一种用于筛选SNP位点的方法及其应用与流程
一种用于筛选snp位点的方法及其应用
技术领域
1.本发明涉及基因工程技术领域,具体而言,涉及一种用于筛选snp位点的方法及其应用。
背景技术:
2.癌症是世界上严重危害人类健康三大疾病之一,2018年最新的全球肿瘤统计结果显示,全球估计有1819万癌症新增病例以及960万癌症死亡病例。肺癌是最常被诊断的癌症(占总病例数的11.6%),并且是癌症死亡的主要原因(占癌症总死亡人数的18.4%)。余下发病率较高的分别为乳腺癌(11.6%),结直肠癌(10.2%),前列腺癌(7.1%)和胃癌(5.7%)。
3.众所周知,肿瘤的发生源于一系列基因变化的积累,进而导致信号通路、细胞分裂周期出现错误,过程中涉及多个关键细胞因子和受体蛋白质,这些细胞因子和受体蛋白质既是导致肿瘤发生和进展的关键要素,也是治疗肿瘤的可能靶点,而免疫逃逸助长了肿瘤的发展,但是患者个体的基因异变存在特异性,并可能随病程进展而改变,呈现出不稳定性。
4.因此,临床需要在诊疗全程就患者个体情况与可选药物、疗法匹配度进行诊断,也就是伴随诊断(companion diagnostic,cd),通过检测人体内特定的基因变异,提供患者针对某种药物、疗法的反应信息,从而协助临床确定最佳的用药和治疗方式,还能够提示治疗的脱靶效应,预测与药物相关的毒副作用,实现精准治疗。
5.目前,免疫治疗已经逐渐证明了它在治疗肿瘤上的价值,旨在激活人体免疫系统,依靠自身免疫机能杀灭癌细胞和肿瘤组织。免疫应答过程需要人体内的hla分子将肿瘤细胞内的新生抗原呈递到细胞表面从而被免疫细胞识别。人体内表达hla的种类影响着可被呈递到细胞表面的新抗原的数量。hla的多样性很高,如果病人本身的hla所在区域发生杂合性缺失(loh),则病人免疫治疗的效果将降低。loh也是伴随诊断的一个重要生物特征。目前检测loh的算法也需要一批均匀分布在基因组范围的杂合性位点作为评估信号。
6.现在已经有越来越多的人基于基因测序来进行伴随诊断。基因测序检测体细胞突变需要用病人本身的正常细胞做为对照来排除胚系突变。如果取到的肿瘤样本混入其它人的dna造成污染,将导致突变检测结果混有其他人的体细胞突变和胚系突变,造成肿瘤突变负荷(tmb)等指标偏高。而检测人源污染也可通过杂合性位点进行评估。
7.综上,筛选出一批均匀分布在基因组中并且性能稳定的杂合性位点将非常重要。目前基于全基因组或全外显子组的方法进行基因检测,可先用正常样本进行胚系突变检测,从而获得杂合突变位点。此种方法虽然天然的可以获得数量较多,针对性很强的杂合性位点,但由于采用全基因组和全外显子组测序,价格昂贵,患者受益有限,并且目前的全基因组或全外显子组测序的测序深度均较低,无法稳定突变频率,导致后续分析产生较大误差。目前肿瘤基因检测广泛采用目标捕获测序,通过筛选更有意义的基因区域,在控制成本的条件下能使病人获得较大的收益。然而目标捕获测序的目标范围普遍较小,且在基因组
中的分布不够均匀,无法得到足够数量且均匀的杂合性位点。
8.鉴于此,特提出本发明。
技术实现要素:
9.本发明的目的在于提供一种用于筛选多用途snp位点的方法及其应用。
10.本发明是这样实现的:
11.第一方面,实施例提供了一种用于筛选多用途snp位点的方法,其包括:基于获取的n例样本基因组中的snp候选位点的突变频率信息,将满足筛选标准的位点作为多用途snp位点,n≥3;
12.所述筛选标准包括:n例样本基因组中的杂合突变型在位点处的平均突变丰度为40%~60%,n例样本基因组中的纯合突变型在位点处的平均突变丰度大于90%;n例样本基因组中的野生型在位点处的平均突变丰度小于5%;
13.对满足所述筛选标准的多用途snp位点进行判断,若单一染色体上相邻多用途snp位点之间的距离≤预设距离,则去除其中任意1个,以使得每条染色体上任意相邻的两个多用途snp位点之间的距离》所述预设距离,所述预设距离为250kb~350kb。
14.第二方面,实施例提供了一种用于筛选多用途snp位点的装置,其包括:
15.获取模块,用于获取的n例样本基因组中的snp候选位点的突变频率信息,n≥3;
16.筛选模块,用于根据获取的突变频率信息,将满足筛选标准的位点作为多用途snp位点;并对满足所述筛选标准的多用途snp位点进行判断,若单一染色体上相邻多用途snp位点之间的距离≤预设距离,则去除其中任意1个,以使得每条染色体上任意相邻的两个多用途snp位点之间的距离》所述预设距离,所述预设距离为250kb~350kb;
17.其中,所述筛选标准包括:n例样本基因组中的杂合突变型在位点处的平均突变丰度为40%~60%,n例样本基因组中的纯合突变型在位点处的平均突变丰度大于90%;n例样本基因组中的野生型在位点处的平均突变丰度小于5%。
18.第三方面,实施例提供了一种电子设备,其包括存储器、处理器以及存储在所述存储器上并可在所述处理器上运行的计算机程序,所述处理器执行所述计算机程序时,实现如前述实施例所述的用于筛选多用途snp位点的方法。
19.第四方面,实施例提供了一种计算机可读存储介质,所述计算机可读存储介质上存储有计算机程序,当所述计算机程序被处理器执行时,实现如前述实施例所述的用于筛选多用途snp位点的方法。
20.第五方面,实施例提供了一种试剂盒,其包括用于检测如前述实施例所述的用于筛选多用途snp位点的方法筛选得到的多用途snp位点的试剂。
21.第六方面,实施例提供了如前述实施例所述的用于筛选多用途snp位点的方法或如前述实施例所述的试剂盒或前述实施例所述的用于筛选多用途snp位点的装置或前述实施例所述的电子设备或前述实施例所述的计算机可读存储介质在样本污染水平检测中的应用。
22.优选地,所述应用不以疾病的诊断或治疗为目的。
23.第七方面,实施例提供了如前述实施例所述的用于筛选多用途snp位点的方法或如前述实施例所述的试剂盒或前述实施例所述的用于筛选多用途snp位点的装置或前述实
施例所述的电子设备或前述实施例所述的计算机可读存储介质在基因杂合性缺失loh检测中的应用。
24.优选地,所述应用不以疾病的诊断或治疗为目的。
25.本发明具有以下有益效果:
26.本发明实施例提供了一种用于筛选snp位点的方法及其应用,该方法包括根据获取的n例样本基因组中的snp候选位点的突变频率信息,将满足筛选标准的位点作为多用途snp位点,对满足上述筛选标准的多用途snp位点进行判断,若单一染色体上相邻多用途snp位点之间的距离>预设距离,则去除其中任意1个,以使得每条染色体上任意相邻的两个多用途snp位点之间的距离》所述预设距离,所述预设距离为250kb~350kb。
27.该方法基于已知存在于群体基因组中的单核苷酸多态性(snp)信息,筛选出一批均匀分布于基因组中并且性能稳定的杂合性位点的合集。目前全基因组检测或全外显子组检测的方法价格昂贵,且测序深度较低,无法稳定突变频率,可能导致一些检测的后续分析产生较大误差。本发明筛选出的位点合集更有针对性,能够应用于很多免疫治疗相关的检测中,如样本污染水平的检测、基因杂合性缺失的检测以及肿瘤基因组倍性检测,具有检测成本更低,检测时间快且检测有效性更高等优点。
附图说明
28.为了更清楚地说明本发明实施例的技术方案,下面将对实施例中所需要使用的附图作简单地介绍,应当理解,以下附图仅示出了本发明的某些实施例,因此不应被看作是对范围的限定,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他相关的附图。
29.图1为实施例2中36个样本的所有杂合突变位点突变丰度标准差在snp panel和全基因组测序中的差异结果图;
30.图2为实施例2中snp panel和传统gene panel中突变位点的分布图;
31.图3为实施例3中对有污染的样本的两组评估结果和真实结果的相关性分析结果图;
32.图4为实施例3中无污染样本的两组评估结果图;
33.图5为实施例4中两组肿瘤纯度的相关性结果;
34.图6为实施例4中两组肿瘤倍性的相关性结果;
35.图7为实施例4中两组肿瘤大片段杂合性缺失数量的相关性结果。
具体实施方式
36.为使本发明实施例的目的、技术方案和优点更加清楚,下面将对本发明实施例中的技术方案进行清楚、完整地描述。实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市售购买获得的常规产品。
37.名词定义
38.本文中的“snp”指单核苷酸多态性,主要指在基因组水平上由单个核苷酸的变异所引起的dna序列多态性,为人类可以遗传的变异中最常见的一种。
39.本文中的“突变丰度”的英文为vaf,variant allele fraction,也称为variant allel frequency(变异等位基因频率),可以指测序过程中突变reads(读长)占总reads的比例,即计算公式可以为:
40.vaf=allele depth/total depth。其中,allele depth为基因组每个位点支持突变基因型的reads(读段)覆盖深度,total depth为这个位点总reads覆盖深度。
41.本文中的“杂合突变型”可以指一对等位基因中,其中一个基因发生了突变,另一个未发生突变;“纯合突变型”可以指:一对等位基因均发生了突变;“野生型”可以指未突变的基因型。
42.本文中的“读段”指高通量测序中的reads。
43.本文中的“测序深度”可以指:测序得到的碱基总量(bp)与基因组大小的比值。
44.本文中的“串联重复区”,又称串联重复序列,是指以相对恒定的短序列为重复单位,首尾相接,串联连接形成的重复序列;又称卫星dna(satellite dna)。在人类基因组中,串联重复序列约占10%,主要分布在非编码区,少数位于编码区。
45.技术方案
46.首先,实施例提供了一种用于筛选多用途snp位点的方法,其包括:基于获取的n例样本基因组中的snp候选位点的突变频率信息,将满足筛选标准的位点作为多用途snp位点,n≥3;
47.所述筛选标准包括:n例样本基因组中的杂合突变型在位点处的平均突变丰度为40%~60%,n例样本基因组中的纯合突变型在位点处的平均突变丰度大于90%;n例样本基因组中的野生型在位点处的平均突变丰度小于5%。基于该筛选标准筛选出的位点,稳定性高,其检测的结果相对于其他排除的位点,更具有代表性。具体地,“n例样本基因组中的杂合突变型在位点处的平均突变丰度为40%~60%”是指n例样本基因组中,在位点处为杂合突变型的样本基因组,在该位点的平均突变丰度为40%~60%,纯合突变型和野生型的情况依此类推。
48.对满足所述筛选标准的多用途snp位点进行判断,若单一染色体上相邻多用途snp位点之间的距离≤预设距离,则去除其中任意1个,以使得每条染色体上任意相邻的两个多用途snp位点之间的距离》所述预设距离,所述预设距离为250kb~350kb。该预设距离是发明人经一系列创造性劳动得出的,在有效缩小snp位点的数量的情况下,保持检测有效性的距离。具体地,预设距离可以选自250kb、260kb、270kb、280kb、290kb、300kb、310kb、320kb、330kb、340kb和350kb中的任意一个数值。
49.本技术的发明人经研究,发明了上述用于筛选多用途snp位点的方法,该方法基于snp候选位点的突变频率信息,针对性地筛选出一批均匀分布于基因组中并且性能稳定的杂合性位点的合集,为现有的免疫治疗提供了一种新的、更有效的检测或辅助检查的手段,相对于全基因组测序和全外显子测序而言,检测成本更低,检测时间更快。
50.在一些优选的实施方式中,若单一染色体上相邻的多用途snp位点之间的距离≤预设距离,则去除其中1个位点,以使得每条染色体上任意相邻的两个多用途snp位点之间的距离》上述预设距离。具体地,该去除的标准包括:(1)若这两个多用途snp位点分别与其另一端相邻位点的距离≤或≥预设距离,则任意去除其中一个;需要说明的是,当这两个多用途snp位点或其中之一为端点时,即其另一端并不存在相邻位点,则视为其与另一端相邻
位点的距离≥预设距离;(2)若这两个多用途snp位点中的其中一个位点与另一端相邻位点的距离>预设距离,另一个位点与另一端相邻位点的距离<预设距离,则去除与另一端相邻位点的距离<预设距离的位点。
51.在一些优选的实施方式中,所述筛选标准还包括:含snp候选位点的读段在n例样本基因组中的比对位置有且只有一个。
52.在一些优选的实施方式中,所述方法还包括对满足所述预设距离的多用途snp位点再次判断:
53.若单一染色体上相邻多用途snp位点之间的距离>2mb的区域,则将该区域划分为多个第一选择区域,并在每个所述第一选择区域内,任意挑选1个位点作为多用途snp位点;所述第一选择区域的长度为80kb~120kb;
54.若单一染色体上存在相邻候选位点之间的距离为1.5mb~2mb的区域,则将该区域划分为多个第二选择区域,并在每个所述第二选择区域内,任意挑选1个位点作为多用途snp位点;所述第二选择区域的长度为250kb~350kb。
55.再次判断的目的在于防止基因组上存在一些空缺snp候选位点的区域,从而导致该区域内无对应的检测信号。第一选择区域内和第二选择区域的长度是特定的,在该两区中选择加入位点的数量及其间隔也是发明人经一系列创造性劳动得出的,从而使得选择得到的最终的多用于snp位点的检测稳定性更高,检测结果有效。
56.具体地,所述第一选择区域的长度可以选自80kb、90kb、100kb、110kb和120kb中的任意一个数值。第二选择区域的长度可以选自250kb、260kb、270kb、280kb、290kb、300kb、310kb、320kb、330kb、340kb和350kb中的任意一个数值。
57.优选地,在所述第一选择区域或所述第二选择区域内挑选位点的标准为:选取gc含量在35%~75%之间的位点,并排除位于连续5~8bp串联重复区的位点。
58.在一些优选的实施方式中,所述snp候选位点为n例样本中等位基因频率为40%~60%的位点。
59.n例样本基因组可以指现有的基因数据库,如千人基因组、exac、gnomad和rmsk等数据库;本发明对n的数值不作限制,可以根据实际情况,选择性设置。优选地,n≥100;优选地,n≥300;优选地,n≥1000。
60.其次,本发明实施例提供了一种用于筛选多用途snp位点的装置,其包括:
61.获取模块,用于获取的n例样本基因组中的snp候选位点的突变频率信息,n≥3;
62.筛选模块,用于根据获取的突变频率信息,将满足筛选标准的位点作为多用途snp位点;并对满足所述筛选标准的多用途snp位点进行判断,若单一染色体上相邻多用途snp位点之间的距离≤预设距离,则去除其中任意1个,以使得每条染色体上任意相邻的两个多用途snp位点之间的距离》所述预设距离,所述预设距离为250kb~350kb;
63.其中,所述筛选标准包括:n例样本基因组中的杂合突变型在位点处的平均突变丰度为40%~60%,n例样本基因组中的纯合突变型在位点处的平均突变丰度大于90%;n例样本基因组中的野生型在位点处的平均突变丰度小于5%。
64.具体地,该实施例中的位点的筛选标准同上述任意实施方式所述,不再赘述。
65.本发明实施例还提供了一种电子设备,其包括存储器、处理器以及存储在所述存储器上并可在所述处理器上运行的计算机程序,所述处理器执行所述计算机程序时,实现
如前述任意实施方式所述的用于筛选多用途snp位点的方法。
66.具体地,该电子设备可以包括存储器、处理器、总线和通信接口,该存储器、处理器和通信接口相互之间直接或间接地电性连接,以实现数据的传输或交互。例如,这些元件相互之间可通过一条或多条总线或信号线实现电性连接。处理器可以处理与目标识别有关的信息和/或数据,以执行本技术中描述的一个或多个功能。
67.存储器可以是但不限于,随机存取存储器(random access memory,ram),只读存储器(read only memory,rom),可编程只读存储器(programmable read-only memory,prom),可擦除只读存储器(erasable programmable read-only memory,eprom),电可擦除只读存储器(electric erasable programmable read-only memory,eeprom)等。
68.处理器可以是一种集成电路芯片,具有信号处理能力。该处理器可以是通用处理器,包括中央处理器(central processing unit,cpu)、网络处理器(network processor,np)等;还可以是数字信号处理器(digital signal processing,dsp)、专用集成电路(application specific integrated circuit,asic)、现场可编程门阵列(field-programmable gate array,fpga)或者其他可编程逻辑器件、分立门或者晶体管逻辑器件、分立硬件组件。
69.该电子设备中的各组件可以采用硬件、软件或其组合实现。在实际应用中,该电子设备可以是服务器、云平台、手机、平板电脑、笔记本电脑、超级移动个人计算机(ultra-mobile personal computer,umpc)、手持计算机、上网本、个人数字助理(personal digital assistant,pda)、可穿戴电子设备、虚拟现实设备等设备,因此本技术实施例对电子设备的种类不做限制。
70.本发明实施例提供了一种计算机可读存储介质,所述计算机可读存储介质上存储有计算机程序,当所述计算机程序被处理器执行时,实现如前述任意实施方式所述的用于筛选多用途snp位点的方法。
71.需要说明的是,本实施例中的计算机可读存储介质与前述实施例中的存储器相互等同。
72.本发明实施例提供了一种试剂盒,其包括用于检测如前述任意实施方式所述的用于筛选多用途snp位点的方法筛选得到的多用途snp位点的试剂。
73.优选地,试剂可以为探针和/或引物。
74.本发明实施例还提供了如前述任意实施方式所述的用于筛选多用途snp位点的方法或如前述任意实施方式所述的试剂盒或前述实施例所述的用于筛选多用途snp位点的装置或前述实施例所述的电子设备或前述实施例所述的计算机可读存储介质在样本污染水平检测中的应用。
75.本发明实施例还提供了如前述任意实施方式所述的用于筛选多用途snp位点的方法或如前述任意实施方式所述的试剂盒或前述实施例所述的用于筛选多用途snp位点的装置或前述实施例所述的电子设备或前述实施例所述的计算机可读存储介质在基因杂合性缺失loh检测中的应用。
76.以下结合实施例对本发明的特征和性能作进一步的详细描述。
77.实施例1
78.一种用于筛选多用途snp位点的方法,其包括以下步骤。
79.(1)获得snp候选位点:
80.选取千人基因组、exac和gnomad等数据库中高频出现的位点,即选取等位基因频率(af)为40%~60%的点作为候选位点,以使所选的位点能在不同人中实现较大利用率且稳定(即在人群病例中有较多的位点呈现杂合状态并且不同人间波动较小)。
81.然后,根据人类rmsk数据库记录的重复序列信息,去除掉位于重复序列内的突变位点以构成snp候选位点的预集合。
82.(2)制定初选panel:根据上述snp候选位点前后60bp的序列信息设计120nt的探针,去掉能够在人类基因组上比对到两个位置以上的探针序列,合成测试探针pool panel。
83.(3)制定位点性能测试数据集:利用上述探针pool panel对测试人群样本进行建库测序。收集临床患者白细胞样本(本实施例中为36例),使用以上设计的探针pool进行捕获建库,建库流程简述如下:
84.3.1将样本进行打断及末端修复。
85.3.2.将上述修复后的dna进行接头连接。
86.3.3.将上述接头连接后的产物进行pcr扩增,得到足量带接头的dna片段,即为预文库。
87.3.4.对上述预文库进行磁珠纯化并进行浓度测定和片段质检。
88.3.5.对预文库进行探针杂交。
89.3.6.使用链霉亲合素磁珠对探针结合的样品进行捕获。
90.3.7.将磁珠捕获到的dna片段进行pcr扩增,得到足量的加上标签的dna片段,即为终文库。
91.3.8.对上述终文库进行磁珠纯化并进行浓度测定和片段质检,利用qpcr进行定量。
92.3.9.用于panel分析建库测序。panel建库测序方法为探针捕获建库,使用基因测序仪(novaseq 6000),按照仪器标准操作规程进行150bp pair-end模式测序(read1:151,read2:151;index1:8,index2:8),最终得到fastq格式二代测序数据作为原始数据(raw data)。
93.(4)测试数据处理:使用第三方软件fastp软件对测序下机数据进行数据过滤,包括减去测序接头序列,去除测序读长小于50bp的dna片段,去除测序质量较低的dna片段,去除含未知碱基数较多的dna片段;使用第三方软件bwa将过滤后的数据与hg19参考基因组进行比对,得到每个dna片段基因组上对应的具体位置信息及详细比对情况。使用第三方软件sambamba去除比对结果中的冗余序列,即双端比对后起始终止比对位置相同的dna片段对只保留一对。使用内部自建软件mutationinfo搜索上述snp位点的突变频率信息。
94.(5)筛选:基于获取的n例样本基因组中的snp候选位点的突变频率信息,将满足筛选标准的位点作为多用途snp位点,n为36;
95.所述筛选标准包括:n例样本基因组中的杂合突变型在位点处的平均突变丰度为0.4~0.6(40%~60%),且标准差小于0.1,n例样本基因组中的纯合突变型在位点处的平均突变丰度大于0.99,且标准差小于0.1;n例样本基因组中的野生型在位点处的平均突变丰度小于0.01,且标准差小于0.1;以及含snp候选位点的读段在n例样本基因组中的比对位置有且只有一个。
96.对满足所述筛选标准的多用途snp位点进行判断,对单一染色体上第一个多用途snp位点开始判断,若其与后续相邻的多用途snp位点≤预设距离,去除后一位点,保留第一个多用途snp位点,直至满足第一个多用途snp位点与保留的第二个位点之间的距离>预设距离;然后以保留的第二个位点作为基准,判断其与后续相邻的位点(第三个位点)之间的距离,直至满足第二个位点与第三个位点之间的距离>预设距离;后续位点的选择依此类推,以单一方向判断的方式对单一染色体上存在的多个多用途snp位点进行判断筛选,以使得每条染色体上任意相邻的两个多用途snp位点之间的距离》所述预设距离,所述预设距离为300kb。
97.(6)最终确定:所述方法还包括对满足所述预设距离的多用途snp位点再次判断:若单一染色体上相邻多用途snp位点之间的距离>2mb的区域,则将该区域划分为多个第一选择区域,并在每个所述第一选择区域内,任意挑选1个位点作为多用途snp位点;所述第一选择区域的长度为100kb;
98.若单一染色体上存在相邻候选位点之间的距离为1.5mb~2mb的区域,则将该区域划分为多个第二选择区域,并在每个所述第二选择区域内,任意挑选1个位点作为多用途snp位点;所述第二选择区域的长度为200kb;
99.其中,在所述第一选择区域或所述第二选择区域内挑选位点的标准为:选取gc含量在35%~75%之间的位点,并排除位于连续5~8bp串联重复区的位点。
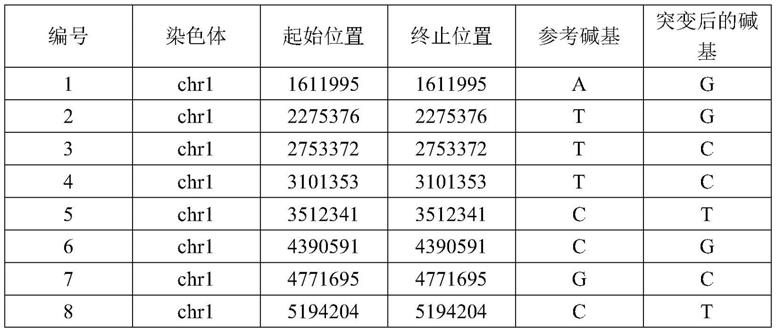
100.基于上述方法,得到最终的多用途snp位点合集(snp panel),共为3905个,snp panel大小468.6kb,位点的部分具体信息如表1所示。
101.表1多用途snp位点
102.103.104.105.106.107.108.109.110.111.[0112][0113]
与全基因组测序(3gb)和全外显子组测序(大约40mb~60mb不等)相比较,目标测序区域大小减小6402倍和85倍多,可以显著降低成本,提高患者受益率。
[0114]
目前全基因组测序深度普遍采用30x深度,全外显子组一般是200x,而利用snp panel在节省测序成本的情况下可提高测序深度至1000x以上,深度增高可显著增加突变频率计算的稳定性,使结果更加准确。
[0115]
实施例2
[0116]
检测36个样本中所有杂合突变位点突变丰度标准差在snp panel和全基因组测序中的差异,结果如图1所示。
[0117]
本发明测试本发明筛选出的snp panel,杂合突变频率的标准差为0.05957,一般panel基因区为0.1247,wgs数据为0.0710。结合图1可知,本发明筛选的snp panel的突变频率标准差显著低于全基因组测序,反应出用snp panel的方法可以使突变位点的突变频率更加稳定。
[0118]
图2为snp panel和传统gene panel中突变位点的分布情况,其中,图2中a为snp panel的结果,图2中b为传统gene panel的结果。从图2可以看出snp panel的位点明显比一般gene panel的位点更密集更均匀。
[0119]
实施例3
[0120]
使用第三方软件conpair(bergmann e a,bo-juen c,kanika a,et al.conpair:concordance and contamination estimator for matched tumor
–
normal pairs[j].bioinformatics(20):3196-3198.)进行污染评估,评估样本为88个污染程度从0.6%到27%的有污染的样本和95个无污染的样本,所有样本数据均为包含我们snp panel位点的目标捕获测序数据。
[0121]
使用conpair软件对这些样本进行污染检测,设定markers参数为默认或本发明实施例1提供的snp panel产生两组评估结果。
[0122]
图3是对有污染的样本的两组评估结果和真实结果的相关性分析,其中,图3中a为conpair的相关性分析结果,图3中b为snp panel的相关性分析结果。结果显示使用本发明提供的snp panel位点的结果与真实值的相关性更好。
[0123]
图4是无污染样本的两组评估结果,可以看出使用snp panel评估的污染数值明显小于使用conpair软件默认位点的结果。
[0124]
实施例4
[0125]
使用78个wgs测序样本进行评估使用实施例1筛选得到的snp panel位点检测肿瘤纯度、倍性、大片段杂合性缺失的性能。
[0126]
利用第三方检测软件purple(priestley p,baber j,lolkema m,et al.pan-cancer whole genome analyses of metastatic solid tumors[j].nature.)进行检测。purple检测流程采用默认参数,在amber步骤的输入snp位点参数-loci分别提供默认使用的1344545个位点和本发明snppanel的3905个位点来得到两组检测结果。
[0127]
图5、图6和图7分别是两组肿瘤纯度、倍性、大片段杂合性缺失数量的相关性结果。从图中可以看出,本发明仅使用3905个snp位点即可得到与使用1344545个位点相关性很高的结果,证明本发明的snp panel可为检测肿瘤纯度、倍性、大片段杂合性缺失提供良好的检测信号。
[0128]
以上所述仅为本发明的优选实施例而已,并不用于限制本发明,对于本领域的技术人员来说,本发明可以有各种更改和变化。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1