一种富集目标区域再酶切构建单倍型的方法与流程
1.本发明涉及分子生物学技术领域,特别是涉及一种富集目标区域再酶切构建单倍型的方法。
背景技术:
2.人是二倍体生物,即含有两组染色体,单组染色体即为单倍体。在单倍体中,紧密连锁的多个等位基因的线性组合,每种组合方式即为一种单倍型。单倍型可以由多个snp位点构成,包含丰富的遗传信息,研究单倍型比单个snp位点具有更好的分析效果,更能有效反映出疾病的遗传机制,其在遗传病检测领域有着广泛的需求。
3.遗传信息的变异是所有基因组的共同特征,而单碱基对的差异,也称单核苷酸多态性(snp)是变异中最常见的一种形式,占所有已知多态性的90%以上。snp位点并不是独立遗传的,而是在染色体上成组地遗传。一般来说,一个snp位点只有两种等位基因,因此又叫双等位基因。单核苷酸多态性是研究人类家族和动植物品系遗传变异的重要依据,因此被广泛用于群体遗传学研究和疾病相关基因的研究,在药物基因组学、诊断学和生物医学研究中起重要作用。
4.基因分型(phasing)还称为基因定相、单倍体分型或单倍体构建。基因分型是指把二倍体(甚至是多倍体)基因组上的等位基因(包括杂合位点,例如snp),按照其亲本正确地定位到父亲或者母亲的染色体上,最终使得所有来自同一个亲本的等位基因都能够排列在同一条染色体里。
5.目前单倍型分析技术主要分为两大类,间接推断法和直接实验法。间接推断法是借助计算机通过统计学方法,从参考基因组中推断出样本单倍型。随着新一代测序技术的快速发展,人们可以比较容易获得大量的基因组信息,这是间接推断法的基础。间接推断法根据研究对象的不同又可分为两类:群体推断法和家族推断法。群体推断法是通过构建一些关联群体的基因池并用统计学方法对预测结果进行分析推断样本的单倍型。如果群体中存在一些突变频率较低的个体,它受连锁不平衡程度的影响往往会被遗漏而无法获得其单倍型信息。家族推断法是根据同一家族众多个体的基因型信息对待测样本进行推断获得其单倍型信息,在使用前要确保同一家族中这些样本基因型信息的可靠性。总之,间接推断法需要依靠大量样本的支持,并不是针对个体样本的单倍型分析,准确性受到不同算法的影响较大。
6.直接实验法是指用单分子稀释、染色体微切割和流式分离法等特殊实验方法在一段有限的染色体区域或单染色体获得精确的单倍型信息。直接实验法又可分为两大类:稠密位点单倍型(dense)法和稀疏位点单倍型(sparse)法。
7.稠密位点单倍型法能精确检测到单染色体局部区域的单倍型,组装结果更完整,在染色体上的排布较密集,是目前最为常用的方法。它主要包括单分子稀释法(single-molecule dilution)、长片段插入克隆法(long-insert cloning)、保留邻近性转座酶测序法(contiguity-preserving transposition sequencing,cpt-seq)、目标位点扩增
(targeted locus amplification,tla)等。然而这些方法多是针对全基因组的单倍型组装,需要大量的测序数据,成本非常高,同时实验操作复杂,流程长,在数据分析阶段依赖如hapcut2这类软件的复杂算法。
8.而稀疏位点单倍型法能获得单染色体上几乎所有区域的单倍型信息,它们包括单染色体测序法(single chromosome sequencing)、单倍型测序法(haploseq)、乳液pcr法(emulsion pcr-based methods)等。但是获得的位点在染色体上的排布比较稀疏,有时不能准确定位该样本单倍型在染色体上的物理位置,甚至会遗漏一些位点。这些方法同样存在着实验操作复杂,涉及特殊的仪器设备,成本高等问题,且很难靶向较小区域的单倍型信息。
9.可见现有的方法(三代测序、tla、10x genmoics等)只能构建全基因组范围的单倍型,而且在数据分析阶段也都依赖复杂的生信算法,因此单倍型分析领域目前缺少一种可以靶向基因组上一个较小区域、且实验操作和数据分析均简便、成本低的技术方案。
技术实现要素:
10.鉴于以上所述现有技术的缺点,本发明的目的在于提供一种构建单倍型的方法,用于解决现有技术中的问题。
11.为实现上述目的及其他相关目的,本发明提供一种构建单倍型的方法,所述方法包括以下步骤:
12.1)富集目标核酸区域;
13.2)设计仅能与目标核酸区域中的一个单倍体结合的向导序列,利用向导序列以及限制性核酸内切酶酶切步骤1)富集的目标核酸区域,分别回收酶切片段和/或未被酶切的片段;
14.3)利用步骤2)回收的酶切片段和/或未被酶切的片段分别制备测序文库并测序,数据分析分别得到酶切片段和/或未被酶切的片段的核酸序列的snp信息,即为目标核酸区域的单倍型信息。
15.所述向导序列满足以下条件中的一项或几项:
16.1)向导序列的长度为10nt~40nt;
17.2)向导序列的5’端第一个碱基为t;
18.3)杂合snp位点对应向导序列的第8~14位;
19.4)与所述向导序列互补的核酸模板片段上仅有一个杂合snp位点。
20.如上所述,本发明的构建单倍型的方法,具有以下有益效果:在单倍型研究的必须步骤即对目标基因所在区域进行富集的步骤中即可实现提高酶切特异性的目的,无需增加额外的实验步骤,方法简便且节约成本;可以靶向基因组上一个较小区域构建其单倍型,相对于现有的方法显著降低了实验操作难度和测序成本。单倍型构建的准确度高,无需复杂的生信分析算法,直接根据数据中杂合snp两个等位基因的覆盖深度即可判断单倍型。
附图说明
21.图1显示为本发明构建单倍型的方法流程图。
22.图2显示为经过pfago酶切的结果图,其中1为pcr扩增产物,2为经过pfago酶切的
产物,有两条明显的短片段,分别对应4891bp和5451bp两个片段。
23.图3显示为目标区域snp的比例变化图。
具体实施方式
24.如图1所示,本发明提供一种构建单倍型的方法,所述方法包括以下步骤:
25.1)富集目标核酸区域;
26.2)设计仅能与目标核酸区域中的一个单倍体结合的向导序列,利用向导序列以及限制性核酸内切酶酶切步骤1)富集的目标核酸区域,分别回收酶切片段和/或未被酶切的片段;
27.3)利用步骤2)回收的酶切片段和/或未被酶切的片段分别制备测序文库并测序,数据分析分别得到酶切片段和/或未被酶切的片段的核酸序列的snp信息,即为目标核酸区域的单倍型信息。
28.所述富集目标核酸区域的方法即从核酸的初始集合中选择性pcr扩增目标核酸区域。所述目标核酸区域为含有杂合snp位点的区域。所述目标核酸区域中是否含有杂合snp位点可以通过测序方法获知。
29.步骤1)中,所述目标核酸区域的最短长度可达5kb。所述目标核酸区域的最长长度取决于pcr扩增的长度,例如目标核酸区域的长度选自以下范围中的一个:5~10kb、10~15kb、15~20kb、20~25kb、25~30kb。
30.pcr扩增时设计的引物只要能够在pcr实施条件下特异性地识别各自的特异性识别区域(优选在单一的反应容器内使用的引物之间不发生退火及自退火)即可。对于各引物的长度而言,只要能够特异性地识别对应的特异性识别区域、且引物之间不发生杂交即可,没有特别限制。
31.在一种实施方式中,所述向导序列的对数为一对或多对。例如两对、三对、四对或更多对。当使用多对向导序列时,可以酶切多个基因。所述向导序列包括一条正链和一条负链。所述正链和负链均为5’端磷酸化的单链dna。
32.设计仅能与目标核酸区域中的一个单倍体结合的向导序列,一种可行的方法是:设计的向导序列对应的核酸模板上仅有一个杂合snp位点。该向导序列对应的核酸模板的杂合snp位点可为野生型序列或突变型序列之中的任一。杂合snp位点可以通过对包含目标核酸区域的核酸片段测序获得。具体的,例如当测序结果中,针对一个snp位点的野生型或突变型占比接近50%时,可确定该snp位点为杂合型snp位点。
33.所述向导序列(或称guide序列)满足以下条件中的一项或几项:
34.1)正链和负链的长度均为10nt~40nt;
35.2)正链和负链的5’端第一个碱基为t;
36.3)杂合snp位点对应向导序列的第8~14位;
37.所述向导序列的长度选自以下范围中的一种或几种:10~13nt、13~16nt、16~20nt、20~25nt、25~30nt、30~35nt或35~40nt。在一较佳实施方式中,所述向导序列长度优选为13nt~25nt。
38.正链和负链的5’端第一个碱基为t时,无论这个t是否与模板互补,均能提高限制性核酸内切酶酶活性。
39.在一较佳实施方式中,杂合snp位点对应向导序列的第10~12号位置。在这个区间内,限制性核酸内切酶对guide非特异性结合的容错率更低,即限制性核酸内切酶的特异性更高。
40.向导序列对应的核酸模板上若有多个snp位点会使guide的结合效率降低,导致限制性核酸内切酶的酶活性降低。
41.在一种实施方式中,步骤2)中酶切时的酶切体系中包括:目标核酸区域的扩增产物、限制性核酸内切酶、向导序列、缓冲液、水。
42.在一种实施方式中,以酶切体系的总体积为基准,所述酶切体系中扩增产物的浓度为4ng/μl~8ng/μl。优选的,扩增产物的浓度为5~7ng/μl。
43.在一种实施方式中,以酶切体系的总体积为基准,所述酶切体系中限制性核酸内切酶的终浓度为0.045~3.84μm。优选的,限制性核酸内切酶的终浓度为0.225-1.92μm。
44.在一种实施方式中,以酶切体系的总体积为基准,所述酶切体系中向导序列的正链和负链的终浓度分别为0.45μm~38.4μm。优选的,正链和负链的终浓度分别为2.25~19.2μm。
45.在一种实施方式中,酶切条件为85~99℃、10~15min,再将缓慢降至10℃。
46.所述限制性核酸内切酶是可以识别并附着特定的脱氧核苷酸序列,并对每条链中特定部位的两个脱氧核糖核苷酸之间的磷酸二酯键进行切割的一类酶。在一种实施方式中,所述限制性核酸内切酶为pfago。
47.pfago是一种从强烈炽热球菌提取出的argonaute蛋白(pfago)构建而成的人工限制酶。用pfago特异性地切割基因组上的一个杂合位点(snp),被切割的基因型所在的dna序列发生断裂,而未被切割的基因型所在的dna序列完整的被保留,对未被切割的dna序列进行回收,可获得该杂合位点附近的单倍型信息。pfago的识别序列可达到16bp,但是这个长度在基因组上依然存在非常多的识别位置,直接对基因组进行切割会产生很多的非特异片段。发明人经过大量实验发现在酶切前先对目标核酸区域进行富集,可以极大改善切割非特异性片段的问题。
48.在一种实施方式中,测序文库的制备和测序的方法可采用本领域常用的方法。在一种实施方式中,使用illumina平台的nextseq 500进行测序。
49.本领域技术人员均了解,数据分析可以采用现有软件的功能实现。例如现有的软件bwa,samtools,gatk。
50.数据分析时,针对一个杂合snp,观察其酶切后突变型或野生型出现的比例相对其酶切前的比例发生的变化。具体的,可以统计酶切片段或未被酶切的片段中突变型基因测序时出现的次数(alt depth),再分别除以该位点各基因型出现次数的总和(total depth),再乘以100%,得到一个alt的占比,自然状态下,杂合snp上alt的占比接近50%,经过酶切后,alt的占比发生变化,可能会大于50%或小于50%,考虑到测序带来的误差,通常认为一个区域内连续的几个snp上的alt的占比都超过60%或者低于40%,判定为该区域的单倍体被成功分离,由于酶切时仅切断二倍体中的其中一个单倍体,因此未被酶切的片段即为其中一个单倍体,而被酶切的片段构成另一个单倍体。同理,也可以按照上述方法统计野生型的占比进行数据分析。
51.以上数据分析方法无需复杂的生信分析算法,直接根据数据中杂合snp两个等位
基因的覆盖深度即可判断单倍型。
52.本技术的目标核酸区域来源于各种分离或获至对象的生物样本。例如羊水、血液或血液制品、脐带血、绒毛、脑脊液、脊髓液中的一种或几种。
53.以下通过特定的具体实例说明本发明的实施方式,本领域技术人员可由本说明书所揭露的内容轻易地了解本发明的其他优点与功效。本发明还可以通过另外不同的具体实施方式加以实施或应用,本说明书中的各项细节也可以基于不同观点与应用,在没有背离本发明的精神下进行各种修饰或改变。
54.在进一步描述本发明具体实施方式之前,应理解,本发明的保护范围不局限于下述特定的具体实施方案;还应当理解,本发明实施例中使用的术语是为了描述特定的具体实施方案,而不是为了限制本发明的保护范围;在本发明说明书和权利要求书中,除非文中另外明确指出,单数形式“一个”、“一”和“这个”包括复数形式。
55.当实施例给出数值范围时,应理解,除非本发明另有说明,每个数值范围的两个端点以及两个端点之间任何一个数值均可选用。除非另外定义,本发明中使用的所有技术和科学术语与本技术领域技术人员通常理解的意义相同。除实施例中使用的具体方法、设备、材料外,根据本技术领域的技术人员对现有技术的掌握及本发明的记载,还可以使用与本发明实施例中所述的方法、设备、材料相似或等同的现有技术的任何方法、设备和材料来实现本发明。
56.实施例1
57.1、目标区域扩增
58.测试所选择的酶切位点snp所在位置为chr11:5220001,rs号为rs6578582,扩增区域为chr11:5215110-5225452。
59.所用试剂:takara la hot start version,货号:rr042q。引物为生工生物工程(上海)股份有限公司合成。
60.引物序列:
61.f:5
‘-
gctctggtaatcccaaaaggctgatagtc-3’(seq id no.1)
62.r:5
‘-
tcattcctcctgtcttgggttgttcatc-3’(seq id no.2)
63.扩增体系:
64.试剂体积/总量takara la taq hs(5u/μl)0.5μl10
×
la pcr buffer ii(mg2+plus)5μldntp mixture(2.5mm each)8μldna50ng引物(10μm)1μl水up to 50μl
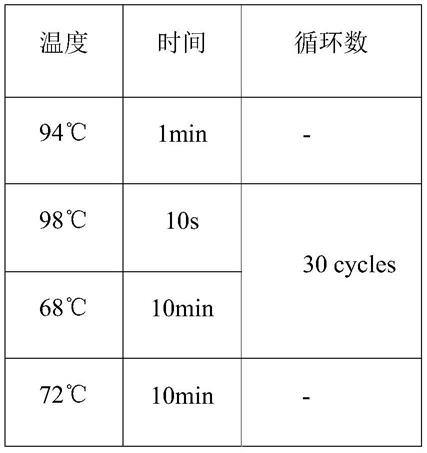
65.pcr反应条件:
[0066][0067]
2、酶切
[0068]
本实施例所挑选标准品na12878在人基因组chr11:5220001位置为杂合基因型t/c,即其中一个单倍体基因型为t/a型,另一个单倍体基因型为c/g。针对其中一个单倍体t/a设计guide序列。dna guide序列如下:
[0069]
guide-top:5
‘-
ttgcatagtgttgtac-3
‘
(seq id no.3)
[0070]
guide-down:5
‘-
ttcaggtacgaaacta-3’(seq id no.4)
[0071]
根据酶切位点和pcr的扩增区域,理论上酶切产生的片段如下表:
[0072]
富集区域起始/结束位置酶切位置酶切后产生片段长度chr11:5215110chr11:52200014891bpchr11:5225452chr11:52200015451bp
[0073]
所用试剂:pfago(45μm),10x reaction buffer(20mm hepes,250mm nacl,0.5mm mncl2),dna guide。
[0074]
dna guide由生工生物工程(上海)股份有限公司合成。
[0075]
酶切体系:
[0076][0077][0078]
酶切条件:
[0079]
温度时间
87℃15min10℃缓慢降温至10℃
[0080]
酶切结果如图2所示。
[0081]
3、片段回收
[0082]
使用胶回收试剂盒分别对步骤2中的2号孔的10k的片段及5k附近的两个片段进行回收。
[0083]
所用试剂:takara minibest agarose gel dna extraction kit ver.4.0,货号:9762。
[0084]
在紫外灯下切出含有10k及5k附近的琼脂糖凝胶,用纸巾吸尽凝胶表面液体。
[0085]
称量胶块重量,以1mg=1μl进行计算,向胶块加入等质量体积的buffer gm体积,均匀混合后室温溶解胶块。
[0086]
当凝胶完全溶解后,将全部溶液转移至spin column中,12000rpm离心1min,弃滤液。
[0087]
将700ul的buffer wb加入spin column中,12000rpm离心30s,弃滤液。重复此步骤1次。
[0088]
将spin column进行12000rpm离心1min,除去残留的buffer wb。
[0089]
将spin column置于1.5ml离心管中,在spin column膜中央加入30ul灭菌水,室温静置1min。
[0090]
室温12000rpm离心1min洗脱dna。
[0091]
4、二代测序文库制备
[0092]
所用试剂:yeasen hiefffast-pacetm dna fragmentation reagent快速片段化/末端修复/a尾添加模块,货号:12609es24;
[0093]
yeasen hiefffast-pace dna ligation module快速dna连接模块,货号:12607es08;
[0094]
yeasen hieffdna selection beads分选磁珠kapa hifi hot start ready mix,货号:kk2602。
[0095]
快速片段化/末端修复/a尾反应体系:
[0096][0097][0098]
反应条件:
[0099]
温度时间4℃1min30℃18min72℃20min
4℃∞
[0100]
接头连接反应体系:
[0101]
试剂体积dna60ul5x fast-pace ligation buffer20μlfast pace t4 dna ligase5μldna adapter1μl水up to 100ul
[0102]
反应条件:
[0103]
温度时间20℃15min
[0104]
磁珠纯化:
[0105]
向连接产物所在离心管中加入100μl磁珠,室温孵育5min。将离心管置于磁力架上直至溶液澄清,用移液器吸弃上清。将离心管保持在磁力架上,加入80%乙醇洗涤磁珠后用移液器吸弃上清,重复此步骤一次,切勿干扰到磁珠。用移液器吸弃残留的80%乙醇,室温晾干磁珠。将离心管从磁力架上取下,加入25μl灭菌水重悬浮磁珠,室温孵育2min。将离心管置于磁力架上直至溶液澄清,用移液器转移全部上清至新的离心管,获得纯化产物。
[0106]
文库扩增体系:
[0107]
试剂体积dna24μl2x kapa hifi hot start reaction mix25μlprimer1μl
[0108]
反应条件:
[0109][0110][0111]
文库纯化:
[0112]
向连接产物所在离心管中加入50μl磁珠,室温孵育5min。将离心管置于磁力架上直至溶液澄清,用移液器吸弃上清。将离心管保持在磁力架上,加入80%乙醇洗涤磁珠后用移液器吸弃上清,重复此步骤一次,切勿干扰到磁珠。用移液器吸弃残留的80%乙醇,室温晾干磁珠。将离心管从磁力架上取下,加入30ul灭菌水重悬浮磁珠,室温孵育2min。将离心
管置于磁力架上直至溶液澄清,用移液器转移全部上清至新的离心管,获得纯化产物。
[0113]
5、二代测序
[0114]
使用illumina平台的nextseq 500进行测序,每个样本数据量50mbps。
[0115]
6、单倍型分析
[0116]
标准品na12878在目标区域的所有杂合snp点的信息如下表,数据参考自hapmap/1000genomes ceu female na12878:
[0117]
ref代表野生基因型,即自然界人群中最高频率的表型,alt代表突变型,即相对于野生型突变而来的基因型。depth即测序平台测到的该基因型的次数,当野生型和突变型测到的次数接近1:1时,即认为该点为杂合snp点。0代表野生型,1代表突变型,”|”两边分别构成一种单倍型。
[0118][0119][0120]
经过酶切实验后,10k片段相同位点的杂合snp信息,如下表:
[0121]
位置refref depthaltalt depthtotal depthchr11:5216780a614g15562173chr11:5217884c665g19822650chr11:5217920c1993t6582652chr11:5219224t523g10661591chr11:5219262g639a17622402chr11:5220001t228c10761305chr11:5221132g2168a44346605chr11:5221645c2229g49607199
chr11:5221825g1679a37405423chr11:5222215c415t8421258chr11:5222379g567a12991866chr11:5222992t1255c28484103chr11:5223435t122c297419chr11:5223750t539c12101751chr11:5223822c556t10981655chr11:5223871c612g12251840chr11:5224660g744t19372685chr11:5224733c1475t31014578chr11:5224783g1658t35105175chr11:5225120g1700c32314937
[0122]
经过酶切后5k附近片段相同位点的杂合snp信息,如下表:
[0123][0124][0125]
将3组数据中的alt depth除以total depth,计算出alt depth占total depth比例并进行对比,获得如下结果,0代表野生型,1代表突变型:
[0126][0127]
根据比例做图,结果如图3所示。
[0128]
最终获得单倍型如下:
[0129]
[0130][0131]
实验结果:
[0132]
目标区域共计20个snp,在10k片段当中,19个杂合子的alt depth占total depth比例发生明显提高,1个杂合子(chr11:5217920)alt depth占total depth比例发生明显下降。5k片段当中,19个杂合子alt depth占total depth比例发生明显下降,1个杂合子(chr11:5217920)alt depth占total depth比例发生明显提高。酶切产生的片段(5k)和未被酶切片段(10k)构成两个单倍型,且与原始数据结果一致,证明本技术方案可以有效富集人基因组目标区域两个单倍体,构建该目标区域两个单倍型。
[0133]
以上的实施例是为了说明本发明公开的实施方案,并不能理解为对本发明的限制。此外,本文所列出的各种修改以及发明中方法的变化,在不脱离本发明的范围和精神的前提下对本领域内的技术人员来说是显而易见的。虽然已结合本发明的多种具体优选实施例对本发明进行了具体的描述,但应当理解,本发明不应仅限于这些具体实施例。事实上,各种如上所述的对本领域内的技术人员来说显而易见的修改来获取发明都应包括在本发明的范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1