一种构建MSH2基因敲除细胞系的方法与流程
【技术领域】
本发明涉及基因工程技术领域,具体涉及一种构建msh2基因敲除细胞系的方法。
背景技术:
人类muts同源蛋白2(humanmutshomologue2,hmsh2)是dna错配修复蛋白家族的一个重要成员,通常定位于细胞核,与msh3或msh6形成异源二聚体,参与修复dna合成中的碱基错配。hmsh2异位表达多见于各种肿瘤细胞,且可以被ebv病毒感染、h2o2刺激所诱导。异位表达的hmsh2可作为γδt细胞受体(γδtcellreceptor,tcrγδ)的配体分子,被tcrγδ和nk细胞受体成员2d(naturalkillergroup2memberd,nkg2d)双重识别,介导γδt细胞对肿瘤细胞的杀伤作用。
在dna错配修复中,hmsh2蛋白质首先与hmsh6形成一种称为hmutsα二聚体复合物,并结合到dna碱基错配区,然后与hmlh1和hpsm2基因形成的二聚体hmutlα结合,其中异源二聚体hmutsα起到识别带有缺口的新生dna链的作用。hmutlα与hmutsα结合到dna错配区后,即可激活与muth蛋白有关的gatc核酸内切酶,这种酶可切出未修饰的甲基化gatc序列。依赖于muts、mutl及dna解旋酶(mutu)的协同作用,随着新生链错配区的切出,整个修复反应即被启动。
msh2蛋白几乎出现于所有结肠正常粘膜,阳性率达94%。阳性产物定位于核内,部分细胞同时出现胞浆染色。阳性细胞分布于粘膜全层,腺窝下1/3-2/3中排列更为密集。
杯状细胞中偶见msh2蛋白的表达。msh2蛋白也表达在间质纤维母细胞、血管内皮细胞以及淋巴细胞上,呈核型或核浆型,但浆细胞内表达较少。
msh2基因的突变与微卫星不稳定性和一些癌症有关。研究发现msh2基因蛋白阳性表达率从正常组织、腺瘤不典型增生、腺瘤癌到腺癌逐渐降低,提示在腺瘤阶段已经有mmr的基因的突变或功能异常,并随其恶性程度的增加,mmr基因的突变频率也逐渐升高。研究表明mmr基因的突变,尤其是msh2的突变可能是结直肠癌发生的早期事件之一。遗传性非息肉病性结直肠癌(hnpcc),有时称为lynch综合征,以常染色体显性遗传方式遗传,其中仅突变错配修复基因的一个拷贝的遗传就足以引起疾病表型,约占结直肠癌总数的15%~18%,占家族性结直肠癌的50%。msh2基因的突变占该疾病相关基因改变的40%,与hnpcc相关的突变广泛分布在msh2的所有结构域中,并且是mlh1突变的主要原因。另外msh2和其他错配修复基因的突变也会导致dna损伤未修复,导致突变频率增加。
已有学者已经证实错配修复蛋白msh2蛋白在脑、肺脏、结肠、心脏、肌肉、脾脏、胃和前列腺等大部分脏器组织中的表达水平随着年龄的增长而逐渐下降,说明其与细胞衰老有着密切关联。
crispr/cas9是一种基于细菌天然免疫机制开发的基因编辑技术。该系统是原核生物用来抵抗外源遗传物质入侵的免疫系统。经简化改造的crispr/cas9系统是继锌指核酸内切酶(zfn)和类转录激活因子效应物核酸酶(talen)之后出现的第三代基因组精确编辑技术。它由核酸内切酶cas9和grna(guiderna,grna)两部分组成,cas9能识别并结合基因组上的原间隔序列临近基序(protospaceradjacentmotif,pam)并形成dna双链解旋,如果此时grna的crrna部分能与pam上游序列成功互补配对,cas9将激活其核酸内切酶活性,在pam上游特定位置形成双链dna断裂(doublestrandbreak,dbs)。dbs可激活细胞的dna损伤修复机制,非同源性末端结合(non-homologousendjoining,nhej)或同源重组介导的修复(homologydirectedrepair,hdr)。nhej,即易错修复,会在修复位点引起随机插入或缺失而造成移码突变使得基因不再表达,由此形成基因敲除。hdr,即精准修复,能借助外源引入的单链或双链dna为模板介导基因替换或插入,这种方式可以将一段dna序列精准地插入特定的基因组位点,由此完成基因敲入或替换。
目前,尚未见有关利用crispr/cas9系统敲除msh2基因的报道,因此有必要基于crispr/cas9系统研究一种构建msh2基因敲除细胞系的方法。
技术实现要素:
本发明目的在于提供一种构建msh2基因敲除细胞系的方法,该方法可以高效、快速、方便地在细胞中敲除msh2基因,构建msh2基因敲除细胞系,可用于研究msh2基因的功能、相关通路和药物开发。
鉴于此,本发明为实现上述目的,提供一种敲除msh2基因的sgrna,包括如seqidno:1所示的sgrna1以及如seqidno:2所示的sgrna2。
本发明另一个目的在于,提供上述sgrna的重组质粒px459m-msh2-sgrnas。
本发明还有一个目的在于,提供上述sgrna的重组质粒px459m-msh2-sgrnas的构建方法,包括如下步骤:
1.基于所述sgrna1和所述sgrna2,合成带酶切位点的sgrnadna序列及其互补序列,分别退火处理获得msh2-sgrna1和msh2-sgrna2两个双链dna片段;
2.分别酶切回收px459m、ez-guidexh两个载体;
3.两个双链dna片段分别与酶切回收的px459m、ez-guidexh载体连接,并进行产物转化和测序鉴定,获得px459m-msh2-sgrna1和ez-guidexh-msh2-sgrna2;
4.对px459m-msh2-sgrna1和ez-guidexh-msh2-sgrna2进行酶切,前者回收含msh2-sgrna1的载体骨架,后者回收含msh2-sgrna2的dna片段,然后进行产物连接构建重组质粒px459m-msh2-sgrnas。
进一步地,所述步骤1中:所述sgrna1带酶切位点的sgrnadna序列为seqidno:3,互补序列为seqidno:4;所述sgrna2带酶切位点的sgrnadna序列为seqidno:5,互补序列为seqidno:6。
进一步地,所述步骤3中dna连接产物的转化和鉴定方法为:将酶切的载体骨架与双链dna片段的连接产物加入到感受态细菌中,补加培养基培养半小时后涂布于抗性平板,过夜培养后,挑取单菌落进行扩大培养,收获菌液抽提重组质粒,然后进行测序鉴定,保存鉴定正确的重组质粒px459m-msh2-sgrna1和ez-guidexh-msh2-sgrna2。
进一步地,所述步骤4为:对px459m-msh2-sgrna1回收含msh2-sgrna1的载体骨架,对ez-guidexh-msh2-sgrna2回收含msh2-sgrna2的dna片段,使用dna连接酶连接回收后的酶切产物;然后同步骤3进行连接产物的转化以及鉴定,将测序鉴定正确的重组质粒命名为px459m-msh2-sgrnas。
本发明的另一个目的在于提供上述的重组质粒px459m-msh2-sgrnas在制备msh2基因敲除的hela细胞系中的应用。
进一步地,所述的sgrna的重组质粒px459m-msh2-sgrnas构建msh2基因敲除hela细胞系的方法包括如下步骤:
1).用重组质粒px459m-msh2-sgrnas对hela细胞进行转染;
2).提取转染后的细胞基因组,针对msh2基因进行pcr扩增鉴定,检测sgrna的活性;
3).对转染后的细胞进行嘌呤霉素药物筛选处理,然后进行细胞单克隆化处理;
4).提取单克隆细胞株基因组dna,进行msh2基因测序鉴定,获得msh2基因敲除hela细胞系。
进一步地,所述步骤2)中sgrna的活性检测方法为:重组质粒px459m-msh2-sgrnas转染hela细胞后,采用鼠尾基因组dna检测试剂盒检测提取细胞的基因组dna作为模板,采用如seqidno:7和seqidno:8所示的引物进行pcr扩增,对扩增产物进行琼脂糖凝胶电泳检测,通过观察和比较对照组和实验组的dna片段大小,判定sgrna的活性。
11.进一步地,所述步骤3)中药筛处理方法为:在所述重组质粒px459m-msh2-sgrnas转染hela细胞后,添加含有嘌呤霉素的完全培养基培养细胞。
相比现有技术,本发明的有益效果为:
1.本发明成功构建msh2基因敲除hela细胞系,由于msh2基因片段缺失,导致基因失活,故不能正确表达msh2蛋白。
2.本发明利用crispr-cas9系统构建的msh2基因敲除hela细胞系,经基因测序鉴定和蛋白质免疫印迹验证,确定成功构建msh2基因敲除hela细胞系1株。
3.本发明构建的msh2基因敲除hela细胞系是在基因组水平上造成的基因大片段缺失,故可以随着细胞的分裂增殖稳定遗传到子代细胞。
4.本发明构建的px459m-msh2-sgrnas质粒具有嘌呤霉素抗性,添加嘌呤霉素筛选细胞,有利于提高获得msh2基因敲除单克隆细胞株的阳性率。
5.本方法可用于msh2基因的定向敲除,致使其功能失活,具有简便、高效、快速、低成本等特点,对于msh2基因功能和相关通路的研究具有重要意义。
【附图说明】
为了更清楚地说明本发明实施例的技术方案,下面将对实施例中所需要使用的附图作简单地介绍,应当理解,以下附图仅示出了本发明的某些实施例,因此不应被看作是对范围的限定,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他相关的附图。
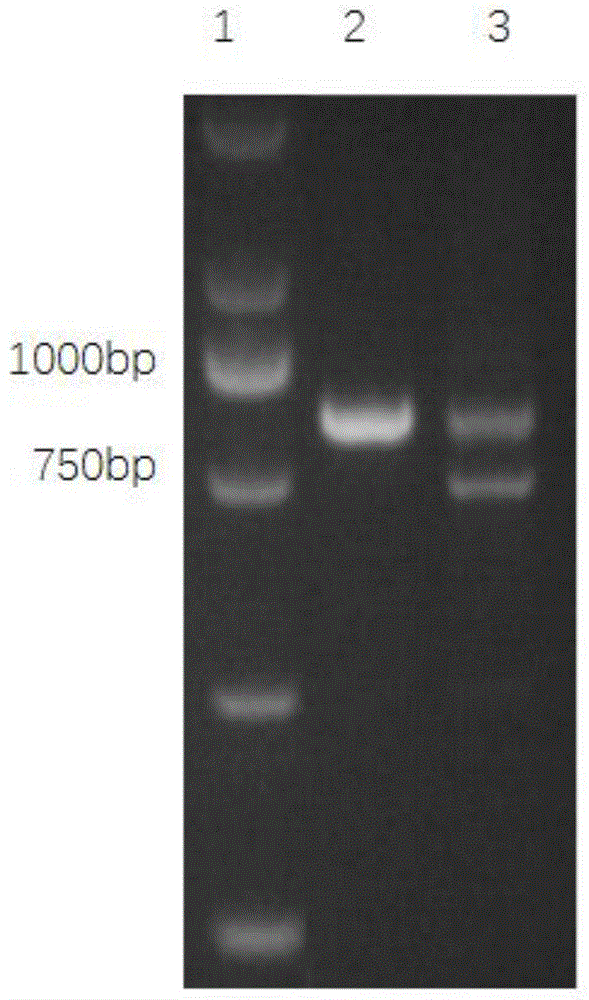
图1是本发明px459m-msh2-sgrnas质粒转染至hela细胞后sgrna活性检测的pcr鉴定结果。
图2是本发明px459m-msh2-sgrnas质粒转染至hela细胞后单克隆细胞株msh2基因的pcr鉴定结果。
图3是本发明px459m-msh2-sgrnas质粒转染至hela细胞后单克隆细胞株msh2基因的测序比对结果。
图4是本发明px459m-msh2-sgrnas质粒转染至hela细胞后单克隆细胞株msh2蛋白表达的western-blotting检测结果。
【具体实施方式】
以下实例用于说明本发明,但不限制本发明的范围。在不背离本发明精神和实质的前提下,对本发明的方法、步骤或条件所作的修改或替换,均属于本发明的范围。
实施例1:载体构建
1)msh2基因的sgrna设计
针对msh2基因(基因名:msh2,基因id号:4436,基因详细信息见https://www.ncbi.nlm.nih.gov/gene/?term=4436),在genebank网站检索并下载获得msh2部分基因组序列(seq-a):
aagctgattgggtgtggtcgccgtggccggacgccgctcgggggacgtgggaggggaggcgggaaacagcttagtgggtgtggggtcgcgcattttcttcaaccaggaggtgaggaggtttcgacatggcggtgcagccg;在mrna介绍中,链接到ensemble网站,找到其外显子序列并标记。
利用在线软件(http://crispor.tefor.net/)设计sgrna,输入上述外显子序列,设置并检索获得若干sgrna序列,通过分析sgrna在基因序列上的位置以及该sgrna的off-target(脱靶)信息,从中分别选择1个最优的上游sgrna序列,如seq-1所示;1个最优的下游sgrna序列,如seq-2所示,具体如表1所示:
表1:靶序列
2)合成sgrnadna片段并退火处理
上述设计优化的sgrna拟分别克隆至px459m和ez-guidexh载体中。将sgrnadna序列加上bbs1限制性内切酶的粘性末端,送奥科生物合成单核苷酸链。
在seq-1序列加上接头,合成得到插入片段msh2-sgrna1:
seq-1f:5’-caccgtcagcttccattggtgttg(seqidno:3)
seq-1r:5’-aaaccaacaccaatggaagctgac(seqidno:4);
在seq-2序列加上接头,得到插入片段msh2-sgrna2:
seq-2f:5’-caccggtatgtggattccatacag(seqidno:5)
seq-2r:5’-aaacctgtatggaatccacatacc(seqidno:6);
将每组seq-f和seq-r分别等体积混合进行退火反应,退火反应体系包括浓度为100μm的seq-f和seq-r所示引物各4.5μl、10×nebbuffer21μl,放置pcr仪中反应,反应程序为95℃反应5min,再以5℃/30s的速率,降温至25℃,即获得sgrna的双链dna片段,此时可与酶切载体连接,或-20℃保存备用。
3)载体酶切并回收
取px459m载体2μg、ez-guidexh载体4μg,分别用bbs1酶切,酶切体系为:px459m2μg、bbs11μl、10×bufferg2μl,加ddh2o补足体系到20μl,以及ez-guidexh4μg、bbs11μl、10×bufferg2μl,加ddh2o补足体系到20μl。分别混匀后于37℃水浴反应2-4小时。使用琼脂糖dna回收试剂盒对上述酶切产物进行回收纯化,具体实验步骤见琼脂糖dna回收试剂盒说明书。
4)两个msh2-sgrnadna片段分别与px459m、ez-guidexh载体的dna连接
分别在无菌的1.5mlep管中加入以下试剂:酶切后的px459m50ng、2×quickligationbuffer10μl、msh2-sgrna137.5ng、dnadna连接酶1μl,加ddh2o补足体系到20μl,以及ez-guidexh50ng、2×quickligationbuffer10μl、msh2-sgrna237.5ng、dna连接酶1μl,加ddh2o补足体系到20μl,混匀后于25℃水浴反应5min。
5)连接产物转化及菌落pcr鉴定
分别取上述连接产物5μl加入到50ul感受态细菌中,冰上放置30min;42℃水浴热激90s后冰上放置5min;加入1ml无抗生素培养基后于37℃摇床振荡培养半小时;将菌液涂布于抗性平板,放置37℃恒温培养箱倒置培养过夜;挑取单菌落进行扩大培养,收获菌液抽提重组质粒,然后进行测序鉴定,保存鉴定正确的重组质粒px459m-msh2-sgrna1和ez-guidexh-msh2-sgrna2。
6)构建重组质粒px459m-msh2-sgrnas
分别使用spe1和kpn1双酶切重组质粒px459m-msh2-sgrna1和ez-guidexh-msh2-sgrna2。酶切体系为:px459m-msh2-sgrna12μg、spe11μl、kpn11μl、10×buffertango2μl,加ddh2o补足体系到20μl,以及ez-guidexh-msh2-sgrna25μg、spe12μl、kpn12μl、10×buffertango2μl,加ddh2o补足体系到20μl,分别于37℃水浴反应2-4小时,跑1%琼脂糖凝胶,切胶回收酶切载体。对于px459m-msh2-sgrna1质粒,切胶回收含sgrna1双链dna片段的载体骨架;对于ez-guidexh-msh2-sgrna2质粒,切胶回收sgrna2双链dna片段。然后使用dna连接酶试剂盒连接胶回收后的酶切产物,连接反应体系包括酶切后的px459m-msh2-sgrna150ng、酶切后的ez-guidexh-msh2-sgrna237.5ng、2×quickligationbuffer10μl、dna连接酶1μl,加ddh2o补足体系到20μl,混匀后于25℃水浴反应5min。然后同步骤5)进行连接产物的转化以及鉴定,保存测序鉴定正确的重组质粒px459m-msh2-sgrnas。
实施例2.制备msh2基因敲除hela细胞系
1.px459m-msh2-sgrnas转染hela细胞
1)细胞接种12孔板,取hela细胞(来自武汉普诺赛生命科技有限公司,货号:cl-0101)悬液,按2.5×105/孔细胞数接种,均匀铺入12孔板的2个孔中,分别补加1ml完全培养基(含10%胎牛血清,1%双抗),放置细胞培养箱培养12-16小时;
2)制备转染复合物,取出2个无菌ep管,分别加入50μl无血清无抗生素dmem基础培养基,第1个无菌ep管中加入px459m-msh2-sgrnas重组质粒3μg(实验组),第2个无菌ep管中加入px459m-gfp质粒3μg(对照组),混合均匀,称为a液;另外取出1个无菌ep管,加入100μl无血清无抗生素dmem培养基,然后加入highgene转染试剂12μl,混合均匀,称为b液;
3)分别取出50μlb液轻轻滴加入a液中,总体积为100μl,混合均匀(用枪吹打3-5次即可),室温静置15-20min;
4)将a/b混合液轻轻滴加到相应的12孔板中,来回轻柔摇晃细胞培养板,然后将12孔板放回细胞培养箱继续培养。
5)细胞转染4-6小时后,半量更换新鲜完全培养基,继续培养24小时。
2.嘌呤霉素药物筛选
1)细胞转染24小时后,在荧光显微镜下观察对照组是否有绿色荧光,若有,则说明本次细胞转染是成功的;
2)实验组和对照组细胞分别加入含有终浓度1.6μg/ml嘌呤霉素药物的新鲜完全培养基;
3)嘌呤霉素筛选24h后,更换含有同上浓度嘌呤霉素药物新鲜完全培养基继续细胞培养;
4)嘌呤霉素筛选48h后,在显微镜下观察到对照组细胞大量死亡漂浮,实验组死亡漂浮40%-50%,停止嘌呤霉素药物筛选。
3.sgrna活性检测
1)使用胰酶消化对照组和实验组的细胞,收集30%-50%的细胞悬液;
2)使用鼠尾基因组检测试剂盒提取对照组和实验细胞的基因组;
3)以提取的细胞基因组dna作为模板,用下列引物进行pcr扩增:
seq-f:5’-tggtggtgtgcgcctgtatt(seqidno:7)
seq-r:5’-ttccccatgtctccagcagtc(seqidno:8);
pcr反应体系包括2×mix10μl、浓度为10μm的seq-f和seq-r所示引物各0.5μl、模板dna1μl及ddh2o8μl;pcr反应程序为95℃预变性反应5min;95℃变性反应10s,60℃退火反应10s,72℃延伸反应20s,30个循环;72℃再延伸5min;
4)对扩增产物进行琼脂糖凝胶电泳检测,通过观察和比较对照组和实验组扩增产物条带大小,判定sgrna的活性,如图1所示,泳道1为dl2000marker,泳道2为正常hela细胞(对照组),泳道3为已转染px459m-msh2-sgrnas质粒的hela细胞。
4.细胞单克隆化培养
1)弃去12孔板实验组的细胞培养基,pbs润洗细胞,加入0.5ml胰酶消化细胞,然后加入1ml完全培养基终止消化,重悬细胞;
2)细胞重悬均匀后,取出200μl细胞悬液到ep管中,使用血球计数板进行计数;
3)从细胞悬液中取出500个细胞,加入到20ml完全培养基中,同时补加1mlclone-easy生长因子溶液,混匀细胞;
4)将上述细胞悬液用排枪铺种到2个96孔培养板中,100μl/孔;
5)细胞培养10天后,在显微镜下观察细胞的生长情况,标记单克隆细胞集落。
5.单克隆细胞株基因水平验证
1)待单克隆细胞密度生长到孔底50%-60%面积时,使用胰酶消化细胞,取出50%细胞到ep管中,提取细胞基因组进行基因测序验证,剩余50%细胞继续在96孔板中培养扩增;
2)以提取的细胞基因组dna作为模板,用下列引物进行pcr扩增:
seq-f:5’-tggtggtgtgcgcctgtatt(seqidno:7)
seq-r:5’-ttccccatgtctccagcagtc(seqidno:8);
pcr反应体系包括2×mix10μl、浓度为10μm的seq-f和seq-r所示引物各0.5μl、模板dna1μl及ddh2o8μl;pcr反应程序为:95℃预变性反应5min;95℃变性反应10s,60℃退火反应10s,72℃延伸反应20s,30个循环;72℃再延伸5min;
4)对扩增产物进行琼脂糖凝胶电泳检测,通过与对照组扩增产物条带大小进行比较,可以判断单克隆细胞株是否发生打靶;如图2所示,泳道1为dl2000marker,泳道2为正常hela细胞(对照组),泳道3为msh2基因敲除的hela-msh2-ko单克隆细胞。
5)切胶回收有打靶的pcr扩增产物,送测序,通过测序比对确认阳性细胞后,将阳性细胞扩大培养,冻存保种,命名为hela-msh2-ko,测序结果比对图见图3。
6.westernblot检测单克隆细胞株msh2蛋白表达情况
为了验证msh2基因的敲除效果,我们采用westernblot实验检测了msh2基因敲除hela细胞系中msh2蛋白的表达情况。
1)细胞收集:将hela-msh2-ko单克隆细胞和正常hela细胞用胰酶消化后,收集到1.5ml离心管中,离心弃上清,保留细胞沉淀;
2)裂解细胞:加入含蛋白酶抑制剂的ripa裂解缓冲液,冰上裂解30分钟,12000r/min,离心15min,取上清转移至新的1.5ml离心管中,测定蛋白浓度;
3)制备细胞样本:将细胞总蛋白上清液与适量5×sds蛋白上样缓冲液混匀,使细胞总蛋白终浓度为2mg/ml,95℃煮沸5min;
4)电泳:配制10%的sds-page胶,进行蛋白的电泳分离,浓缩胶时,电压为80v,时间为30分钟,分离胶时,电压为120v,时间为1小时;
5)转膜:采用湿转法,恒流240ma,转膜2小时;
6)封闭:使用终浓度为5%脱脂奶粉的tbst溶液,室温将膜孵育2小时;
7)一抗孵育:将一抗msh2mab(abclonal:a8740)按1:1000稀释于终浓度为5%脱脂奶粉的tbst溶液中,4℃孵育过夜;
8)二抗孵育:孵育二抗之前,使用tbst将膜洗涤3次,每次10分钟;二抗peroxidaseaffinipuregoatanti-rabbitigg(h+l)(jackson:111-035-003)1:5000稀释于终浓度为5%脱脂奶粉的tbst溶液中,室温下孵育1小时;
9)曝光成像:使用tbst将膜洗涤3次,每次10分钟。取等量ecl显色液a液和b液混合均匀后,利用凝胶成像仪成像。
结果显示:hela-msh2-ko单克隆细胞中没有检测到msh2蛋白信号,表明该单克隆细胞在蛋白水平上实现了msh2基因敲除。具体见图4,泳道1为正常hela细胞,检测到msh2蛋白信号;泳道2为msh2基因敲除的hela-msh2-ko单克隆细胞,没有检测到msh2蛋白信号。
本发明利用cricpr/cas9系统构建的msh2基因敲除hela细胞系,在dna水平对msh2基因产生了编辑,完全沉默了msh2蛋白的表达,为msh2基因功能研究方面提供了一种实用工具。
序列表
<110>武汉爱博泰克生物科技有限公司
<120>一种构建msh2基因敲除细胞系的方法
<141>2020-11-25
<160>8
<170>siposequencelisting1.0
<210>1
<211>20
<212>dna
<213>artificialsequence
<400>1
gtcagcttccattggtgttg20
<210>2
<211>20
<212>dna
<213>artificialsequence
<400>2
ggtatgtggattccatacag20
<210>3
<211>24
<212>dna
<213>artificialsequence
<400>3
caccgtcagcttccattggtgttg24
<210>4
<211>24
<212>dna
<213>artificialsequence
<400>4
aaaccaacaccaatggaagctgac24
<210>5
<211>24
<212>dna
<213>artificialsequence
<400>5
caccggtatgtggattccatacag24
<210>6
<211>24
<212>dna
<213>artificialsequence
<400>6
aaacctgtatggaatccacatacc24
<210>7
<211>20
<212>dna
<213>artificialsequence
<400>7
tggtggtgtgcgcctgtatt20
<210>8
<211>21
<212>dna
<213>artificialsequence
<400>8
ttccccatgtctccagcagtc21
- 还没有人留言评论。精彩留言会获得点赞!