一类取代苯甲酰哌嗪类化合物及其应用的制作方法
[0001]
本发明属于医药技术领域,具体地说,涉及一类取代苯甲酰哌嗪类化合物及其应用。
背景技术:
[0002]
天然产物的抑菌作用受到了普遍关注,天然产物以其结构新颖,生物活性强,毒性低的特点,成为了近年寻找抗真菌新药及其先导化合物的研究热点。没食子酸(gallic acid),又称五倍子酸或棓酸,即3,4,5-三羟基苯甲酸,是一种存在于五倍子、漆树、茶等植物中的有机酸。文献报道其具有抗菌、抗病毒、抗肿瘤等作用。以此为基础,设计合成了系列取代苯甲酰哌嗪类化合物,评价其单独及协同抗真菌作用,为进一步研究和开发抗真菌药物提供基础。
技术实现要素:
[0003]
本发明的第一个目的是提供一类取代苯甲酰哌嗪类化合物。
[0004]
本发明的第二个目的是提供一种所述取代苯甲酰哌嗪类化合物在制备抗真菌药物中的应用。
[0005]
为了实现上述目的,本发明采用的技术方案如下:
[0006]
本发明的第一个方面提供了一类取代苯甲酰哌嗪类化合物或其药用盐,具有以下结构通式:
[0007][0008]
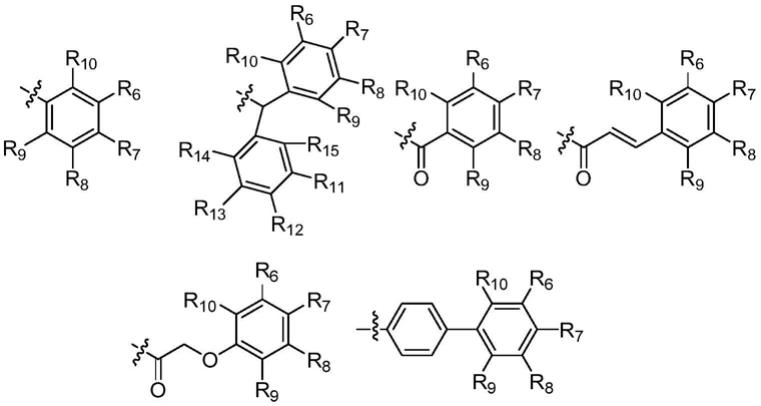
r1、r2、r3、r4、r5各自独立的选自氢、羟基、c1~c10烷基、c1~c10烷氧基、卤素;
[0009]
x选自c或n;
[0010]
y选自以下基团中的一种:
[0011][0012]
r6、r7、r8、r9、r
10
、r
11
、r
12
、r
13
、r
14
、r
15
各自独立的选自氢、羟基、卤素(氟、氯、溴、碘)、c1~c10烷基、c1~c10烷氧基。
[0013]
较优选的,所述取代苯甲酰哌嗪类化合物为以下结构中的一种:
[0014][0015]
r1、r2、r3、r4、r5各自独立的选自氢、羟基、c1~c10烷氧基;
[0016]
r6、r7、r8、r9、r
10
、r
11
、r
12
、r
13
、r
14
、r
15
各自独立的选自氢、羟基、卤素(氟、氯、溴、碘)、c1~c10烷基、c1~c10烷氧基。
[0017]
比较优选的,所述取代苯甲酰哌嗪类化合物中:
[0018]
r1、r2、r3同时为羟基,r4、r5同时为氢;
[0019]
或,r1、r2同时为羟基,r3、r4、r5同时为氢;
[0020]
或,r2、r3同时为羟基,r1、r4、r5同时为氢;
[0021]
或,r2、r3、r4同时为羟基,r1、r5同时为氢;
[0022]
或,r2为甲氧基,r1、r4、r5、r3同时为氢;
[0023]
进一步优选的,所述取代苯甲酰哌嗪类化合物中:
[0024]
r6、r7、r8、r9、r
10
、r
11
、r
12
、r
13
、r
14
、r
15
各自独立的选自氢、氟、氯。
[0025]
最优选的,所述取代苯甲酰哌嗪类化合物为以下结构中的一种:
[0026]
[0027]
[0028][0029]
本发明的第三个方面提供了一种所述取代苯甲酰哌嗪类化合物或其药用盐在制备抗真菌药物中的应用。
[0030]
本发明的第四个方面提供了一种所述取代苯甲酰哌嗪类化合物或其药用盐在制备协同氟康唑抗真菌药物中的应用。
[0031]
所述真菌为白念珠菌。
[0032]
本发明的取代苯甲酰哌嗪类化合物可按照常规方法制备为药用盐的形式。
[0033]
本发明所述取代苯甲酰哌嗪类化合物的药用盐是由药学上可接受的无机酸和有机酸所形成的盐,其中较优的无机酸包括:盐酸、氢溴酸、磷酸、硝酸、硫酸;较优的有机酸包括:甲酸、乙酸、丙酸、丁二酸、萘二磺酸(1,5)、亚细亚酸、甘珀酸、甘草次酸、齐墩果酸、山楂酸、熊果酸、科罗索酸、白桦酸、乳香酸、草酸、酒石酸、乳酸、水杨酸、苯甲酸、戊酸、二乙基乙酸、丙二酸、琥珀酸、富马酸、庚二酸、己二酸、马来酸、苹果酸、氨基磺酸、苯丙酸、葡糖酸、抗坏血酸、烟酸、异烟酸、甲磺酸、乙磺酸、对甲苯磺酸、柠檬酸,以及氨基酸。
[0034]
由于采用上述技术方案,本发明具有以下优点和有益效果:
[0035]
本发明的取代苯甲酰哌嗪类化合物,具有较好的抗真菌活性,尤其是对耐药性白念珠菌,在单独使用的时候,即具有显著的效果。而且,与氟康唑联合使用时,可以使原来对唑类耐药的真菌重新对唑类药物敏感,降低唑类药物的用量,提高唑类药物的疗效,起到增效的作用。
具体实施方式
[0036]
为了更清楚地说明本发明,下面结合优选实施例对本发明做进一步的说明。本领域技术人员应当理解,下面所具体描述的内容是说明性的而非限制性的,不应以此限制本发明的保护范围。
[0037]
本发明实施例中所使用的材料如下:采用bruker spectmspin ac-p300(瑞士bruker公司);ac-p600型核磁共振仪(瑞士bruker公司);agilent 6120型lc-ms联用质谱仪(美国agilent公司);硅胶板gf254(烟台黄海化学,中国);暗箱式紫外分析仪(zf-20d型号);旋转蒸发仪(buchi rotavapor r-3型号);shb-iii循环式多用真空泵;真空干燥箱(dzf-6021型);超声波清洗器(kq-500e型);实验所用的试剂(分析纯)和原料购于安耐吉试剂有限公司、百灵威科技有限公司、毕得医药科技有限公司、探索平台等。
[0038]
实施例1
[0039]
化合物1b-1的制备:
[0040][0041]
将3,4,5-三甲氧基苯甲酸(化合物1)(100.0mg,0.5mmol)置于100ml圆底烧瓶中,加入dcm(10.0ml,二氯甲烷)使其溶解,然后依次加入1-(2-氯苯基)哌嗪(120.0mg,0.6mmol),dcc(124.0mg,0.6mmol,n,n'-二环己基碳酰亚胺),dmap(244.0mg,2.0mmol),室温条件下搅拌6h,观察到溶液由澄清变浑浊,tlc检测(dcm:meoh=10:1),待反应完毕。过滤,滤液减压蒸干,用95%etoh重结晶后得到化合物1a-1(111.0mg,收率:56%)。
[0042]
将化合物1a-1(100.0mg,0.3mmol)置于100ml圆底三口瓶中,在无水无氧氩气保护的条件下加入无水dcm(10.0ml),在-30℃的温度下以每秒钟两滴的恒定速度滴加bbr3的dcm溶液(6.0ml,3.0mmol),滴加完将反应装置移到室温条件下搅拌过夜,tlc检测(dcm:meoh=10:1),待反应完毕,加入水100.0ml进行淬灭,室温条件下搅拌1h,然后观察是否有
固体析出,若有固体析出,过滤,滤饼置于烘箱内烘干;若没有固体析出,ea萃取三次,合并有机相,无水硫酸钠干燥,将有机相蒸干,用dcm或etoh重结晶后得到化合物1b-1。
[0043]
实施例2
[0044]
化合物1b-2的制备:
[0045][0046]
以1-(3-氯苯基)哌嗪(120.0mg,0.6mmol)替换实施例1中所用的1-(2-氯苯基)哌嗪,其他参照实施例1,得到化合物1a-2(123.0mg,收率:61%)。化合物1b-2的制备参考实施例1中化合物1b-1的制备。
[0047]
实施例3
[0048][0049]
以1-(2,3-二氯苯基)哌嗪(140.0mg,0.6mmol)替换实施例1中所用的1-(2-氯苯基)哌嗪,其他参照实施例1,得到化合物1a-3(107.0mg,收率:58%)。化合物1b-3的制备参考实施例1中化合物1b-1的制备。
[0050]
实施例4
[0051][0052]
将3,4,5-三甲氧基苯甲酸(化合物1)(100.0mg,0.5mmol)置于100ml圆底烧瓶中,加入dmf(10.0ml)使其溶解,然后依次加入4,4'-二氟二苯甲哌嗪(173.0mg,0.6mmol),pybop(312.2mg,0.6mmol),再加入diea(1.0ml,2.0mmol),加料完毕后室温条件下搅拌4h,tlc检测(dcm:meoh=10:1),待反应完毕。加入水100.0ml进行淬灭,室温条件下搅拌1h,观察是否有固体析出,有固体析出,过滤,滤饼置于烘箱内烘干,用95%etoh重结晶后得到化合物1a-4(118.0mg,收率:60%)。
[0053]
化合物1b-4的制备参考实施例1中化合物1b-1的制备。
[0054]
实施例5
[0055][0056][0057]
将3,4,5-三甲氧基苯甲酸(化合物1)(100.0mg,0.5mmol),n-boc-哌嗪(150.0mg,0.6mmol),pybop(312.2mg,0.6mmol),再加入diea(1.0ml,2.0mmol),加料完毕后室温条件下搅拌4h,tlc检测(dcm:meoh=10:1),待反应完毕。加入水100.0ml进行淬灭,室温条件下搅拌1h,过滤,滤饼用95%etoh重结晶后得到中间体a。称重,按照1:3的当量比将产物溶解于盐酸-乙酸乙酯溶液中,室温条件下搅拌,tlc检测(dcm:meoh=10:1),待反应完毕,过滤,用95%etoh重结晶后得到中间体b。
[0058]
中间体b(140.0mg,0.5mmol),2,3-二氯苯甲酸(115.0mg,0.6mmol),pybop(312.2mg,0.6mmol)和diea(1.0ml,2.0mmol),方法同化合物1a-4,得化合物1a-5121.0mg(收率:59%)。
[0059]
化合物1b-5的制备参考实施例1中化合物1b-1的制备。
[0060]
实施例6
[0061][0062]
以3,4-二氯苯甲酸(115.0mg,0.6mmol)替换实施例5中所用的2,3-二氯苯甲酸,其他参照实施例5,得到化合物1a-6(115.0mg,收率:58%)。化合物1b-6的制备参考实施例1中化合物1b-1的制备。
[0063]
实施例7
[0064]
[0065]
以3,4,5-三氟苯甲酸(110.0mg,0.6mmol)替换实施例5中所用的2,3-二氯苯甲酸,其他参照实施例5,得到化合物1a-7(120.0mg,收率:65%)。化合物1b-7的制备参考实施例1中化合物1b-1的制备。
[0066]
实施例8
[0067][0068]
以反式肉桂酸(100.0mg,0.6mmol)替换实施例5中所用的2,3-二氯苯甲酸,其他参照实施例5,得到化合物1a-8(116.0mg,收率:59%)。化合物1b-8的制备参考实施例1中化合物1b-1的制备。
[0069]
实施例9
[0070][0071]
以苯氧乙酸(100.0mg,0.6mmol)替换实施例5中所用的2,3-二氯苯甲酸,其他参照实施例5,得到化合物1a-9(130.0mg,收率:75%)。化合物1b-9的制备参考实施例1中化合物1b-1的制备。
[0072]
实施例10
[0073][0074]
以2,4-二氯苯氧乙酸(135.0mg,0.6mmol)替换实施例5中所用的2,3-二氯苯甲酸,其他参照实施例5,得到化合物1a-10(109.0mg,收率:58%)。化合物1b-10的制备参考实施例1中化合物1b-1的制备。
[0075]
实施例11
[0076][0077]
将化合物c(163.0mg,0.6mmol)置于100ml圆底烧瓶中,加入dmf(10.0ml)使其溶解,然后依次加入3,4,5-三甲氧基苯甲酸(化合物1)(100.0mg,0.5mmol),pybop(312.2mg,0.6mmol),再加入diea(1.0ml,2.0mmol),加料完毕后室温条件下搅拌2h,tlc检测(dcm:meoh=10:1),待反应完毕。加入水100.0ml进行淬灭,室温条件下搅拌1h,观察是否有固体析出,有固体析出,过滤,滤饼置于烘箱内烘干,用95%etoh重结晶后得到1a-11(138.0mg,收率:78%)。
[0078]
化合物1b-11的制备参考实施例1中化合物1b-1的制备。
[0079]
实施例12
[0080][0081]
2,3,4-三甲氧基苯甲酸(化合物4)(100.0mg,0.5mmol),1-(2-氯苯基)哌嗪(120.0mg,0.6mmol),dcc(124.0mg,0.6mmol),dmap(244.0mg,2.0mmol),方法同实施例1中1a-1,得到化合物1c-1(102.0mg,收率:48%)。
[0082]
化合物1c-1(100.0mg,0.3mmol),bbr3的dcm溶液(6.0ml,3.0mmol),方法同实施例1中化合物1b-1,得到化合物1d-1。
[0083]
实施例13
[0084][0085]
以1-(3-氯苯基)哌嗪(120.0mg,0.6mmol)代替实施例12中所用的1-(2-氯苯基)哌嗪,其他参照实施例12,得到化合物1c-2(110.0mg,收率:54%)。
[0086]
化合物1d-2的制备参考实施例12中化合物1d-1的制备。
[0087]
实施例14
[0088][0089]
以1-(2,3-二氯苯基)哌嗪(140.0mg,0.6mmol)代替实施例12中所用的1-(2-氯苯基)哌嗪,其他参照实施例12,得到化合物1c-3(109.0mg,收率:50%)。
[0090]
化合物1d-3的制备参考实施例12中化合物1d-1的制备。
[0091]
实施例15
[0092][0093]
2,3,4-三甲氧基苯甲酸(化合物4)(100.0mg,0.5mmol),4,4'-二氟二苯甲哌嗪(173.0mg,0.6mmol),pybop(312.2mg,0.6mmol),diea(1.0ml,2.0mmol),方法同实施例4中1a-4,得到化合物1c-4(117.0mg,收率:58%)。
[0094]
化合物1d-4的制备参考实施例12中化合物1d-1的制备。
[0095]
实施例16
[0096][0097][0098]
取2,3,4-三甲氧基苯甲酸(化合物4)(100.0mg,0.5mmol),n-boc-哌嗪(150.0mg,0.6mmol),pybop(312.2mg,0.6mmol),diea(1.0ml,2.0mmol),方法同实施例5中中间体b,得中间体e。
[0099]
称取中间体e(140.0mg,0.5mmol),2,3-二氯苯甲酸(115.0mg,0.6mmol),pybop(312.2mg,0.6mmol),diea(1.0ml,2.0mmol),方法同实施例4中1a-4,得到1c-5(135.0mg,收
率:79%)。
[0100]
化合物1d-5的制备参考实施例12中化合物1d-1的制备。
[0101]
实施例17
[0102][0103]
以3,4-二氯苯甲酸(115.0mg,0.6mmol)代替实施例16中所用的2,3-二氯苯甲酸,其他参照实施例16,得到化合物1c-6(124.0mg,收率:68%)。
[0104]
化合物1d-6的制备参考实施例12中化合物1d-1的制备。
[0105]
实施例18
[0106][0107]
以3,4,5-三氟苯甲酸(110.0mg,0.6mmol)代替实施例16中所用的2,3-二氯苯甲酸,其他参照实施例16,得到化合物1c-7(132.0mg,收率:70%)。
[0108]
化合物1d-7的制备参考实施例12中化合物1d-1的制备。
[0109]
实施例19
[0110][0111]
以反式肉桂酸(100.0mg,0.6mmol)代替实施例16中所用的2,3-二氯苯甲酸,其他参照实施例16,得到化合物1c-8(130.0mg,收率:78%)。
[0112]
化合物1d-8的制备参考实施例12中化合物1d-1的制备。
[0113]
实施例20
[0114][0115]
以苯氧乙酸(100.0mg,0.6mmol)代替实施例16中所用的2,3-二氯苯甲酸,其他参照实施例16,得到化合物1c-9(139.0mg,收率:75%)。
[0116]
化合物1d-9的制备参考实施例12中化合物1d-1的制备。
[0117]
实施例21
[0118][0119]
以2,4-二氯苯氧乙酸(135.0mg,0.6mmol)代替实施例16中所用的2,3-二氯苯甲酸,其他参照实施例16,得到化合物1c-10(125.0mg,收率:70%)。
[0120]
化合物1d-10的制备参考实施例12中化合物1d-1的制备。
[0121]
实施例22
[0122][0123]
称取中间体c(163.0mg,0.6mmol),2,3,4-三甲氧基苯甲酸(化合物4)(100.0mg,0.5mmol),pybop(312.2mg,0.6mmol),diea(1.0ml,2.0mmol),方法同化合物1a-4,得到1c-11(134.0mg,收率:70%)。
[0124]
化合物1d-11的制备参考实施例12中化合物1d-1的制备。
[0125]
实施例23
[0126][0127]
3,4-二甲氧基苯甲酸(化合物5)(100.0mg,0.5mmol),1-(2-氯苯基)哌嗪(120.0mg,0.6mmol),pybop(312.2mg,0.6mmol),diea(1.0ml,2.0mmol),方法同化合物1a-4,得到化合物2a-1(126.0mg,收率:71%)。
[0128]
化合物2a-1(100.0mg,0.3mmol),bbr3的dcm溶液(6.0ml,3.0mmol),方法同实施例1中化合物1b-1,得到2b-1。
[0129]
实施例24
[0130][0131]
以1-(3-氯苯基)哌嗪(120.0mg,0.6mmol)代替实施例23中所用的1-(2-氯苯基)哌嗪,其他参照实施例23,得到化合物2a-2(121.0mg,收率:60%)。
[0132]
化合物2b-2的制备参考实施例23中化合物2b-1的制备。
[0133]
实施例25
[0134][0135]
以1-(2,3-二氯苯基)哌嗪)(140.0mg,0.6mmol)代替实施例23中所用的1-(2-氯苯基)哌嗪,其他参照实施例23,得到化合物2a-3(135.0mg,收率:72%)。
[0136]
化合物2b-3的制备参考实施例23中化合物2b-1的制备。
[0137]
实施例26
[0138][0139]
以4,4'-二氟苯甲哌嗪(173.0mg,0.6mmol)代替实施例23中所用的1-(2-氯苯基)哌嗪,其他参照实施例23,得到化合物2a-4(125.0mg,收率:50%)。
[0140]
化合物2b-4的制备参考实施例23中化合物2b-1的制备。
[0141]
实施例27
[0142]
[0143][0144]
3,4-二甲氧基苯甲酸(化合物5)(100.0mg,0.5mmol),n-boc-哌嗪(150.0mg,0.6mmol),pybop(312.2mg,0.6mmol),diea(1.0ml,2.0mmol),方法同化合物1a-4,用95%etoh重结晶后得到中间体g。
[0145]
中间体g(125.0mg,0.5mmol),2,3-二氯苯甲酸(115.0mg,0.6mmol),pybop(312.2mg,0.6mmol),diea(1.0ml,2.0mmol),方法同化合物1a-4,得到2a-5(128.0mg,收率:65%)。
[0146]
化合物2b-5的制备参考实施例23中化合物2b-1的制备。
[0147]
实施例28
[0148][0149]
以3,4-二氯苯甲酸(115.0mg,0.6mmol)代替实施例27中所用的2,3-二氯苯甲酸,其他参照实施例27,得到化合物2a-6(112mg,收率:70%)。
[0150]
化合物2b-6的制备参考实施例23中化合物2b-1的制备。
[0151]
实施例29
[0152][0153]
以3,4,5-三氟苯甲酸(110.0mg,0.6mmol)代替实施例27中所用的2,3-二氯苯甲酸,其他参照实施例27,得到化合物2a-7(126.0mg,收率:66%)。
[0154]
化合物2b-7的制备参考实施例23中化合物2b-1的制备。
[0155]
实施例30
[0156][0157]
以反式肉桂酸(100.0mg,0.6mmol)代替实施例27中所用的2,3-二氯苯甲酸,其他参照实施例27,得到化合物2a-8(104.0mg,收率:50%)。
[0158]
化合物2b-8的制备参考实施例23中化合物2b-1的制备。
[0159]
实施例31
[0160][0161]
以苯氧乙酸(100.0mg,0.6mmol)代替实施例27中所用的2,3-二氯苯甲酸,其他参照实施例27,得到化合物2a-9(130.0mg,收率:66%)。
[0162]
化合物2b-9的制备参考实施例23中化合物2b-1的制备。
[0163]
实施例32
[0164][0165]
以2,4-二氯苯氧乙酸(135.0mg,0.6mmol)代替实施例27中所用的2,3-二氯苯甲酸,其他参照实施例27,得到化合物2a-10(116.0mg,收率:60%)。
[0166]
化合物2b-10的制备参考实施例23中化合物2b-1的制备。
[0167]
实施例33
[0168][0169]
2,3-二甲氧基苯甲酸(化合物6)(100.0mg,0.5mmol),1-(2-氯苯基)哌嗪(120.0mg,0.6mmol),pybop(312.2mg,0.6mmol),diea(1.0ml,2.0mmol),方法同实施例4中化合物1a-4,得到2c-1(116.0mg,收率:65%)。
[0170]
称取2c-1(100.0mg,0.3mmol),bbr3的dcm(6.0ml,3.0mmol)溶液,方法同实施例1中化合物1b-1,得到2d-1。
[0171]
实施例34
[0172][0173]
以1-(3-氯苯基)哌嗪(120.0mg,0.6mmol)代替实施例33中所用的1-(2-氯苯基)哌嗪,其他参照实施例33,得到化合物2c-2(113.0mg,收率:60%)。
[0174]
化合物2d-2的制备参考实施例33中化合物2d-1的制备。
[0175]
实施例35
[0176][0177]
以1-(2,3-二氯苯基)哌嗪(140.0mg,0.6mmol)代替实施例33中所用的1-(2-氯苯基)哌嗪,其他参照实施例33,得到化合物2c-3(136.0mg,收率:77%)。
[0178]
化合物2d-3的制备参考实施例33中化合物2d-1的制备。
[0179]
实施例36
[0180][0181]
以4,4'-二氟二苯甲哌嗪(173.0mg,0.6mmol)代替实施例33中所用的1-(2-氯苯基)哌嗪,其他参照实施例33,得到化合物2c-4(126.0mg,收率:50%)。
[0182]
化合物2d-4的制备参考实施例33中化合物2d-1的制备。
[0183]
实施例37
[0184][0185]
2,3-二甲氧基苯甲酸(化合物6)(100.0mg,0.5mmol),n-boc-哌嗪(150.0mg,0.6mmol),pybop(312.2mg,0.6mmol),diea(1.0ml,2.0mmol),方法同实施例4中化合物1a-4,得到中间体i。
[0186]
称取中间体i(150.0mg,0.5mmol),2,3-二氯苯甲酸(化合物5)(115.0mg,0.6mmol),pybop(312.2mg,0.6mmol),diea(1.0ml,2.0mmol),方法同实施例4中化合物1a-4,得到2c-5(129.0mg,收率:70%)。
[0187]
化合物2d-5的制备参考实施例33中化合物2d-1的制备。
[0188]
实施例38
[0189][0190]
以3,4,5-三氟苯甲酸(110.0mg,0.6mmol)代替实施例35中所用的2,3-二氯苯甲
酸,其他参照实施例35,得到化合物2c-7(114.0mg,收率:72%)。
[0191]
化合物2d-7的制备参考实施例33中化合物2d-1的制备。
[0192]
实施例39
[0193][0194]
以反式肉桂酸(100.0mg,0.6mmol)代替实施例35中所用的2,3-二氯苯甲酸,其他参照实施例35,得到化合物2c-8(126.0mg,收率:66%)。
[0195]
化合物2d-8的制备参考实施例33中化合物2d-1的制备。
[0196]
实施例40
[0197][0198]
以2,4-二氯苯氧乙酸(135.0mg,0.6mmol)代替实施例35中所用的2,3-二氯苯甲酸,其他参照实施例35,得到化合物2c-10(123.0mg,收率:60%)。
[0199]
化合物2d-10的制备参考实施例33中化合物2d-1的制备。
[0200]
实施例41
[0201]
化合物(4-(2,3-dichlorophenyl)piperazin-1-yl)(3-methoxyphenyl)methanone(3b-3)的合成方法
[0202][0203]
3-甲氧基苯甲酸(化合物9)(100.0mg,0.65mmol),1-(2,3-二氯苯基)哌嗪(139.0mg,0.8mmol),pybop(416.3mg,0.8mmol),diea(1.0ml,2.0mmol),方法同实施例4中化合物1a-4,得到3b-3(126mg,收率:79%)。
[0204]
实施例1~41制备的化合物的结构、nmr和ms数据如表1所示:
[0205]
表1
[0206]
[0207]
[0208]
[0209]
[0210]
[0211]
[0212]
[0213]
[0214]
[0215]
[0216][0217]
实施例42
[0218]
药理学实验材料与方法
[0219]
仪器与试剂:采用multiskan mk3型酶标检测仪(芬兰labsystems产品);sw-ct-if型超净化工作台(苏州安泰空气技术有限公司);倒置显微镜(amersham pharmacia产品);微量加样器(芬兰finnpette产品);96孔细胞培养板(丹麦nunclon产品);mjx型智能霉菌培养箱(宁波江南仪器厂);隔水式电热恒温培养箱(上海跃进医疗器械厂);thz-82a台式恒温震荡器(上海跃进医疗器械厂)。
[0220]
氟康唑(fluconazole,flc)注射液(规格:5ml:0.2g;美国辉瑞(pfizer)制药公司);二甲基亚砜(dmso,中国国药集团化学试剂有限公司);黄芩素(be,安耐吉试剂有限公司);没食子酸(ga,adamas公司)。
[0221]
受试菌株:耐药白念珠菌(c.albicans 901、c.albicans 904)获赠于长海医院菌群保存中心,并经过生物学、形态学鉴定。所有受试菌株经沙氏葡萄糖琼脂培养基(sda)划板活化,在30℃恒温培养2周后再次进行单克隆划板活化,选择二次活化后所得的单克隆沙氏葡萄糖琼脂培养基斜面,采用以上培养方法培养后,在4℃冰箱保存待用。
[0222]
培养液
[0223]
1.沙氏葡萄糖琼脂固体培养基(sda)
[0224]
沙氏葡萄糖琼脂培养基(sda)成分(g/l):葡萄糖(40.0g)、蛋白胨(10.0g)、琼脂(15.0g),调节培养基ph值至7.0
±
0.1,加热搅拌溶解于900.0ml无菌三蒸水中,定容至1.0l,121℃高压灭菌20min,冷却至30℃左右,转移至无菌培养皿,在4℃冰箱保存待用。
[0225]
2.rpmi 1640液体培养基
[0226]
rpmi-1640液体培养基(g/l):吗啡啉丙磺酸(17.25g)、rpmi 1640(5.0g)、nahco3(1.0g)加热搅拌溶解于450.0ml无菌三蒸水中,定容至500.0ml,经0.22μm微孔滤膜微滤分装于250.0ml三角瓶,在4℃冰箱保存待用。
[0227]
3.yepd培养液
[0228]
yepd培养液(g/l):蛋白胨(20.0g)、葡萄糖(20.0g)、酵母浸膏(10.0g)加热搅拌溶解于900.0ml无菌三蒸水中,定容至1.0l,121℃高压灭菌20min,冷却至30℃左右,转移至无菌培养皿,在4℃冰箱保存待用。
[0229]
实验方法
[0230]
菌株活化:用接种环沾取少量的-80℃冻存菌液接种于4℃冰箱保存待用的无菌sda培养基上,在30℃培养箱中培养48h,放回4℃冰箱保存待用。
[0231]
真菌悬液的制备:实验前,用接种环从4℃冰箱中保存待用的无菌sda培养基上挑取活化后的白念珠菌,接种至1.0ml无菌yepd液体培养液,在30℃培养箱中,经200rpm振荡活化培养16h,使真菌处于指数生长期后期。使用移液枪吸取该菌液至1.0ml无菌yepd培养液中,重复上述方法,再次活化培养16h,用血球计数板计算真菌数目,可用无菌rpmi-1640液体培养基进行稀释,调整菌液浓度在1
×
10
3-5
×
103cfu/ml的范围内即可。
[0232]
药敏反应板的制备:在无菌96孔板的每排1号孔加入无菌rpmi 1640液体培养基(100.0μl),作为空白对照;在2号孔依次加入受试化合物溶液(40.0μl)和菌液(160.0μl);3-12号孔加入菌液(100.0μl),其中12号孔不加药物,只加菌液(100.0μl)作为阳性对照。通过比例2-11号孔依次稀释,使其每个孔的最终药物浓度分别为64.0、32.0、16.0、8.0、4.0、2.0、1.0、0.5、0.25和0.125μg/ml,其中dmso的含量全部低于1.0%。氟康唑溶液的终浓度为8.0μg/ml,获得受试化合物与氟康唑(8.0μg/ml)合用时的mic值。同等条件下需要制备一批质控菌的药敏板,所用药敏板均在恒温箱30℃中培养24h。
[0233]
最低抑菌浓度(mic)的判定:用酶标分析仪在620nm波长下测定恒温条件培养的药敏板各孔的od值。其中阳性对照12号孔的od值约为0.2。再与其对照,其他各孔的od值减少80%以上时,所对应的最低浓度,即为mic值(80%真菌生长受到抑制的最小药物浓度)。如果药物的mic值不在测定范围时,按如下规则进行计数:当mic值低于最低浓度0.125μg/ml时,计为“≤0.125μg/ml”,当mic值超出最高浓度64.0μg/ml时,计为“>64.0μg/ml”。重复三次上述平行操作,被采纳的mic值能被准确重复或者两者间相差一个浓度单位时,以相对较高的浓度作为mic值;如果测试中mic值相差两个浓度单位以上时,就需要重新进行以上操作,直到符合要求为止。
[0234]
质控菌:本实验参考采用近平滑念珠菌atcc18062为质控菌,其mic值参考值:flc:0.25-1.0μg/ml;amb:0.5-2.0μg/ml。每次实验均以此类菌株作为参照,当实际测量mic值保持在上述范围内,则会确认实验操作准确,结果可靠。因此只有相同条件下实验菌株和对照菌株生长良好时,实验认定成功,结果可靠。
[0235]
联合用药的药效评价:本实验参考采用clsi的m27-a3和m38-a2标准测试规定的部分抑菌浓度指数(fici)对两个药物的联合作用进行评价。部分抑菌浓度指数(fractional inhibitory concentration index,ficii)作为联合用药的两药相互作用方式的主要参数,可用来解释抗真菌药物相互作用的方式。fici指数的计算公式:fici指数=mic(a组联用)/mic(a组单用)+mic(b组联用)/mic(b组单用)(当mic
80
值高于检测最高限度时以最高限浓度的两倍值用以计算fici)。目前国外期刊采用的最新标准:fici≤0.5时两药作用为协同作用;0.5<fici≤1时两药作用为相加作用;1<fici≤4时两药作用为无关作用;当fici>4时两药作用为拮抗作用。
[0236]
化合物抗真菌活性与构效关系:测试实施例1~41制备的目标化合物体外单独作用以及协同氟康唑抗耐药白念珠菌的活性,结果见表2所示。
[0237]
表2.目标化合物与氟康唑联用抗耐药白念珠菌活性测试结果
[0238]
[0239]
[0240][0241]
a
与氟康唑(flc,8.0μg/ml)联合作用于耐药白念珠菌901;
b
与氟康唑(flc,8.0μg/ml)联合作用于耐药白念珠菌904.
[0242]
共有25个目标化合物(1b-1、1d-1、2d-1、1b-2、1d-2、2d-2、1b-3、1d-3、2d-3、1b-4、1d-4、2b-4、1b-5、1d-5、1b-6、1d-6、1d-7、1b-8、1d-8、2d-8、1b-9、1d-9、1b-10、1d-10、2d-10、)显示出单独的抗耐药真菌活性(mic<128.0μg/ml),其中化合物(2d-2,2d-10)显示出最强的单用抗耐药真菌活性,mic=2.0μg/ml。共有37个目标化合物(1b-1、1d-1、2d-1、1b-2、1d-2、2d-2、1b-3、1d-3、2d-3、1b-4、1d-4、2b-4、1b-5、1d-5、1b-6、1d-6、1d-7、1b-8、1d-8、2d-8、1b-9、1d-9、1b-10、1d-10、2d-10、2b-1、2b-2、2b-3、2b-5、2d-5、2b-6、1b-7、2b-7、2d-7、2b-8、1b-11、1d-11)显示出与氟康唑协同抗耐药真菌活性,其中化合物(2d-5,2b-5,1b-10)显示出最强的协同氟康唑抗耐药活性,mic=0.5μg/ml,fici=0.066。此外,还有2个目标化合物(1b-10、1d-10)显示出加合活性。
[0243]
以上所述仅是本发明的较佳实施例而已,并非对本发明作任何形式上的限制,虽然本发明已以较佳实施例揭露如上,然而并非用以限定本发明,任何熟悉本专利的技术人员在不脱离本发明技术方案范围内,当可利用上述提示的技术内容作出些许更动或修饰为等同变化的等效实施例,但凡是未脱离本发明技术方案的内容,依据本发明的技术实质对以上实施例所作的任何简单修改、等同变化与修饰,均仍属于本发明方案的范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1