作为BCL-2抑制剂的N-(苯基磺酰基)苯甲酰胺及相关化合物的制作方法
作为bcl
‑
2抑制剂的n
‑
(苯基磺酰基)苯甲酰胺及相关化合物
背景技术:
1.细胞凋亡是程序性细胞死亡的过程,是组织稳态的重要生物学过程。在哺乳动物中,已证明它可以调节早期胚胎发育。在生命的后期,细胞死亡是一种默认机制,通过该机制可以清除潜在危险的细胞,例如携带癌性缺陷的细胞。几种凋亡途径是已知的。最重要的细胞凋亡途径之一涉及bcl
‑
2蛋白家族,它是细胞凋亡的线粒体(也称为“内在或固有”)途径的关键调节因子。同源结构域bh1,bh2,bh3和bh4是bcl
‑
2蛋白家族的特征。bcl
‑
2蛋白家族可以进一步分为三个亚家族,这取决于每种蛋白包含多少个同源结构域及其生物学活性,即它是否具有促凋亡或抗凋亡功能。
2.下调的细胞凋亡(更特别是bcl
‑
2蛋白家族)可能与癌性恶性肿瘤的发生有关。抗凋亡蛋白bcl
‑
2和bcl
‑
xl在许多癌细胞类型中过表达。这种失调的结果是病变细胞得以存活,否则这些细胞在正常条件下会发生凋亡。这些与失控增殖相关的缺陷的重复被认为是癌变的起点。另外,当在患病动物中表达时,仅bh3蛋白可以充当肿瘤抑制因子。
3.抗凋亡bcl
‑
2蛋白家族成员的天然表达水平在不同细胞类型中是不同的。例如,在新生的血小板中,bcl
‑
xl蛋白高表达并在调节血小板的细胞死亡(寿命)中起重要作用。而且,在特定癌细胞类型中,癌细胞的存活归因于由一种或多种抗凋亡bcl
‑
2蛋白家族成员的过表达引起的凋亡通路的失调。鉴于bcl
‑
2蛋白家族在调节癌细胞和正常细胞(即非癌细胞)凋亡中的重要作用,以及公认的bcl
‑
2蛋白家族表达的细胞间类型的变化性,对于具有选择性靶向并优选结合一种抗凋亡bcl
‑
2蛋白或其亚型的小分子抑制剂,例如,结合至在特定癌症类型中过表达的抗凋亡bcl
‑
2家族成员。这种选择性化合物还可以在临床中赋予一定优势,通过提供例如选择给药方案的灵活性,在正常细胞中降低的靶上毒性作用等,尤其在bcl
‑
2缺陷小鼠中观察到淋巴细胞减少。
4.选择性抑制一种bcl
‑
2蛋白或其亚型的化合物的需求正在持续增长,以治疗过度增生性疾病,例如癌症,包括血液系统恶性肿瘤。
技术实现要素:
5.本发明提供例如在有需要的患者中治疗血液系统恶性肿瘤的方法,其包括以逐步给药方案(step
‑
wise dosing regimen)向患者施用bcl
‑
2抑制剂。
6.例如,本文部分地描述了在有需要的患者中治疗血液系统恶性肿瘤的方法,该方法包括:以一个每日逐步给药方案给予化合物n
‑
((4
‑
(((1,4
‑
二恶烷
‑2‑
基)甲基)氨基)
‑3‑
硝基苯基)磺酰基)
‑2‑
((1h
‑
吡咯并[2,3
‑
b]吡啶
‑5‑
基)氧基)
‑4‑
(4
‑
((6
‑
(4
‑
氯苯基)螺[3.5]壬
‑6‑
烯
‑7‑
基)甲基)哌嗪
‑1‑
基)苯甲酰胺或其药学上可接受的盐;其中每日逐步给药方案的给药包括:向患者给药第一剂量20mg的化合物,持续一天;在第一剂量之后的第二天向患者施用第二剂量50mg化合物,持续一天;在第二剂量之后的第二天向患者施用第三剂量100mg的化合物,持续一天。
[0007]
在另一个实施方案中,本文描述了一种在有需要的患者中治疗血液系统恶性肿瘤的方法,该方法包括:以一个每日逐步给药方案给予化合物n
‑
((4
‑
(((1,4
‑
二恶烷
‑2‑
基)甲
基)氨基)
‑3‑
硝基苯基)磺酰基)
‑2‑
((1h
‑
吡咯并[2,3
‑
b]吡啶
‑5‑
基)氧基)
‑4‑
(4
‑
((6
‑
(4
‑
氯苯基)螺[3.5]壬
‑6‑
烯
‑7‑
基)甲基)哌嗪
‑1‑
基)苯甲酰胺或其药学上可接受的盐;其中给予每日逐步给药方案包括向患者给予第一剂量的20mg至100mg化合物,持续一天;在第一剂量之后的第二天向患者施用第二剂量50mg至200mg的化合物,持续一天。
[0008]
在另一个实施方案中,本文还描述了一种在有需要的患者中治疗血液系统恶性肿瘤的方法,其中还向所述患者施用cyp2c8抑制剂,另一种cyp抑制剂(例如cyp3a4抑制剂)或特定食物,例如柚子汁,包括给予有效量的化合物n
‑
((4
‑
(((1,4
‑
二恶烷
‑2‑
基)甲基)氨基)
‑3‑
硝基苯基)磺酰基)
‑2‑
((1h
‑
吡咯[2,3
‑
b]吡啶
‑5‑
基)氧基)
‑4‑
(4
‑
((6
‑
(4
‑
氯苯基)螺[3.5]壬
‑6‑
烯
‑7‑
基)甲基)哌嗪
‑1‑
基)苯甲酰胺或对患者而言是药学上可接受的盐,其中相较不给予cyp2c8抑制剂的患者的有效量,患者服用cyp2c8抑制剂的有效量为约60%或以下,约50%或以下,约40%或以下,或约20%或以下。
附图说明
[0009]
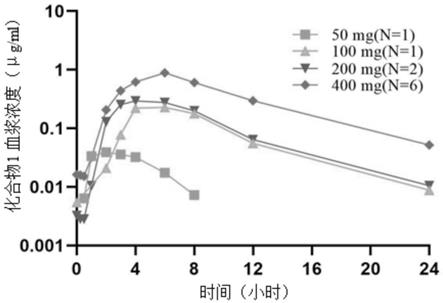
图1显示在人类患者中单次口服施用50mg,100mg,200mg和400mg化合物1后的示例性血浆浓度
‑
时间曲线。
[0010]
图2显示在人类患者中单次口服施用50mg,100mg,200mg,300mg和400mg化合物z后的示例性血浆浓度
‑
时间曲线。
[0011]
图3显示在患有华氏巨球蛋白血症的患者中单独或与其他治疗剂组合的化合物1的安全性,耐受性和有效性的ib/ii期研究的示例性给药方案。
[0012]
图4显示在患有复发和/或难治性慢性淋巴细胞性白血病(cll)/小淋巴细胞性淋巴瘤(sll)的患者中作为单一药物或与其他治疗药物组合的化合物1的ib/ii期研究的示例性给药方案。
[0013]
图5显示针对患有复发和/或难治性慢性淋巴细胞白血病(cll)或复发和/或难治性小淋巴细胞淋巴瘤(sll)的患者的作为单一药物或与依鲁替尼或利妥昔单抗组合的化合物1的示例性ib/ii期研究的研究设计。该研究包括(a)使用化合物1作为单一药物的剂量扩展研究,以评估化合物1单药治疗在患有复发性或难治性cll/sll的患者中的安全性和抗肿瘤活性以及药代动力学特征(图5a)和(b)对化合物1与依鲁替尼或利妥昔单抗联合进行的研究,以探讨在所研究的联合治疗方案中化合物1的剂量限制性毒性(dlt),最大耐受剂量(mtd),ii期推荐剂量(rp2d)和安全性,以及药代动力学研究和有效性评估(图5b)。
[0014]
图6显示化合物1与pd或drd组合用于治疗复发和/或难治性多发性骨髓瘤(rrmm)患者的示例性ib/ii期临床研究的研究设计。该研究包含一个治疗组,用于化合物1与pd组合的剂量递增和剂量扩展研究,以及另一个治疗组,用于化合物1与drd组合的剂量递增和剂量扩展研究。
[0015]
图7显示化合物1单药治疗或与rd组合用于治疗复发和/或难治性多发性骨髓瘤患者的示例性ib/ii期临床研究的研究设计。该研究包含一个治疗组,用于化合物1单药治疗的剂量递增和剂量扩展研究的;以及另一个治疗组,用于化合物1与rd组合的剂量递增和剂量扩展研究。
具体实施方式
[0016]
如本文所述,化合物n
‑
((4
‑
(((1,4
‑
二恶烷
‑2‑
基)甲基)氨基)
‑3‑
硝基苯基)磺酰基)
‑2‑
((1h
‑
吡咯并[2,3
‑
b]吡啶
‑5‑
基)氧基)
‑4‑
(4
‑
((6
‑
(4
‑
氯苯基)螺[3.5]壬
‑6‑
烯
‑7‑
基)甲基)哌嗪
‑1‑
基)苯甲酰胺具有以下结构:
[0017][0018]
如本文所述的“化合物1”是指(s)
‑
n
‑
((4
‑
(((1,4
‑
二恶烷
‑2‑
基)甲基)氨基)
‑3‑
硝基苯基)磺酰基)
‑2‑
((1h
‑
吡咯并[2,3
‑
b]吡啶
‑5‑
基)氧基)
‑4‑
(4
‑
((6
‑
(4
‑
氯苯基)螺[3.5]壬
‑6‑
烯
‑7‑
基)甲基)哌嗪
‑1‑
基)苯甲酰胺,具有以下结构:
[0019][0020]
如本文所述,化合物(r)
‑
n
‑
((4
‑
(((1,4
‑
二恶烷
‑2‑
基)甲基)氨基)
‑3‑
硝基苯基)磺酰基)
‑2‑
((1h
‑
吡咯并[2,3
‑
b]吡啶
‑5‑
基)氧基)
‑4‑
(4
‑
((6
‑
(4
‑
氯苯基)螺[3.5]壬
‑6‑
烯
‑7‑
基)甲基)哌嗪
‑1‑
基)苯甲酰胺具有以下结构:
[0021][0022]“化合物z”是指具有以下结构的化合物:
[0023][0024]
化合物z也称为venetoclax。
[0025]
用于本文所述方法的化合物,例如n
‑
((4
‑
(((1,4
‑
二恶烷
‑2‑
基)甲基)氨基)
‑3‑
硝基苯基)磺酰基)
‑2‑
((1h
‑
吡咯并[2,3
‑
b]吡啶
‑5‑
基)氧基)
‑4‑
(4
‑
((6
‑
(4
‑
氯苯基)螺[3.5]壬
‑6‑
烯
‑7‑
基)甲基)哌嗪
‑1‑
基)苯甲酰胺,(r)
‑
n
‑
((4
‑
(((1,4
‑
二恶烷
‑2‑
基)甲基)氨基)
‑3‑
硝基苯基)磺酰基)
‑2‑
((1h
‑
吡咯并[2,3
‑
b]吡啶
‑5‑
基)氧基)
‑4‑
(4
‑
((6
‑
(4
‑
氯苯基)螺[3.5]壬
‑6‑
烯
‑7‑
基)甲基)哌嗪
‑1‑
基)苯甲酰胺和(s)
‑
n
‑
((4
‑
(((1,4
‑
二恶烷
‑2‑
基)甲基)氨基)
‑3‑
硝基苯基)磺酰基)
‑2‑
((1h
‑
吡咯并[2,3
‑
b]吡啶
‑5‑
基)氧基)
‑4‑
(4
‑
((6
‑
(4
‑
氯苯基)螺[3.5]壬
‑6‑
烯
‑7‑
基)甲基)哌嗪
‑1‑
基)苯甲酰胺。基于化合物1在患者中表现出的抗增殖,抗肿瘤和凋亡诱导活性,预期比其他已知的bcl
‑
2抑制剂(例如venetoclax(化合物z))具有更好的疗效。此外,化合物1的药代动力学特性优于化合物z,例如较短的半衰期和较高的cmax系统性暴露与肿瘤组织中相似的半衰期和更高的cmax。这种优选的药代动力学性质可以导致较低的毒性而不影响化合物的治疗功效。
[0026]
本文所述的方法可以降低在血液恶性肿瘤治疗期间患者发生肿瘤溶解综合征(tls)的风险。tls是一种患者在服用bcl
‑
2抑制剂期间会遇到的常见但危险的副作用。为了降低tls的风险,在给予bcl
‑
2抑制剂venetoclax的患者中,应采用每周递增的剂量方案达到建议的治疗剂量。然而,这种每周递增的剂量方案可能导致低的药物暴露并且对bcl
‑
2抑制剂的功效产生负面影响。本文所述的方法包括治疗上更有效的每日增加剂量方案,该方案允许在达到给药化合物标准治疗剂量的情况下缩短所需时间,同时降低tls的风险。
[0027]
使用方法
[0028]
在一个实施方案中,本文描述了一种在有需要的患者中治疗血液系统恶性肿瘤的方法,其包括:以每日逐步给药方案施用化合物n
‑
((4
‑
(((1,4
‑
二恶烷
‑2‑
基)甲基)氨基)
‑3‑
硝基苯基)磺酰基)
‑2‑
((1h
‑
吡咯并[2,3
‑
b]吡啶
‑5‑
基)氧基)
‑4‑
(4
‑
((6
‑
(4
‑
氯苯基)螺[3.5]壬
‑6‑
烯
‑7‑
基)甲基)哌嗪
‑1‑
基)苯甲酰胺或其药学上可接受的盐;其中每日逐步给药方案的给药包括:向患者给药第一剂量20mg的化合物,持续一天;在第一剂量之后的第二天向患者施用第二剂量50mg化合物,持续一天;在第二剂量之后的第二天向患者施用第三剂量100mg的化合物,持续一天。在一些实施方案中,该方法进一步包括每天向患者施用第四剂量的200mg化合物,持续1
‑
5天或更长时间。在一些实施方案中,向患者施用第四剂量,持续一天。在一些实施方案中,每日逐步给药方案进一步包括在施用第四剂量之后向患者施用400mg的第五剂量。在一些实施方案中,该方法进一步包括在第三或第四剂量之后每天向患者施用400mg至800mg剂量的化合物。在一些实施方案中,该方法进一步包括在每日逐步给药方案给药后,向患者施用400mg,600mg或800mg化合物的日剂量,持续1周或更长时间,1个月或更长时间。在一些实施方案中,该方法进一步包括在每日逐步给药方案给药后向患者施用400mg,600mg,800mg或1000mg化合物的每日剂量,持续1周或更长时间,或1个月或更长时间。在一些实施方案中,该化合物为(r)
‑
n
‑
((4
‑
(((1,4
‑
二恶烷
‑2‑
基)甲基)氨基)
‑3‑
硝基苯基)磺酰基)
‑2‑
((1h
‑
吡咯[2,3
‑
b]吡啶
‑5‑
基)氧基)
‑4‑
(4
‑
((6
‑
(4
‑
氯苯基)螺[3.5]壬
‑6‑
烯
‑7‑
基)甲基)哌嗪
‑1‑
基)苯甲酰胺。在一些实施方案中,该化合物是(s)
‑
n
‑
((4
‑
(((1,4
‑
二恶烷
‑2‑
基)甲基)氨基)
‑3‑
硝基苯基)磺酰基)
‑2‑
((1h
‑
吡咯并[2,3
‑
b]吡啶
‑5‑
基)氧基)
‑4‑
(4
‑
((6
‑
(4
‑
氯苯基)螺[3.5]壬
‑6‑
烯
‑7‑
基)甲基)哌嗪
‑1‑
基)苯甲酰胺。
[0029]
本文还考虑了在有需要的患者中治疗血液系统恶性肿瘤的方法,该方法包括:每天以每日逐步给药方案施用化合物n
‑
((4
‑
(((1,4
‑
二恶烷
‑2‑
基)甲基)氨基)
‑3‑
硝基苯基)磺酰基)
‑2‑
((1h
‑
吡咯并[2,3
‑
b]吡啶
‑5‑
基)氧基)
‑4‑
(4
‑
((6
‑
(4
‑
氯苯基)螺[3.5]壬
‑6‑
烯
‑7‑
基)甲基)哌嗪
‑1‑
基)苯甲酰胺或其药学上可接受的盐;其中每日逐步给药方案包括向患者给予第一剂量的20mg至100mg化合物,持续一天。在第一剂量之后的第二天向患者施用第二剂量50mg至200mg的化合物,持续一天。在一些实施方案中,每日逐步给药方案包括向患者施用第一剂量的100mg化合物,持续一天。在一些实施方案中,每日逐步给药方案还包括在第二剂量之后的第二天向患者施用100至400mg化合物的第三剂量。在一些实施方案中,每日逐步给药方案还包括在第三剂量之后一到七天向患者施用第四剂量200mg至800mg的化合物。在一些实施方案中,该方法进一步包括在每日逐步给药方案之后,每日向患者施用约400mg至800mg的化合物,持续1周或更长时间。在一些实施方案中,该方法进一步包括在每日逐步给药方案之后,每日向患者施用约400mg至1000mg的化合物,持续1周或更长时间。在一些实施方案中,患者在每日逐步给药方案的给药期间或之后,具有降低的肿瘤溶解
综合征风险。在一些实施方案中,血液恶性肿瘤选自由慢性淋巴细胞性白血病,急性髓细胞性白血病,多发性骨髓瘤,淋巴浆细胞性淋巴瘤,非霍奇金淋巴瘤和小淋巴细胞性白血病组成的组。在一些实施方案中,血液恶性肿瘤选自由慢性淋巴细胞性白血病,急性髓性白血病,多发性骨髓瘤,淋巴浆细胞性淋巴瘤,非霍奇金淋巴瘤和原发性轻链型淀粉样变组成的组。在一些实施方案中,血液恶性肿瘤选自慢性淋巴细胞性白血病和小淋巴细胞性白血病。在一些实施方案中,血液系统恶性肿瘤是华氏巨球蛋白血症。在一些实施方案中,患者患有原发性难治性急性髓细胞性白血病。在一些实施方案中,患者患有难治性小淋巴细胞白血病或难治性慢性淋巴细胞性白血病。在一些实施方案中,患者在先前的血液恶性肿瘤治疗后已经复发。在一些实施方案中,先前的血液系统恶性疗法是用于慢性淋巴细胞性白血病的疗法。在一些实施方案中,先前的血液系统恶性疗法是小淋巴细胞白血病的疗法。在一些实施方案中,先前的血液系统恶性疗法是急性骨髓性白血病的疗法。在一些实施方案中,患者已经接受了至少一种血液恶性肿瘤的先前治疗,例如,患者先前已经接受过依鲁替尼,奥比妥单抗(obinutuzumab),苯达莫司汀,泼尼松,氟达拉滨,环磷酰胺,喷司他丁或其两种或更多种的组合;和/或患者先前曾接受过放射治疗或手术治疗。在一些实施方案中,该方法进一步包括在每日逐步给药方案之前,之中或之后将利妥昔单抗施用于患者。在一些实施方案中,该方法还包括在每日逐步给药方案之后向患者施用利妥昔单抗。在一些实施方案中,在每日逐步给药方案后约1周或更长时间将利妥昔单抗施用于患者。在一些实施方案中,该方法进一步包括在每日逐步给药方案之前,之中或之后,向患者施用阿扎胞苷,地西他滨或低剂量阿糖胞苷。在一些实施方案中,该方法进一步包括在每日逐步给药方案之前,之中或之后向患者施用依鲁替尼,拓扑替康,mdm2抑制剂,cd20抑制剂,cdk9i抑制剂或高三尖杉酯碱抑制剂。在一些实施方案中,该方法进一步包括在每日逐步给药方案之后向患者施用依鲁替尼,拓扑替康,mdm2抑制剂,cd20抑制剂,cdk9i抑制剂或高三尖杉酯碱抑制剂。在一些实施方案中,该方法进一步包括在每日逐步给药方案之前,之中或之后向患者施用btk抑制剂。在一些实施方案中,该方法进一步包括在每日逐步给药方案之后向患者施用btk抑制剂。在一些实施方案中,在每日逐步给药方案后约1周或更长时间将btk抑制剂施用于患者。在一些实施方案中,btk抑制剂是依鲁替尼。在一些实施方案中,该方法进一步包括在每日逐步给药方案之前,之中或之后向患者施用阿扎胞苷。在一些实施方案中,该方法进一步包括在每日逐步给药方案之后向患者施用阿扎胞苷。在一些实施方案中,该方法进一步包括在每日逐步给药方案之前,之中或之后,向患者施用高三尖杉酯碱。在一些实施方案中,该方法进一步包括在每日逐步给药方案之后向患者施用高三尖杉酯碱。在一些实施方案中,该化合物为(r)
‑
n
‑
((4
‑
(((1,4
‑
二恶烷
‑2‑
基)甲基)氨基)
‑3‑
硝基苯基)磺酰基)
‑2‑
((1h
‑
吡咯并[2,3
‑
b]吡啶
‑5‑
基)氧基)
‑4‑
(4
‑
((6
‑
(4
‑
氯苯基)螺[3.5]壬
‑6‑
烯
‑7‑
基)甲基)哌嗪
‑1‑
基)苯甲酰胺。在一些实施方案中,该化合物是(s)
‑
n
‑
((4
‑
(((1,4
‑
二恶烷
‑2‑
基)甲基)氨基)
‑3‑
硝基苯基)磺酰基)
‑2‑
((1h
‑
吡咯并[2,3
‑
b]吡啶
‑5‑
基)氧基)
‑4‑
(4
‑
((6
‑
(4
‑
氯苯基)螺[3.5]壬
‑6‑
烯
‑7‑
基)甲基)哌嗪
‑1‑
基)苯甲酰胺。
[0030]
在另一个实施方案中,本文提供了在有需要的患者中治疗多发性骨髓瘤和/或原发性轻链型淀粉样变的方法,所述方法包括:向所述患者施用200mg或更多的化合物n
‑
((4
‑
(((1,4
‑
二恶烷
‑2‑
基)甲基)氨基)
‑3‑
硝基苯基)磺酰基)
‑2‑
((1h
‑
吡咯并[2,3
‑
b]吡啶
‑5‑
基)氧基)
‑4‑
(4
‑
((6
‑
(4
‑
氯苯基)螺[3.5]壬
‑6‑
烯
‑7‑
基)甲基)哌嗪
‑1‑
基)苯甲酰胺或其药
学上可接受的盐。在一些实施方案中,该方法进一步包括向患者施用泊马度胺和地塞米松。在一些实施方案中,该方法还包括向患者施用来那度胺和地塞米松。在一些实施方案中,该方法进一步包括将达雷木单抗(daratumumab),来那度胺和地塞米松施用于患者。在一些实施方案中,该方法包括向患者施用200mg,400mg,600mg或800mg的化合物。在一些实施方案中,患者患有难治性多发性骨髓瘤。在一些实施方案中,患者在先前的多发性骨髓瘤治疗后已经复发。在一些实施方案中,该化合物为(r)
‑
n
‑
((4
‑
(((1,4
‑
二恶烷
‑2‑
基)甲基)氨基)
‑3‑
硝基苯基)磺酰基)
‑2‑
((1h
‑
吡咯并[2,3
‑
b]吡啶
‑5‑
基)氧基)
‑4‑
(4
‑
((6
‑
(4
‑
氯苯基)螺[3.5]壬
‑6‑
烯
‑7‑
基)甲基)哌嗪
‑1‑
基)苯甲酰胺。在一些实施方案中,该化合物是(s)
‑
n
‑
((4
‑
(((1,4
‑
二恶烷
‑2‑
基)甲基)氨基)
‑3‑
硝基苯基)磺酰基)
‑2‑
((1h
‑
吡咯并[2,3
‑
b]吡啶
‑5‑
基)氧基)
‑4‑
(4
‑
((6
‑
(4
‑
氯苯基)螺[3.5]壬
‑6‑
烯
‑7‑
基)甲基)哌嗪
‑1‑
基)苯甲酰胺。
[0031]
在另一个实施方案中,本文描述了在有需要的患者中治疗血液系统恶性肿瘤的方法,其中所述患者还被给予cyp2c8抑制剂(和/或可能引起药物/药物相互作用的另一种药物,例如cyp3a4抑制剂或食品(例如柚子汁),包括给予有效量的化合物n
‑
((4
‑
(((1,4
‑
二恶烷
‑2‑
基)甲基)氨基)
‑3‑
硝基苯基)磺酰基)
‑2‑
((1h
‑
吡咯并[2,3
‑
b]吡啶
‑5‑
基)氧基)
‑4‑
(4
‑
((6
‑
(4
‑
氯苯基)螺[3.5]壬
‑6‑
烯
‑7‑
基)甲基)哌嗪
‑1‑
基)苯甲酰胺或其药学上可接受的盐,其中较未给予cyp2c8抑制剂(或在某些实施方案中,其他药物或食品,例如柚子汁或cyp3a4抑制剂)的患者的有效量,服用cyp2c8抑制剂患者的有效量为约60%或更少,约50%或更少,约40%或更少,20%或更少。在一些实施方案中,服用cyp2c8抑制剂的患者的有效量为每天约20mg至约100mg化合物。在一些实施方案中,服用cyp2c8抑制剂的患者的有效量为每天约10mg,20mg,30mg,40mg,50mg,100mg或200mg。在一些实施方案中,该方法包括在通过最初的每日或每周的逐步给药方案施用化合物之后,对服用cyp2c8抑制剂的患者施用有效量。在一些实施方案中,cyp2c8抑制剂是强cyp2c8抑制剂。在一些实施方案中,cyp2c8抑制剂选自由吉非贝齐,甲氧苄氨嘧啶,噻唑烷二酮,孟鲁司特和槲皮素。在一些实施方案中,该化合物为(r)
‑
n
‑
((4
‑
(((1,4
‑
二恶烷
‑2‑
基)甲基)氨基)
‑3‑
硝基苯基)磺酰基)
‑2‑
((1h
‑
吡咯并[2,3
‑
b]吡啶
‑5‑
基)氧基)
‑4‑
(4
‑
((6
‑
(4
‑
氯苯基)螺[3.5]壬
‑6‑
烯
‑7‑
基)甲基)哌嗪
‑1‑
基)苯甲酰胺。在一些实施方案中,该化合物是(s)
‑
n
‑
((4
‑
(((1,4
‑
二恶烷
‑2‑
基)甲基)氨基)
‑3‑
硝基苯基)磺酰基)
‑2‑
((1h
‑
吡咯并[2,3
‑
b]吡啶
‑5‑
基)氧基)
‑4‑
(4
‑
((6
‑
(4
‑
氯苯基)螺[3.5]壬
‑6‑
烯
‑7‑
基)甲基)哌嗪
‑1‑
基)苯甲酰胺。
[0032]
定义
[0033]“有效量”包括将引起研究者,兽医,医生或其他临床医生所寻求的引起组织,系统,动物或人的生物学或医学反应的主题化合物的量。以有效量施用本文所述的化合物,例如化合物1,以治疗病症,例如血液系统恶性肿瘤。或者,化合物的有效量是达到期望的治疗和/或预防作用所需的量,例如导致预防或减轻与该病症有关的症状的量。
[0034]“个体”,“患者”或“受试者”在本文中可互换使用,并且包括任何动物,包括哺乳动物,包括小鼠,大鼠,其他啮齿动物,兔,狗,猫,猪,牛,绵羊,马或灵长类动物和人类。本文所述的化合物可以施用于哺乳动物,例如人,但是也可以施用于其他哺乳动物,例如需要兽医治疗的动物,例如家畜(例如狗,猫等),农场动物(例如,牛,绵羊,猪,马等)和实验动物(例如,大鼠,小鼠,豚鼠等)。用本文所述方法治疗的哺乳动物理想地是需要治疗本文所述病症的哺乳动物,例如人。
[0035]
本文所用的术语“药学上可接受的盐”是指组合物中所使用的化合物的酸性或碱性基团的盐。本发明组合物中包括的化合物本质上是碱性的,其能够与各种无机和有机酸形成各种盐。可用于制备此类碱性化合物的药学上可接受的酸加成盐的酸是形成无毒酸加成盐的酸,即含有药学上可接受的阴离子的盐,包括但不限于苹果酸盐,草酸盐,氯离子,溴离子,碘化物,硝酸盐,硫酸盐,硫酸氢盐,磷酸盐,磷酸,异烟酸盐,乙酸盐,乳酸盐,水杨酸盐,柠檬酸盐,酒石酸盐,油酸盐,单宁酸盐,泛酸盐,酒石酸氢盐,抗坏血酸盐,琥珀酸盐,马来酸盐,龙胆酸盐,富马酸盐,葡萄糖酸盐,葡糖醛酸盐,蔗糖酸盐甲酸盐,苯甲酸盐,谷氨酸盐,甲磺酸盐,乙磺酸盐,苯磺酸盐,对甲苯磺酸盐和棕榈酸盐(即1,1'
‑
亚甲基
‑
双
‑
(2
‑
羟基
‑3‑
萘甲酸))盐。
[0036]
如本文所用,“治疗”包括导致状况,疾病,病症等的改善的任何作用,例如减轻,减少,调节或消除。
[0037]
本文描述的化合物,例如化合物1或其药学上可接受的盐,可以使用药学上可接受的载体配制为药物组合物,并通过多种途径给药。在一些实施方案中,此类组合物用于口服施用。在一些实施方案中,此类组合物用于肠胃外(通过注射)施用。在一些实施方案中,此类组合物用于透皮施用。在一些实施方案中,此类组合物用于静脉内(iv)施用。在一些实施方案中,此类组合物用于肌内(im)施用。这样的药物组合物及其制备方法是本领域众所周知的。参见,例如,remington:the science and practice of pharmacy(a.gennaro,et al.,eds.,19
th ed.,mack publishing co.,1995)。
[0038]
实施例
[0039]
本文所述的化合物可以基于本文所包含的教导和本领域已知的合成方法以多种方式制备。应当理解,除非另有说明,否则可以将所有建议的反应条件,包括溶剂的选择,反应气氛,反应温度,实验的持续时间和后处理程序选择为该反应的条件标准。有机合成领域的技术人员应理解,存在于分子各个部分上的官能团应与所提出的试剂和建议的反应相容。与反应条件不相容的取代基对于本领域技术人员将是显而易见的,因此指示了替代方法。实施例的起始原料可以商购获得,也可以通过标准方法容易地由已知材料制备。
[0040]
n
‑
((4
‑
(((1,4
‑
二恶烷
‑2‑
基)甲基)氨基)
‑3‑
硝基苯基)磺酰基)
‑2‑
((1h
‑
吡咯并[2,3
‑
b]吡啶
‑5‑
基)氧基)
‑4‑
(4
‑
((6
‑
(4
‑
氯苯基)螺[3.5]壬
‑6‑
烯
‑7‑
基)甲基)哌嗪
‑1‑
基)苯甲酰胺,(r)
‑
n
‑
((4
‑
(((1,4
‑
二恶烷
‑2‑
基)甲基)氨基)
‑3‑
硝基苯基)磺酰基)
‑2‑
((1h
‑
吡咯并[2,3
‑
b]吡啶
‑5‑
基)氧基)
‑4‑
(4
‑
((6
‑
(4
‑
氯苯基)螺[3.5]壬
‑6‑
烯
‑7‑
基)甲基)哌嗪
‑1‑
基)苯甲酰胺和化合物1可以根据wo 2018/027097中描述的合成方法合成,其通过引用并入本文。
[0041]
缩写:pk:药代动力学;mtd:最大耐受剂量;dlt:剂量限制性毒性;rp2d:推荐的第2阶段剂量;btki:btk抑制剂。
[0042]
实施例1.人类患者血液恶性肿瘤的临床研究。
[0043]
在治疗血液恶性肿瘤的患者的1期研究的剂量递增阶段中评估化合物1。在这项研究中,以递增的时间表进行3+3剂量递增队列给药,包括每日增加剂量的化合物1或安慰剂,直至达到建议的每日剂量。剂量将从20毫克开始。每天的后续剂量增量为50mg,100mg,200mg,400mg,600mg和800mg(基于该队列的推荐每日剂量)。
[0044]
从该给药方案开始,剂量递增直到确定最大耐受剂量(mtd)。在最多20名患者的剂
量扩展队列中,mtd是推荐的每日剂量,其中应用如上所述的每日递增时间表,直到达到mtd。
[0045]
实施例2.化合物1和化合物z在患者中的临床药理作用。
[0046]
在患者中评估了化合物1和化合物z的药代动力学。例如,图1和图2中分别显示了患者单次口服50mg,100mg,200mg,300mg(仅化合物z)和400mg后的化合物1和化合物z的血浆浓度
‑
时间曲线。
[0047]
此外,表1显示了在每日一次400mg的多次口服给药后,化合物1和化合物z在稳态下的药代动力学参数的比较。
[0048]
表1.每天一次400mg多次口服给药后,化合物1和化合物z稳态药代动力学的比较。
[0049][0050]
实施例3.化合物1和化合物z的代谢研究比较。
[0051]
在小鼠和狗肝微粒体测定中都评价了化合物1和化合物z的代谢研究。从这些研究中,发现化合物1主要被cyp3a4和cyp2c8代谢。在测定中未发现化合物1抑制或诱导任何主要的cyp酶。在小鼠肝微粒体试验中,小鼠血浆中的主要代谢物(>10%)是酰胺键水解m2,还有少量的氧化代谢物。在犬肝微粒体中评估的犬血浆中发现了少量的氧化代谢产物。
[0052]
相反,发现化合物z主要被cyp3a4和cyp3a5代谢。对于化合物z,人血浆中的主要代谢物(>10%)是氧化代谢物m27。在评估的小鼠和狗血浆中均发现了其他少量的氧化代谢产物。
[0053]
实施例4.化合物1单独的或与其他治疗剂组合在华氏巨球蛋白血症患者中的安全性,耐受性和功效的研究。
[0054]
进行了评价化合物1作为单一药剂或与其他治疗药剂例如依鲁替尼或利妥昔单抗组合的安全性,耐受性,有效性和pk的临床研究。该研究包括剂量递增和剂量扩展阶段。该临床试验具有多组,能够根据华氏巨球蛋白血症(wm)中化合物1的临床活性,随后添加更多的治疗组。最初,研究包含3个如下所述的小组,所有组都是独立的。
[0055]
组a:将化合物1作为单一药剂施用,以确定在对依鲁替尼或其他btk抑制剂复发/耐药或不耐受的wm患者中的mtd/rp2d。确定mtd/rp2d后,在剂量扩展阶段以rp2d水平再招募多达12名患者,以进一步评估化合物1的安全性和有效性。
[0056]
组b:在先前未经治疗的wm患者中,化合物1与依鲁替尼联合给药。化合物1与依鲁替尼联用的mtd/rp2d将在组b的第1周期中建立。确定mtd/rp2d之后,在剂量扩展阶段将以该剂量水平招募多达15名患者。
[0057]
组c:在患有已复发/耐药的疾病且以前从未用依鲁替尼或其他btki治疗的wm患者
中,化合物1与利妥昔单抗联合给药。在组c第1周期中建立了化合物1与利妥昔单抗联合的mtd/rp2d。确定mtd/rp2d之后,在剂量扩展阶段以该剂量水平再招募15名患者。每个周期为4周(28天长)。
[0058]
作为单药治疗或与依鲁替尼或利妥昔单抗组合的化合物1的剂量递增阶段将遵循mtpi
‑
2剂量递增设计:
[0059]
以28天的周期连续28天每日一次口服给予化合物1,直到疾病进展或出现不可接受的毒性。起始目标剂量(如果需要,可使用递增剂量)为400mg(剂量水平1;dl1),并相应增加至600mg(dl2)和800mg(dl3)。根据研究者和申办者的讨论,可以根据安全性和pk结果将剂量提高到更高水平。从第1个周期的第1天开始,每日以420mg口服依鲁替尼,此后连续给药。每个周期为4周(28天)。利妥昔单抗每周每包静脉注射375mg/m2,连续四个星期,然后在三个月的间隔后(第1
‑
4周的第1天和第17
‑
20周的第1天,共8次注射利妥昔单抗)进行第二个四周的利妥昔单抗疗程。利妥昔单抗的输注从第1个周期的第1天开始。图3中提供了代表研究的给药方案。
[0060]
评估所有患者的肿瘤溶解综合征(tls)的特殊风险,并在化合物1的第一剂量前至少72小时开始接受预防(包括水合作用,抗高尿酸药,严密的实验室监测和住院治疗,如果有的话)。对于具有高tls风险的患者,无论达到最终剂量水平之前分配的剂量水平如何,都可以实施剂量递增方案,对于低/中风险tls患者,他们可以从最终分配的剂量开始。下表2中列出了每日3天固定的递增时间表。
[0061]
表2.化合物1的每日递增方案
[0062][0063][0064]
*第1周期,第1天(c1d1)是分配400mg剂量的第1天。
[0065]
**第1周期,第1天(c1d1)是分配600mg剂量的第1天。
[0066]
***第1周期,第1天(c1d1)是分配800mg剂量的第1天。
[0067]
考虑到dlt的可评估性,受试者必须在dlt评估期(周期1)中接受至少70%的化合物1或其组合的计划剂量。如果患者在评估期间因dlt以外的其他原因接受的总剂量少于计划剂量的70%,则无法评估dlt。如果受试者经历了dlt(在dlt评估期内),则必须中断化合物1单独或联合使用的治疗,并且视dlt的严重程度而定,患者可能会中止研究。但是,在dlt解析为1级或基线值之后,研究人员可以酌情以降低的剂量水平继续接受患者的研究治疗。
[0068]
实施例5.在复发和/或难治性慢性淋巴细胞性白血病(cll)/小淋巴细胞性淋巴瘤(sll)患者中,化合物1作为单一药物或与其他治疗药物组合的临床研究。
[0069]
进行了评估化合物1作为单一药剂或组合疗法的安全性,耐受性,有效性和pk的开放性研究。该研究包括每日逐步期(daily ramp
‑
up period),剂量递增和剂量扩展部分。每个患者在进入治疗周期之前都要经历每日递增期。每日递增期的持续时间将取决于分配的
剂量水平。每日的递增包括每天给予一次化合物1的治疗,从第1天20mg开始,第2天50mg,第3天100mg,第4天200mg,第5天400mg,第6天600mg,第7天800mg,第8天1000mg。用于评估的预定最大队列剂量始于400mg化合物1至最大1000mg化合物1。例如,预定剂量为400mg的患者将有4天的递增期,预定剂量为600mg,有5天的递增期,以此类推,请参见图1。第1部分评估了化合物1单药治疗的四个剂量水平(400mg,600mg,800mg和1,000mg);第2部分是化合物1与利妥昔单抗的组合,在任何安全剂量水平(包括400mg,600mg和800mg的化合物1),剂量逐渐增加和扩大(参见图4)。在这项研究中,最大耐受剂量(mtd)定义为在第一个28天周期中接受治疗的6名患者不会引起不可接受的毒性的剂量,通过零或一名患者出现剂量限制性毒性(dlt)证明,其中下一个更高的剂量显示出不可接受的毒性(≥2/3或≥2/6dlt)。
[0070]
第1部分:化合物1作为单一药物每天口服(qd)一次,以确定复发或难治性cll患者的mtd/rp2d。患者将在28天的周期内接受治疗。首先经历第1至4天(第1天,20mg;第2天,50mg;第3天,100mg;第4天,200mg)的每日递增期;然后在第1周期第1天,化合物1以400mg,600mg,800mg和1000mg平行使用。每个剂量队列的前3名患者均需进行pk分析。在前3名患者后,每剂量队列均被视为无安全问题,可对额外最多15名患者进行治疗,以进一步表征安全性和有效性。化合物1在cll单药治疗中的最佳剂量取决于经研究者和申办者之间讨论后的总体安全性,pk和初步疗效数据。
[0071]
第2部分:化合物1与利妥昔单抗联合给药。患者在28天的周期内接受治疗。化合物1从第1天到第4天(第1天,20mg;第2天,50mg;第3天,100mg和第4天,200mg)开始增加剂量,然后是第1周期第1天,化合物1以三个剂量水平:400mg,600mg和800mg。如上所述,对于每个较高剂量,递增期相应地增加。服用化合物1直至疾病进展,不可接受的毒性,开始替代疗法或撤回同意。组合从第1个周期的第8天开始如下:利妥昔单抗在第1个周期的第8天以375mg/m2 iv输注和在第2
‑
6个周期的第1天以500mg/m2 iv输注给药。
[0072]
评估所有患者的肿瘤溶解综合症(tls)的特定风险,并且所有患者将在第一化合物1剂量之前接受预防(包括水合作用,抗高尿酸药,密切的实验室监测和住院治疗(如果有需要的话))。表3中详细列出了化合物1的每日递增时间表。
[0073]
表3.化合物1剂量递增方案
[0074][0075]
剂量队列:
a
400mg,第1周期,第1天;
b
600mg,第1周期,第1天;
c
800mg,第1周期,第1天;
d
1000mg,第1周期,第1天.
[0076]
实施例6.化合物1作为单一药物或与化学疗法联合用于治疗复发和/或难治性急性髓细胞白血病(r/r aml)患者的临床研究。
[0077]
这是化合物1单剂量递增研究,以评估化合物1单药的dlt,mtd或rp2d,并评估化合物1的体内药代动力学和抗aml功效。根据标准3+3方案增加化合物1剂量。化合物1的初始剂量队列接受200mg。然后在三个剂量递增队列中分别以400mg,600mg和800mg的剂量水平增加剂量。化合物1每天给药一次(qd),每28天为一个给药周期。
[0078]
该研究遵循包括以下内容的每日递增剂量方案。200mg剂量组的每日剂量如下:100mg至200mg。400mg剂量组应按以下每日剂量给药:100mg至200mg至400mg。600mg剂量组应按以下每日剂量给药:100mg至200mg至400mg至600mg。800mg剂量组应按以下每日剂量给药:100mg至200mg至400mg至600mg至800mg。
[0079]
实施例7.在复发和/或难治性慢性淋巴细胞白血病(cll)或复发和/或难治性小淋巴细胞性淋巴瘤(sll)患者中化合物1作为单一药物或与利妥昔单抗或依鲁替尼联用的安全性,pk,pd和有效性的ib/ii期研究
[0080]
这是一个开放的多中心ib/ii期研究,研究化合物1作为单一药物或与利妥昔单抗或伊布鲁替尼联合治疗复发和/或难治性cll和/或sll患者的安全性、pk、pd和疗效。除化合物1和利妥昔单抗,或伊布替尼外,受试者不能接受其他抗恶性肿瘤研究药物、市售药物或治疗方法。对症支持治疗是可以接受的,包括控制由肿瘤引起的症状。研究的示意图如图5所示。
[0081]
本研究由两部分组成。本研究的第一部分是化合物1单药扩展队列研究,目的是评价化合物1单药治疗复发或难治性cll和/或sll患者的安全性、抗肿瘤活性和药代动力学特征。队列扩展在三个剂量水平下进行,即400mg、600mg和800mg,每个剂量水平计划有12
‑
15名患者加入。
[0082]
在研究的第一部分中,为了将受试者患肿瘤溶解综合征(tls)的风险降至最低,所有受试者在接受目标剂量水平的治疗之前,按照每日递增计划接受化合物1。400mgmg剂量水平队列中的受试者以400mgmg为目标剂量,连续每天施用20mg、50mg、100mg、200mg和400mg的化合物1。600mg剂量水平队列中的受试者以600mg为目标剂量,连续每天施用20mg、50mg、100mg、200mg、400mg和600mg的化合物1。800mg剂量水平队列中的受试者以800mg为目标剂量,连续每天施用20mg、50mg、100mg、200mg、400mg、600mg和800mg的化合物1。
[0083]
图5a提供了第一部分研究示意图。将收集每个队列中前三名受试者的样本,以进行药代动力学(pk)数据分析。在前3名患者入选400mg队列后,对这3名患者在第一个周期中的安全性进行评估。如果评估结果表明该药物是安全的,则入选400mg队列中的9
‑
12名患者和600mg队列中的前3名患者。如果前三名服用600mg的患者完成了第一个周期的评估,明该药物是安全的,则随后的9
‑
12名患者被纳入600mg队列中,并且800mg队列中的前三名患者同时被纳入。在对三名800mg的患者进行pk和安全性评估后,最后9
‑
12名患者被纳入800mg队列。如果由于任何原因无法获得3名受试者的完整pk样本,则应将其替换为该队列中的后续受试者,直到收集到3名受试者的完整pk样本为止。
[0084]
连续口服化合物1,每日1次,28天为一个周期。当所有受试者完成6个疗程或停止治疗后(以先发生者为准),对安全性和有效性进行初步分析。第二部分研究是在第一部分小组招募结束后进行的。
[0085]
第二部分研究包含两组:化合物1与利妥昔单抗结合(组a),化合物1与伊布替尼结合(组b)(图5b)。目的是探讨和评价化合物1在所研究的联合治疗方案中的dlt、mtd、rp2d和安全性,药代动力学研究和疗效。化合物1有4种剂量水平:200mg、400mg、600mg和800mg。这两个组包括两个阶段的剂量递增(第一阶段)和剂量扩展(第二阶段)。两组受试者在28天周期内接受治疗。化合物1以口服片剂形式提供,每片含有10mg或50mg化合物1。
[0086]
为了将研究组中第二部分受试者患肿瘤溶解综合征(tls)的风险降至最低,所有受试者在接受目标剂量水平的治疗之前,按照每日递增计划接受化合物1。200mg剂量水平队列中的受试者以200mg为目标剂量,连续每天服用20mg、50mg、100mg和200mg化合物1。400mg剂量水平队列中的受试者以400mg为目标剂量,连续每天服用20mg、50mg、100mg、200mg和400mg化合物1。600mg剂量水平队列中的受试者以600mg为目标剂量,连续每天服用20mg、50mg、100mg、200mg、400mg和600mg化合物1。800mg剂量水平队列中的受试者以800mg为目标剂量,连续每天服用20mg、50mg、100mg、200mg、400mg、600mg和800mg化合物1。
[0087]
第二部分的第一阶段是化合物1联合利妥昔单抗或伊布替尼的剂量递增研究。化合物1的剂量递增遵循标准的3+3方案,初始剂量为200mg,随后剂量递增,共4个剂量水平(200mg、400mg、600mg和800mg)。在达到每日递增方案后的目标剂量水平后,化合物1在28天周期内连续口服,每天一次(图5b)。
[0088]
在利妥昔单抗组(a组)的第一个周期中,利妥昔单抗或伊布替尼的使用从第1周期第8天开始。利妥昔单抗在第1个周期、第8天以375mg/m2的速度静脉注射,在第2
‑
6个周期的第1天以500mg/m2的速度静脉注射,每名受试者总共注射6次。
[0089]
在伊布替尼组,从第1周期的第8天开始,以420mg的剂量连续口服伊布替尼,并在此后连续服用。联合用药的给药期间,在每个单独治疗组施用,口服化合物1后30
‑
90分钟内利妥昔单抗给药,化合物1和伊布替尼同时施用。
[0090]
第二部分研究的第二阶段是rp2d扩展阶段。一旦确定了a组和b组各自的rp2d值,每个组进入研究第二阶段的扩展队列(图5b)。在每个rp2d剂量水平下,多达15名受试者接受化合物1联合利妥昔单抗或伊布替尼治疗,以进一步评估该剂量水平下联合用药方案的安全性和有效性。a组和b组允许受试者自身剂量递增。在研究者和申办方增加病患内剂量之前,受试者需要在最初分配的剂量水平上完成至少3个给药周期,并确定较高剂量水平的安全性。受试者体内剂量递增,以当前剂量为梯度递增的起始剂量,每日递增达到目标剂量,完成逐步递增,达到目标剂量日为用药周期的第1天。从给药化合物1的前一天起,患者住院至达到目标剂量后至少48小时,每天接受静脉补液,同时密切监测并及时处理任何提示肿瘤溶解的代谢变化。
[0091]
实施例8.化合物1与pd或drd组合用于治疗复发和/或难治性多发性骨髓瘤(rrmm)患者的ib/ii期临床研究。
[0092]
这是一项ib/ii期、开放、多中心研究,研究并评估了化合物1与pd(与泊马利度胺和地塞米松联合使用)或drd(与达雷木单抗、来那度胺和地塞米松联合用药)对复发/难治性多发性骨髓瘤(mm)患者的安全性、耐受性、疗效和pk/pd。研究分为剂量递增和剂量扩增两个阶段。这项研究从以下两条组开始,两组是独立的。研究设计的方案如图6所示,并在下文中进一步描述。
[0093]
在本方案中,a组研究治疗定义为泊马度胺(p)、地塞米松(d)和化合物1,以及组b中的达雷木单抗(d)、来那度胺(r)、地塞米松(d)和化合物1。研究药物为化合物1。停止pd或drd治疗的受试者可在研究过程中继续服用化合物1。
[0094]
所有患者在第一次服用化合物1前至少72小时,接受肿瘤溶解综合征(tls)预防治疗(包括补水、抗高尿酸血症、严密的实验室监测和住院治疗,如有需要)。如果在a组或b组的剂量递增阶段,出现临床或实验室设置中的任何tls,可修改方案,以便在随后入组的患者中实施化合物1的每日递增剂量方案。
[0095]
a组:化合物1与pd联合给药,以测定疾患rrmm受试者的mtd/rp2d。患者每天接受一次目标剂量水平的化合物1,并在第1天到第21天与泊马度胺(4mg)联合施用,在第1、8、15和22天使用地塞米松(40mg,对于75岁以上的患者为20mg),重复28天周期。患者可以继续接受治疗,直到疾病进展、无法接受的毒性、同意撤回或继续进行干细胞移植,以先到者为准。可根据毒性/年龄进行剂量调整。这是一种全口服疗法。
[0096]
化合物1与pd的剂量递增阶段采用3+3剂量递增设计。化合物1的起始目标剂量为每天一次400mg(剂量水平1,或dl1),并在随后的队列中相应地增加到每天一次600mg(剂量水平2,或dl2)和每天一次800mg(剂量水平3,或dl3)。如果不能耐受化合物1,400mg的剂量,则可以将每天一次的剂量减少到200mg(剂量水平
‑
1,或dl
‑
1)。这项基于规则的设计首先在dl1对三名患者进行队列研究。如果在dl1入组的三个病人中没有一个经历过dlt,另外三个病人将在dl2接受治疗,依此类推。然而,如果一名患者出现dlt,则将有三名患者接受相同剂量的治疗。剂量持续增加,直到三到六名患者中至少有两名患者出现dlt。mtd通常被定义为最高剂量水平,在该水平下,≤33%的患者经历dlt。在对安全性数据进行综合分析以确定800mg的剂量是否可以耐受后,考虑超过800mg的更高剂量水平。根据化合物1的有效性和安全性来确定组a的rp2d。
[0097]
在确定了a组的mtd和rp2d后,在剂量扩展阶段,在rp2d(组a)增加了12名额外患
者,以进一步评估化合物1联合pd的安全性和有效性。
[0098]
b组:化合物1与drd联合给药,以测定组b中rrmm受试者的mtd和rp2d。在组a600mg剂量水平安全确定之前,不得开展组b研究。然后根据研究者的判断同时招募两组的病人。
[0099]
从第1个周期第1天开始,患者以目标剂量每天接受一次化合物1,为期28天。对于骨干疗法,患者在每28天周期的第1天到第21天每天口服一次来那度胺(po),地塞米松每周给药40mg(或75岁以上的患者20mg)。在第1和第2周期中,每周静脉注射达雷木单抗16mg/kg(或皮下注射1800mg,如有市售),然后在第3至第6周期中每2周注射一次,此后每4周注射一次。患者可以继续接受治疗,直到出现疾病进展、无法接受的毒性、同意撤回或继续进行干细胞移植为止,以先到者为准。可根据毒性/年龄进行剂量调整。
[0100]
化合物1与drd联合使用遵循3+3剂量递增设计。起始目标剂量为每天一次400mg(剂量水平1,或dl1),并在随后的队列中增加到每天一次600mg(剂量水平2,或dl2)和每天一次800mg(剂量水平3,或dl3)。如果化合物1不能耐受400mg的剂量,则可以将剂量减少到200mg(剂量水平
‑
1,或dl
‑
1)。这项基于规则的设计首先在dl1对三名患者进行队列研究。如果在dl1入组的三个病人中没有一个经历过dlt,另外三个病人将在dl2接受治疗,依此类推。然而,如果三个病人中有一个经历了dlt,另外三个病人将接受相同剂量的治疗。剂量持续增加,直到三到六名患者中至少有两名患者出现dlt。mtd通常被定义为最高剂量水平,在该水平下,≤33%的患者经历dlt。超过800mg的更高剂量水平,只有在对安全数据进行综合分析,以确定800mg剂量是否可以耐受后才会考虑。根据化合物1的有效性和安全性来确定组b的rp2d。
[0101]
在确定了b组mtd和rp2d后,将有12名额外患者加入组b剂量扩展阶段rp2d,以进一步评估化合物1与drd联合使用的安全性和有效性。
[0102]
在a组或b组中,在第1个周期内不能进行dlt评估的患者,和/或在定义的dlt评估治疗周期内因dlt以外的任何原因退出的患者,和/或在一个周期内未接受超过70%的计划剂量(化合物1和/或联合研究药物)被认为是不可评估的,应予以替换。根据研究者和ascentage医疗监护者之间的讨论,一旦dlts的症状缓解到小于或等于1级,稳定或有反应的患者可以继续接受治疗。
[0103]
化合物1与新型治疗药剂联合在复发或难,治性多发性骨髓瘤和原发性轻链型淀粉样变患者中的ib/ii期开放标签研究
[0104]
组c:化合物1将与pd联合施用,以确定在患有原发性轻链型淀粉样变的受试者中化合物1的mtd和rp2d。
[0105]
患者将在第1天至第21天每天施用一次化合物1,共28天,再施用泊马度胺(4mg)和地塞米松(≤75岁的患者为40mg,>75岁的患者为20mg),在第1、8天,15和22,每28天为一个周期。患者可能会继续接受治疗,直到疾病进展,不可接受的毒性,撤回同意或继续进行干细胞移植(以先到者为准)为止。可以根据毒性/年龄进行剂量调整,这是一种全口服治疗方案。
[0106]
实施例9.在患有复发和/或难治性多发性骨髓瘤的受试者中,化合物1单一疗法或与来那度胺/地塞米松组合的ib/ii期开放性研究。
[0107]
这是一项ib/ii期、开放、多中心研究,以评估化合物1单药疗法或与rd(来那度胺(r)和地塞米松(d)联合用药)治疗复发性和/或难治性(r/r)多发性骨髓瘤(mm)患者的安全
性、耐受性、疗效以及药代动力学/药效学。本研究的主要目的是评估中国r/r mm患者单用化合物1或联合rd的安全性和耐受性,确定剂量限制性毒性(dlt)、最大耐受剂量(mtd)和推荐剂量(rp2d)。
[0108]
本研究由两组构成,包括化合物1单剂(组a)和化合物1与rd(组b)联合的两组。两组都包括剂量递增和剂量扩展阶段。在a组的剂量递增阶段完成后启动b组。b组化合物1的起始剂量和最大递增剂量,可根据a组化合物1的mtd/rp2d进行调整。所有受试者均接受28天周期的连续治疗。研究总结如下以及图7所示。
[0109]
在组a中,化合物1每天给药一次,周期为28天。在组b中,化合物1每天给药一次,周期为28天。患者在每28天周期中,第1天到第21天口服来那度胺,剂量为25mg(或肌酐清除率(crcl)为30
‑
60毫升/分钟的患者为10mg)。地塞米松每周(第1、8、15和22天)给药一次,剂量为40mg(75岁以上的患者为20mg)。
[0110]
采用3+3设计剂量递增阶段,包括a组化合物1单药治疗,以及b组化合物1与rd联合用药。化合物1的起始目标剂量为400mg(剂量水平1,或dl1),随后相应地递增至600mg(剂量水平2,或dl2)和800mg(剂量水平3,或dl3)。这个基于规则的设计首先从三个病人的队列开始。如果在dl1入组的3名患者中没有一人经历dlt,则其他3名患者将在dl2接受治疗,依此类推。然而,如果1名患者出现dlt,则会有3名患者接受相同剂量的治疗。剂量持续增加,直到至少2名3至6名患者的同一队列中出现dlt。mtd通常被定义为最高剂量水平,在该水平下≤33%的患者经历dlt。根据化合物1单药治疗,或与rd联合用药的总体疗效和安全性来确定rp2d。确定rp2d后,6
‑
9名a组患者和12名b组患者将在剂量扩展阶段rp2d中入组,以进一步评估化合物1单药疗法或与rd联合用药的安全性和有效性。
[0111]
为了将肿瘤溶解综合征(tls)风险降至最低,所有处于剂量递增阶段的患者在第一次服用化合物1之前和之后接受为期3天的监测(tls化学和完整的血细胞检测)。如果在剂量递增过程中未发现tls(临床或实验室),除非研究人员要求,否则剂量扩展的患者不需要在第2天和第3天再进行tls监测。
[0112]
所有受试者持续接受治疗,直到疾病进展、不可接受的毒性或达到方案规定的其他停止治疗标准。所有受试者在停止治疗后完成生存随访,直至研究结束、撤回知情同意书、失去随访或死亡为止。
[0113]
实施例10.在复发和/或难治性急性髓细胞性白血病(r/r aml)患者中,化合物1单药治疗以及与高三尖杉酯碱(hht)或阿扎胞苷(aza)组合的ib期研究。
[0114]
这是一项ib期开放性剂量递增研究,即化合物1单药疗法和化合物1联合hht或aza,治疗复发和难治性急性髓系白血病(aml)的研究。除化合物1、hht和aza外,受试者不能接受其他抗恶性肿瘤研究药物、市售药物或治疗方法。患者可接受对症支持治疗,包括控制由受试者恶性肿瘤引起的症状。本研究可分为三个阶段。
[0115]
第一阶段
‑
化合物1单药治疗剂量递增
[0116]
第一阶段为化合物1单药治疗剂量递增研究,其目的是探索化合物1单药治疗的dlt、mtd和/或rp2d,并评估化合物1的药代动力学和抗aml疗效。根据标准的3+3设计,化合物1的剂量在剂量水平队列中递增。初始剂量为200mg,依次递增至400mg、600mg和800mg。化合物1以研究的目标剂量水平每日口服一次(qd),所有受试者在28天周期内,接受目标剂量水平的连续治疗。
[0117]
为了将第一阶段出现临床或实验室肿瘤溶解综合征(tls)的风险降至最低,所有受试者均接受每日递增方案,每个剂量水平如下所示。200mg剂量水平队列中的受试者,连续每天服用100mg和200mg的化合物1,目标剂量为200mg。400mg剂量水平队列中的受试者以400mg为目标剂量,连续每天服用100mg、200mg和400mg的化合物1。600mg剂量水平队列中的受试者以600mg为目标剂量,连续每天服用100mg、200mg、400mg和600mg的化合物1。800mg剂量水平队列中的受试者以800mg为目标剂量,连续每天服用100mg、200mg、400mg、600mg和800mg的化合物1。见表4。
[0118]
表4.第一阶段剂量递增设计。
[0119][0120]
第二阶段
‑
联合用药方案中化合物1的剂量递增
[0121]
第二阶段是化合物1与hht或aza联合用药剂量递增研究。第二阶段目的是测定化合物1与hht或aza联合治疗的dlt、mtd或rp2d,并评估化合物1在这些联合用药方案中的药代动力学和抗aml疗效。联合用药研究中,化合物1剂量递增遵循标准的3+3设计,研究的剂量水平为第一阶段的mtd/rp2d
‑
1(例如,比第一阶段的mtd/rp2d低一个剂量水平)和第一阶段的mtd/rp2d水平,研究的三个联合用药方案共有两个剂量水平。化合物1以每种剂量水平口服,每天一次(qd),周期28天。
[0122]
为了将第二阶段的临床或实验室肿瘤溶解综合征(tls)风险降至最低,所有受试者首先接受每日递增方案,每个剂量水平如下所示。rp2d
‑
1剂量水平队列中的受试者从100mg开始接受化合物1,然后以每天100mg的增量连续给药,直到达到目标剂量的rp2d
‑
1。rp2d剂量水平队列中的受试者从100mg开始接受化合物1,然后以每天100mg的增量连续施用,直到达到目标剂量的rp2d。见表5。
[0123]
表5.第二阶段剂量递增设计。
[0124][0125]
*n=2
‑5[0126]
在第一天接受初始剂量的受试者,如果耐受,则根据上述方案在指定剂量水平基
础上给予每日递增剂量。每日递增给药的初始剂量固定为100mg,在达到指定剂量的当天达到药物周期的第1天。受试者继续接受治疗,直到疾病进展、无法忍受的毒性或转移到其他适当的治疗,或当研究者认为他们无法受益,或达到方案中指定的终止治疗的其他标准时为止。无论治疗是否终止,受试者都将进行生存随访(除非撤销知情同意或失去随访)。
[0127]
化合物1联合用药的初始剂量和剂量递增方案,将由申办方和研究者根据单药剂量探索的临床结果和pk数据进行讨论和决定,并进行适当的剂量调整。
[0128]
这两种剂量水平的联合用药方案分别在3个队列中进行研究。每一剂量水平的a组,接受化合物1联合低剂量hht治疗。hht的用量为每天1mg,连续14天静脉滴注,共14次输液。每个剂量水平的b组,用化合物1联合标准剂量hht治疗。在b组,hht的用量为2mg/m2,每天一次,连续7天,共7次输液。hht作为静脉输注给药,持续4小时,与口服化合物1在首次给药后不超过30分钟的间隔内给药。每个剂量水平的c组接受化合物1与aza联合用药。aza皮下注射,剂量75mg/m2,每天一次,连续7天。在自首次施用aza起不超过30分钟的间隔内,将aza与口服化合物1同时给药。
[0129]
申办者基于临床数据决定每种联合用药方案中化合物1的rp2d剂量。一旦确定了每种联合用药方案中化合物1的rp2d,研究将进入第三阶段,这是联合用药方案的扩展研究。
[0130]
第三阶段
‑
扩展研究
[0131]
扩展研究包括3组队列。从第二阶段开始,每个组合队列中最多有15名复发性和难治性aml患者接受rp2d剂量水平的hht或aza化合物1的联合治疗。a组接受化合物1的减量hht治疗。连续14天每天静脉滴注1mghht,共14次输注。用化合物1与标准剂量hht联合治疗组b,在组b中,通过静脉输注hht的使用量为2mg/m2,每天一次,连续7天,总共7次输注。hht以静脉内输注的方式给药4小时,在首次给药hht后不超过30分钟的间隔内,与口服化合物1联合给药。群组c接受化合物1与aza的联合给药,连续7天,每天一次以75mg/m2的剂量皮下注射aza。在自首次施用aza起不超过30分钟的间隔内,将aza与口服化合物1同时给药。
[0132]
第三阶段进一步评估在该剂量水平下联合用药方案对复发和难治性aml患者的安全性和有效性。为了将第三阶段临床或实验室肿瘤溶解综合征(tls)的风险降至最低,所有受试者首先接受每日递增方案,每个剂量水平如下所示。rp2d(第二阶段)剂量水平队列中的受试者从100mg开始接受化合物1,然后以每天100mg的增量连续施用,直到达到目标剂量的rp2d。见表6。
[0133]
表6.第三阶段剂量递增设计。
[0134][0135]
*n=2
‑5[0136]
等价方案
[0137]
本领域技术人员仅使用常规试验就将认识到或能够确定本文具体描述的具体实施方案的许多等价方案,这样的等价方案也应包含在所附权利要求的范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1