态湮灭上转换体系可在较低的激发功率密度下实现较高量子产率的上转换发光,并且可以在非相干光、甚至太阳光下工作,因而在光催化、太阳能发电以及生物上成像等领域具有重要的应用前景。
[0003]
三重态-三重态湮灭(triplet-triplet annihilation,简称tta)上转换是光敏剂将激发态能量转移给湮灭剂,使两分子激发三重态湮灭剂(t1)产生一个高能级的激发单重态(s1),并辐射出高能量光子的过程。光敏剂是三重态-三重态湮灭上转换体系的一个重要组成部分,其光物理性质极大地影响了上转换量子效率。近年来,研究者已经开发出多种材料来构建三重态-三重态湮灭上转换体系。许多过渡金属(pt,pd,ir等)的络合物由于具有高的系间窜越速率,被广泛用作上转换光敏剂,其中一些体系的上转换量子产率超过30%。最近,研究者报道了多种非过渡金属光敏剂,如具有重原子效应的有机化合物,热活化延迟荧光分子等,这些光敏剂无需过渡金属参与即可实现上转换发光,为构建上转换体系提供了新的途径。然而,基于这些非过渡金属光敏剂的上转换体系大多数表现出低的上转换量子产率。因此,开发高效的非过渡金属光敏剂具有重要的应用价值。
技术实现要素:[0004]
本发明提供了一种化合物及其制备方法和三重态-三重态湮灭上转换体系,解决了非过渡金属光敏剂的上转换体系大多数表现出低的上转换量子产率和高的激发阈值的问题。
[0005]
其具体技术方案如下:
[0006]
本发明提供了一种化合物,所述化合物具有式(ⅰ)所示结构;
[0007]
[0008]
其中,a为吸电子基团,r为氢或x为卤素原子。
[0009]
本发明中,a为苯并噻唑、苯并噻二唑或苯并二噻二唑。
[0010]
本发明提供的式(ⅰ)所示结构以a为吸电子单元,以卤素取代的9,9-二甲基吖啶为给电子单元。该化合物具有热活化延迟荧光特征和重原子效应。
[0011]
本发明提供的化合物具体为:
[0012][0013]
本发明还提供了上述化合物的制备方法,包括以下步骤:
[0014]
步骤1:将a-x或x-a-x、配体、9,9-二甲基吖啶、碱和溶剂混合,在催化剂的条件下进行buchwald-hartwing反应,得到式(ⅱ)所示结构的化合物;
[0015]
步骤2:将式(ⅱ)所示结构的化合物与卤化剂进行亲电取代反应,得到式(ⅰ)所示的化合物;
[0016]
其中,a为吸电子基团,r为氢或x为卤素原子,r’为氢或
[0017]
本发明步骤1中,优选在氮气或惰性气体的气氛下进行buchwald-hartwing反应;
[0018]
反应原料a-x优选为:
[0019][0020]
原料x-a-x优选为:
[0021][0022]
所述配体为三叔丁基膦四氟硼酸盐;
[0023]
所述碱为叔丁醇钠;
[0024]
催化剂为钯系催化剂为醋酸钯;
[0025]
所述溶剂为甲苯;所述甲苯经除水除氧处理后再加入反应体系。
[0026]
所述a-x与所述9,9-二甲基吖啶的摩尔比为1:1.5;
[0027]
所述x-a-x与所述9,9-二甲基吖啶的摩尔比为1:2.2;
[0028]
所述buchwald-hartwing反应的温度为120℃时间为48小时所述buchwald-hartwing反应结束后,还需对产物进行纯化,所述纯化具体为:将反应倒入冰水中,采用二氯甲烷萃取,浓缩有机相后,采用体积比为2:1的正己烷和二氯甲烷的混合熔液进行柱层析分离,得到式(ⅱ)所示结构的化合物。
[0029]
中间体:式(ⅱ)所示结构的化合物优选为:
[0030][0031]
本发明步骤2中,所述卤化剂为n-卤代丁二酰亚胺;
[0032]
当所述式(ⅱ)所示结构的化合物中r’为氢时,与所述卤化剂的摩尔比为,优选为1:4;当所述式(ⅱ)所示结构的化合物中r’为时,与所述卤化剂的摩尔比为1:8;
[0033]
所述亲电取代反应的温度为0℃,时间为6~12h;
[0034]
所述亲电取代反应结束后,还对产物进行纯化,所述纯化具体为:将反应液倒入冰水中,采用二氯甲烷进行萃取,浓缩有机相,采用体积比为3:1的正己烷和二氯甲烷进行柱层析分离,得到式(ⅰ)所示结构的化合物。
[0035]
本发明提供的化合物具有热活化延迟荧光特征。因此,本发明还提供了上述式(ⅰ)所示结构的化合物或上述制备方法制得的式(ⅰ)所示结构的化合物在热活化延迟荧光材料中的应用。热活化延迟荧光材料也被用作光敏剂构建三重态-三重态湮灭上转换体系,通过
选择合适的湮灭剂,热活化延迟荧光光敏剂吸收能量后,其最低激发单重态激子通过系间窜越到达最低激发三重态,此时,最低激发三重态的激子通过三重态-三重态能量转移过程到达湮灭剂的激发三重态,并最终通过三重态-三重态湮灭过程实现上转换发光。重原子效应可以增强分子的自旋轨道耦合作用,使激子从最低激发单重态向最低激发三重态的系间窜跃速率增加,有利于增强光敏剂最低激发三重态激子浓度,从而提升上转换量子产率。
[0036]
本发明提供的化合物具有热活化延迟荧光特征和重原子效应,因此,本发明还提供了式(ⅰ)所示结构的化合物或上述制备方法制得的式(ⅰ)所示结构的化合物在三重态-三重态湮灭上转换光敏剂中的应用。式(ⅰ)所示结构的化合物是一类新型的上转换光敏剂。
[0037]
本发明还提供了一种三重态-三重态湮灭上转换体系,包括1:5化合物或上述制备方法制得的1:5化合物、湮灭剂和溶剂。
[0038]
本发明中,所述湮灭剂优选为9,10-二苯基蒽;
[0039]
所述溶剂为甲苯。
[0040]
本发明上转换光敏剂具有可见光吸收和红光或近红外光发射性能,其与湮灭剂9,10-二苯基蒽组成三重态-三重态湮灭上转换体系,可以有效的将绿光转换至蓝光,具有高的上转换量子产率。
[0041]
本发明三重态-三重态湮灭上转换体系中,所述1:5化合物的摩尔浓度为1mm,所述湮灭剂的摩尔浓度为5mm;
[0042]
所述式(ⅰ)所示结构化合物与所述湮灭剂的摩尔比为1:5。
[0043]
从以上技术方案可以看出,本发明具有以下优点:
[0044]
本发明提供的具有式(ⅰ)所示结构的化合物具有热活化延迟荧光特征和重原子效应,可以作为热活化延迟荧光材料和三重态-三重态湮灭上转换光敏剂,重原子效应可以显著增加光敏剂最低三重态激子浓度,提高上转换量子效率。本发明提供的式(ⅰ)所示结构的上转换光敏剂具有可见光吸收和红光或近红外光发射性能,其与9,10-二苯基蒽组成三重态-三重态湮灭上转换体系,在有机溶剂中实现高激发光功率密度条件下的上转换发光,可以有效的将绿光转换至蓝光,其上转换量子产率可达到24.4%。
附图说明
[0045]
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动性的前提下,还可以根据这些附图获得其它的附图。
[0046]
图1为本发明实施例7中btdz-2dmac-4br在甲苯溶液中,得到的紫外-可见光吸收光谱和荧光发射光谱图(激发波长520nm);
[0047]
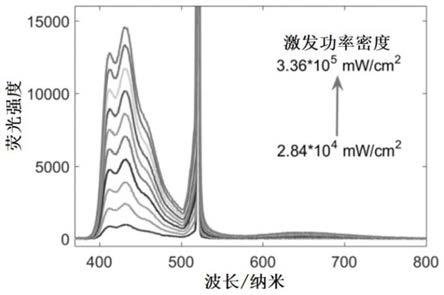
图2为本发明实施例7中btdz-2dmac-4br作为上转换光敏剂,以9,10-二苯基蒽作为湮灭剂,在甲苯溶液中的上转换发光谱图;
[0048]
图3为本发明实施例8中btdz-2dmac-4br在甲苯溶液中,以377nm为激发波长得到的瞬态光谱图;
[0049]
图4为本发明实施例8中btdz-2dmac-4br作为上转换光敏剂,以9,10-二苯基蒽作为湮灭剂,在甲苯溶液中的瞬态光谱图。
具体实施方式
[0050]
为使得本发明的发明目的、特征、优点能够更加的明显和易懂,下面将对本发明实施例中的技术方案进行清楚、完整地描述,显然,下面所描述的实施例仅仅是本发明一部分实施例,而非全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其它实施例,都属于本发明保护的范围。
[0051]
实施例1:式(ⅰ)所示结构的化合物btz-dmac-2cl的制备。
[0052]
(1)中间体:式(ⅱ)所示结构的化合物btz-dmac的合成
[0053][0054]
向100ml三口瓶中加入btz-br(500mg,2.34mmol),9,9-二甲基吖啶(dmac,735mg,3.51mmol),醋酸钯(78mg,0.35mmol)和三叔丁基膦四氟硼酸盐(305mg,1.05mmol),叔丁醇钠(808mg,8.42mmol),在氩气氛围下加入30ml经除水除氧处理的甲苯,在120℃反应48小时。冷却至室温,将反应液倒入50ml冰水中,二氯甲烷萃取三次,浓缩有机相,柱层析分离(正己烷:二氯甲烷,v:v,3:1),得红色粉末601mg,产率75%。ms(ei)m/z:342.12。
[0055]
(2)式(ⅰ)所示结构的btz-dmac-2cl的合成
[0056][0057]
100ml圆底烧瓶中,将btz-dmac(300mg,0.88mmol)溶于三氯甲烷(7ml)中。在0℃下,缓慢滴加n-氯代丁二酰亚胺(ncs,470mg,3.52mmol)的三氯甲烷(28ml)溶液,搅拌8小时。反应结束后,将反应液倒入100ml冰水中,二氯甲烷萃取三次,浓缩有机相,柱层析分离(正己烷:二氯甲烷,v:v,3:1),得到红色粉末145mg,产率40%。ms(ei)m/z:410.04。
[0058]
实施例2:式(ⅰ)所示结构的化合物btz-2dmac-4cl的制备。
[0059]
(1)中间体:式(ⅱ)所示结构的化合物btz-2dmac的合成
[0060][0061]
向100ml三口瓶中加入btz-br(500mg,1.71mmol),9,9-二甲基吖啶(dmac,787mg,3.76mmol),醋酸钯(57mg,0.26mmol)和三叔丁基膦四氟硼酸盐(223mg,0.77mmol),叔丁醇钠(591mg,6.16mmol),在氩气氛围下加入22ml经除水除氧处理的甲苯,在120℃反应48小时。冷却至室温,将反应液倒入50ml冰水中,二氯甲烷萃取三次,浓缩有机相,柱层析分离(正己烷:二氯甲烷,v:v,2:1),得红色粉末658mg,产率70%。ms(ei)m/z:549.22。
[0062]
(2)式(ⅰ)所示结构的化合物btz-2dmac-2cl的合成
[0063][0064]
100ml圆底烧瓶中,将btz-2dmac(300mg,0.55mmol)溶于三氯甲烷(5ml)中。在0℃下,缓慢滴加n-氯代丁二酰亚胺(ncs,588mg,4.4mmol)的三氯甲烷(35ml)溶液,搅拌8小时。反应结束后,将反应液倒入100ml冰水中,二氯甲烷萃取三次,浓缩有机相,柱层析分离(正己烷:二氯甲烷,v:v,2:1),得到红色粉末144mg,产率38%。ms(ei)m/z:685.07。
[0065]
实施例3:式(ⅰ)所示结构的化合物btz-dmac-2br的制备。
[0066]
(1)中间体:式(ⅱ)所示结构的化合物btdz-dmac的合成
[0067][0068]
向100ml三口瓶中加入btdz-br(500mg,2.32mmol),9,9-二甲基吖啶(dmac,728mg,3.48mmol),醋酸钯(78mg,0.34mmol)和三叔丁基膦四氟硼酸盐(303mg,1.04mmol),叔丁醇钠(802mg,8.35mmol),在氩气氛围下加入30ml经除水除氧处理的甲苯,在120℃反应48小时。冷却至室温,将反应液倒入50ml冰水中,二氯甲烷萃取三次,浓缩有机相,柱层析分离(正己烷:二氯甲烷,v:v,3:1),得红色粉末574mg,产率72%。ms(ei)m/z:343.11。
[0069]
(2)式(ⅰ)所示结构的化合物btdz-dmac-2br的合成
[0070][0071]
100ml圆底烧瓶中,将btdz-dmac(300mg,0.87mmol)溶于三氯甲烷(4ml)中。在0℃下,缓慢滴加n-溴代丁二酰亚胺(nbs,619mg,3.48mmol)的三氯甲烷(7ml)溶液,搅拌6小时。反应结束后,将反应液倒入50ml冰水中,二氯甲烷萃取三次,浓缩有机相,柱层析分离(正己烷:二氯甲烷,v:v,3:1),得到红色粉末218mg,产率50%。ms(ei)m/z:498.94。
[0072]
实施例4:式(ⅰ)所示结构的化合物btdz-2dmac-4br的制备。
[0073]
(1)中间体:式(ⅱ)所示结构的化合物btdz-2dmac的合成
[0074][0075]
向100ml三口瓶中加入btdz-2br(500mg,1.70mmol),9,9-二甲基吖啶(dmac,783mg,3.76mmol),醋酸钯(57mg,0.26mmol)和三叔丁基膦四氟硼酸盐(223mg,0.77mmol),
叔丁醇钠(588mg,6.12mmol),在氩气氛围下加入22ml经除水除氧处理的甲苯,在120℃反应48小时。冷却至室温,将反应液倒入50ml冰水中,二氯甲烷萃取三次,浓缩有机相,柱层析分离(正己烷:二氯甲烷,v:v,2:1),得红色粉末609mg,产率65%。ms(ei)m/z:550.22。
[0076]
(2)式(ⅰ)所示结构的化合物btdz-2dmac-4br的合成
[0077][0078]
100ml圆底烧瓶中,将btdz-2dmac(300mg,0.54mmol)溶于三氯甲烷(3ml)中。在0℃下,缓慢滴加n-溴代丁二酰亚胺(nbs,769mg,4.32mmol)的三氯甲烷(9ml)溶液,搅拌6小时。反应结束后,将反应液倒入50ml冰水中,二氯甲烷萃取三次,浓缩有机相,柱层析分离(正己烷:二氯甲烷,v:v,2:1),得到红色粉末239mg,产率51%。ms(ei)m/z:861.86。
[0079]
实施例5:式(ⅰ)所示结构的化合物bdtdz-dmac-2i的制备。
[0080]
(1)中间体:式(ⅱ)所示结构的化合物bdtdz-dmac的合成
[0081][0082]
向100ml三口瓶中加入bdtdz-br(500mg,1.83mmol),9,9-二甲基吖啶(dmac,575mg,2.75mmol),醋酸钯(61mg,0.27mmol)和三叔丁基膦四氟硼酸盐(239mg,0.82mmol),叔丁醇钠(632mg,6.59mmol),在氩气氛围下加入24ml经除水除氧处理的甲苯,在120℃反应48小时。冷却至室温,将反应液倒入50ml冰水中,二氯甲烷萃取三次,浓缩有机相,柱层析分离(正己烷:二氯甲烷,v:v,3:1),得红色粉末514mg,产率70%。ms(ei)m/z:401.08。
[0083]
(2)式(ⅰ)所示结构的化合物bdtdz-dmac-2i的合成
[0084][0085]
100ml圆底烧瓶中,将bdtdz-dmac(300mg,0.75mmol)溶于三氯甲烷(8ml)中。在0℃下,缓慢滴加n-碘代丁二酰亚胺(nis,675mg,3mmol)的三氯甲烷(15ml)溶液,搅拌12小时。反应结束后,将反应液倒入100ml冰水中,二氯甲烷萃取三次,浓缩有机相,柱层析分离(正己烷:二氯甲烷,v:v,3:1),得到红色粉末269mg,产率55%。ms(ei)m/z:652.87。
[0086]
实施例6:式(ⅰ)所示结构的化合物bdtdz-2dmac-4i的制备。
[0087]
(1)中间体:式(ⅱ)所示结构的化合物bdtdz-2dmac的合成
[0088][0089]
向100ml三口瓶中加入bdtdz-2br(500mg,1.42mmol),9,9-二甲基吖啶(dmac,658mg,3.14mmol),醋酸钯(48mg,0.21mmol)和三叔丁基膦四氟硼酸盐(186mg,0.64mmol),叔丁醇钠(494mg,5.14mmol),在氩气氛围下加入18ml经除水除氧处理的甲苯,在120℃反应48小时。冷却至室温,将反应液倒入50ml冰水中,二氯甲烷萃取三次,浓缩有机相,柱层析分离(正己烷:二氯甲烷,v:v,2:1),得红色粉末536mg,产率62%。ms(ei)m/z:608.18。
[0090]
(2)式(ⅰ)所示结构的化合物bdtdz-2dmac-4i的合成
[0091][0092]
100ml圆底烧瓶中,将bdtdz-2dmac(300mg,0.49mmol)溶于三氯甲烷(5ml)中。在0℃下,缓慢滴加n-碘代丁二酰亚胺(nis,882mg,3.92mmol)的三氯甲烷(20ml)溶液,搅拌12小时。反应结束后,将反应液倒入100ml冰水中,二氯甲烷萃取三次,浓缩有机相,柱层析分离(正己烷:二氯甲烷,v:v,2:1),得到红色粉末294mg,产率54%。ms(ei)m/z:1111.77。
[0093]
实施例7:以实施例4化合物btdz-2dmac-4br为光敏剂的三线态-三线态湮灭上转换发光体系
[0094]
配置浓度为1mm的btdz-2dmac-4br甲苯溶液,以520nm为激发波长,通过紫外-可见分光光度计和荧光仪分别得到btdz-2dmac-4br在甲苯溶液中的紫外-可见吸收光谱和荧光发射光谱,如图1所示,btdz-2dmac-4br在甲苯溶液中吸收峰为490纳米,发射峰为650纳米,具有可见光吸收和红光或近红外光发射性能。
[0095]
以摩尔比为1:5配置btdz-2dmac-4br与9,10-二苯基蒽的双组份甲苯溶液,其中btdz-2dmac-4br的浓度为1mm,9,10-二苯基蒽的浓度为5mm,以毛细管吸附混合均匀的双组分甲苯溶液,以具有不同激光功率密度的520纳米波长光源激发该双组分溶液。图2为本实施例混合溶液的上转换发光光谱图,如图2所示,本实施例上转换发光体系得到了具有不同强度的上转换发光,发光范围为410至490纳米,对应蓝光,其上转换量子产率为24.4%。
[0096]
实施例8:以实施例4化合物btdz-2dmac-4br为光敏剂的三线态-三线态湮灭上转换发光体系
[0097]
配置浓度为1mm的btdz-2dmac-4br甲苯溶液,以377纳米为激发波长激发溶液,图3为btdz-2dmac-4br在甲苯溶液中的瞬态光谱。
[0098]
以摩尔比为1:5配置btdz-2dmac-4br与9,10-二苯基蒽的双组份甲苯溶液,其中btdz-2dmac-4br的浓度为1mm,9,10-二苯基蒽的浓度为5mm,以毛细管吸附混合均匀的双组分甲苯溶液,以377纳米为激发波长激发组分溶液。图4为本实施例混合溶液瞬态光谱,混合溶液相对btdz-2dmac-4br溶液寿命变短,说明有光敏剂和湮灭剂间的三线态-三线态能量
转换。
[0099]
以上所述,以上实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的精神和范围。