一种制备卡马替尼的方法与流程
1.本发明属于药物合成领域,具体涉及一种制备卡马替尼或其药学上可接受的盐的方法。
背景技术:
2.卡马替尼(capmatinib),由瑞士诺华公司开发,于2020年05月在美国获批上市,用于治疗metex14突变的成人转移性非小细胞肺癌(nsclc)。该药物为fda批准的首个选择性met抑制剂,具有疗效好,安全可控等优点。其化学名为2-氟-n-甲基-4-[7-[(喹啉-6-基)甲基]咪唑并[1,2-b]-[1,2,4]三嗪-2-基]苯甲酰胺,主要以其二盐酸盐一水合物作为临床产品的活性成分,其结构如下所示,
[0003][0004]
目前,现有技术公开的卡马替尼的制备方法主要采用以下两种合成路线,分别为:
[0005]
1、如专利cn 101641093b记载的合成路线一,
[0006][0007]
该合成路线存在以下缺点:步骤多,生产周期长,不利于工业化生产;.部分中间体
的纯化需要柱层析,工序繁琐;反应过程中使用氰化锌作氰化试剂,存在安全隐患。
[0008]
2、专利cn 108586463 a记载的合成路线二,
[0009][0010]
该合成路线存在以下缺点:(1)路线共计9步反应,溴代步骤收率低于20%,导致工艺路线总收率较低;(2)该合成路线中的氰基水解反应需在强酸(浓盐酸),高温(100℃)条件下回流18h以上,反应剧烈,设备要求高,对环境不友好,不利于工业化生产;(3)从步骤6到步骤8合成化合物6的过程中,需经偶联关环,氰基水解,酰胺缩合等三步反应,合成收率仅为47%,生产成本高。
技术实现要素:
[0011]
有鉴于此,本发明的主要目的在于提供一种易于工业化的制备卡马替尼或其药学可接受的盐的方法,通过合成路线的设计获得关键中间体化合物4,大大缩短了合成路线,简化了工艺步骤,并且各步骤反应条件温和,产物收率高且稳定,而且较现有技术的方法避免了有毒试剂氰化物的使用,更加安全。
[0012]
本发明第一方面,提供所述化合物4的制备方法,包括以下步骤:
[0013][0014]
其中:
[0015]
步骤2:在反应溶剂中,使化合物3在催化剂作用下,与3-氨基-6-溴-1,2,4-三嗪发生偶联反应后,得到化合物4。
[0016]
在部分实施方案中,步骤2中所述反应溶剂的用量与化合物3的体积质量比(以ml/g计)为2:1~100:1;优选为5:1~20:1;进一步优选为10:1;
[0017]
在部分实施方案中,步骤2中所述催化剂选自pd2(dba)3,pd(oac)2,pd[pph3]4和pdcl2(dppf)2中的一种或多种,优选催化剂为pdcl2(dppf)2。
[0018]
在部分实施方案中,步骤2中所述催化剂的用量为化合物3质量的5%-20%;优选8~15%;进一步优选10%;
[0019]
在部分实施方案中,步骤2中所述反应溶剂选自乙腈、四氢呋喃、1,4-二氧六环中的一种或多种与水的混合溶剂;
[0020]
优选地,反应溶剂为四氢呋喃和1,4-二氧六环中的一种或多种与水的混合溶剂;更优选地,反应溶剂为1,4-二氧六环与水的混合溶剂。
[0021]
在部分实施方案中,步骤2中所述反应溶剂用量与化合物3的体积质量比(以ml/g计)为2:1~100:1,优选5:1~20:1;进一步优选为12:1;
[0022]
在部分实施方案中,步骤2中所述水的用量与化合物3的体积质量比(以ml/g计)为0.5:1~10:1,优选为4:1~8:1;进一步优选为6:1;
[0023]
在部分实施方案中,步骤2中还包括加入碱性试剂的步骤,所述碱性试剂选自碳酸钠,氢氧化钠,碳酸氢钠,碳酸钾,氢氧化钾中的一种或者几种,优选碳酸钾;
[0024]
在部分实施方案中,步骤2中所述碱性试剂与化合物3的质量比为10:1~0.1:1;优选为5:1~0.5:1;进一步优选为2:1~1:1;
[0025]
在部分实施方案中,步骤2中所述偶联反应的反应温度为50℃~100℃,优选70℃~90℃。
[0026]
在部分实施方案中,步骤2中的化合物3采用以下步骤1的方法制备:
[0027][0028]
其中:
[0029]
步骤1:在反应溶剂中,使化合物2在催化剂作用下,与联硼酸频那醇酯发生偶联反应,得到化合物3;
[0030]
在部分实施方案中,步骤1中所述催化剂选自pd2(dba)3,pd(oac)2,pd[pph3]4和pdcl2(dppf)2中的一种或多种;优选催化剂为pd2(dba)3;
[0031]
在部分实施方案中,步骤1中所述催化剂的用量为化合物2质量的5%-20%;优选8~15%;进一步优选10%;
[0032]
在部分实施方案中,步骤1中所述反应溶剂选自乙腈,四氢呋喃,1,4-二氧六环中的一种或多种;
[0033]
优选地,反应溶剂为四氢呋喃或1,4-二氧六环;更优选地,反应溶剂为1,4-二氧六环;
[0034]
在部分实施方案中,步骤1中所述偶联反应的反应温度为50℃~100℃;优选为70℃~90℃。
[0035]
本发明第二方面,提供一种制备卡马替尼或其药学上可接受的盐的方法,包括以下路线a~路线c中的任意一种:
[0036]
路线a
[0037][0038]
路线b
[0039][0040]
路线c
[0041][0042]
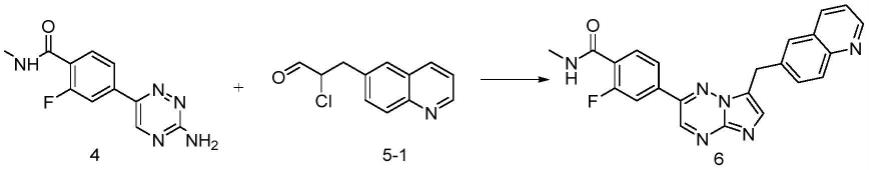
在部分实施方案中,路线a为:化合物4与化合物5-1发生偶联关环反应,得到化合物6。
[0043]
在部分实施方案中,路线a的反应溶剂选自甲醇、乙醇、异丙醇和乙二醇中的一种或多种,优选反应溶剂为乙二醇;
[0044]
在部分实施方案中,路线a中,所述反应溶剂的用量与化合物4的体积质量比(以ml/g计)为2:1~100:1,优选为5:1~15:1;进一步优选为10:1;
[0045]
在部分实施方案中,路线a中,反应温度为100℃~150℃,优选为120℃~130℃。
[0046]
在部分实施方案中,路线b为:化合物4与化合物5-2发生偶联关环反应,得到化合物6。
[0047]
在部分实施方案中,路线b的反应溶剂选自甲醇、乙醇、异丙醇和乙二醇中的一种或多种,优选反应溶剂为乙二醇;
[0048]
在部分实施方案中,路线b中,所述反应溶剂用量与化合物4的体积质量比(以ml/g计)为2:1~100:1;优选为5:1~15:1;进一步优选为10:1;
[0049]
在部分实施方案中,路线b中,反应温度为100℃~150℃,优选为120℃~130℃;
[0050]
在部分实施方案中,路线c为:第一阶段中,化合物5-3与ncs进行反应;第二阶段中,第一阶段得到的反应混合物不经进一步处理、直接与化合物4发生偶联关环反应,得到化合物6。
[0051]
所述第一阶段的反应溶剂选自乙酸乙酯,二氯甲烷,氯仿,乙腈中的一种或几种,优选为氯仿和乙腈中的一种或两种,更优选为氯仿;
[0052]
所述第一阶段反应溶剂用量与化合物5-3的体积质量比(以ml/g计)为2:1~100:1,优选为3:1~10:1;进一步优选为5:1;
[0053]
所述第一阶段反应温度范围选自-10℃~50℃,优选0℃~25℃;
[0054]
所述第一阶段还包括加入l-脯氨酸或d-脯氨酸中的一种或多种有机试剂的步骤,优选地,所述第一阶段还包括加入l-脯氨酸的步骤;
[0055]
所述有机试剂与化合物5-3的质量比为1:1~1:100;优选为1:5~1:20;进一步优选为1:10;
[0056]
所述第二阶段的反应溶剂选自甲醇,乙醇,异丙醇,乙二醇中的一种或几种,优选为乙二醇;
[0057]
所述第二阶段反应溶剂用量与化合物4的体积质量比(以ml/g计)为2:1~100:1;优选为5:1~15:1;进一步优选溶剂用量10:1;
[0058]
所述反应温度范围选自100℃~150℃,优选120℃~130℃。
[0059]
在部分实施方案中,通过本发明提供的制备方法制备得到化合物6后,将化合物6与甲醇混合均匀,再与盐酸发生进一步反应得到卡马替尼二盐酸盐(化合物1),反应路线如下所示:
[0060][0061]
本发明第三方面,提供化合物4在制备卡马替尼或其药学可接受的盐中的用途:
[0062][0063]
本发明提供的技术方案具有以下有益效果:
[0064]
(1)通过合成路线的设计获得关键中间体化合物4,有效缩短了合成路线,具有较好工业化生产潜力;
[0065]
(2)反应条件温和,不使用氰化物等剧毒试剂,提高了工艺安全性;
[0066]
(3)避免了现有技术(例如cn 108586463 a)中氰基水解所需要的强酸(浓盐酸),高温(100℃,反应18小时)等剧烈反应条件,设备适用性好,对环境友好;
[0067]
(3)各步骤所得中间体均可通过析晶方式纯化,产物收率高且稳定,例如化合物6的收率由现有技术的47%提高到65%以上。
具体实施方式
[0068]
下面将结合本发明实施方式,对本发明的技术方案进行清楚、完整地描述,显然,所描述的实施方式仅仅是本发明的一部分实施方式,而不是全部的实施方式。基于本发明中的实施方式,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施方式,都属于本发明保护的范围。
[0069]
仪器
[0070]
化合物的结构是通过核磁共振(1h、
13
c nmr)或质谱(ms)来确定的。
[0071]1h、
13
c nmr波谱法采用bruker超导核磁共振波谱仪(仪器型号:brukeravance 400型核磁共振仪);溶剂dmso-d6或cdcl3,温度20℃;
[0072]
lc-ms检测采用:agilentg 6230tofms磁质谱仪,电离方式:esi+;扫描范围:50~1000da;电子电量:70ev;
[0073]
采用高效液相色谱仪(hplc-uv)测定各步骤制得的化合物纯度,检测器为:紫外(uv)检测器,色谱柱:c18反相柱(4.6*250mm,5μm)。
[0074]
实施例中所涉及试剂缩写具有以下中文含义:
[0075]
缩写中文名称ncsn-氯代丁二酰亚胺pd2(dba)3三(二亚苄基丙酮)二钯pdcl2(dppf)21,1'-双二苯基膦二茂铁二氯化钯pd(pph3)4四三苯基膦钯dmfn,n-二甲基甲酰胺mtbe甲基叔丁基醚9-bbn9-硼二环[3.3.1]壬烷
[0076]
实施例1:2-氟-n-甲基-4-(4,4,5,5-四甲基-1,3,2-二氧杂硼烷-2-基)苯甲酰胺(化合物3)的制备
[0077][0078]
向反应瓶内加入4-溴-2-氟-n-甲基苯甲酰胺(化合物2)11.60g,联硼酸频那醇酯15.24g,pd2(dba)31.10g,三环己基膦0.80g以及乙酸钾14.72g,再向反应瓶中加入1,4-二氧六环116ml。氮气置换三次后,升温至80℃搅拌反应2小时,期间取样tlc监测反应进程。当监测至原料基本反应完全后,将反应体系降至室温,再用硅藻土过滤。所得滤液在减压条件下浓缩至干后,加入乙酸乙酯100ml至完全溶解后,用纯化水100ml分两次洗涤有机相,合并有机相,并用无水硫酸钠干燥,,过滤,所得滤液在减压条件下浓缩至干后,在正己烷中重结晶,得标题化合物10.31g,收率73.9%。
[0079]1h nmr(400mhz,cdcl3)δ8.07(t,j=7.7hz,1h),7.65(d,j=7.7hz,1h),7.51(d,j=12.5hz,1h),6.81(br,1h),3.02(dd,j=4.8,1.0hz,3h),1.34(s,12h).
[0080]
实施例2 4-(3-氨基-1,2,4-三嗪-6-基)-2-氟-n-甲基苯甲酰胺(化合物4)的制备
[0081][0082]
向反应瓶内加入2-氟-n-甲基-4-(4,4,5,5-四甲基-1,3,2-二氧杂硼
烷-2-基)苯甲酰胺(化合物3)14.37g和1,4-二氧六环74ml,在室温条件下搅拌溶清后,再加入3-氨基-6-溴-1,2,4-三嗪6.15g,碳酸钾水溶液(19.42g碳酸钾溶于37ml水中)和pdcl2(dppf)20.61g。反应体系氮气置换后,升温至80℃搅拌反应2小时,当原料基本反应完全后,将反应体系置于冰水浴中降温至10℃以下,向反应体系滴加117ml盐酸水溶液。滴加完毕后,反应体系升至室温搅拌1小时后过滤,所得滤液用乙酸乙酯洗涤,收集水相。将水相置于冰水浴中,向水相中滴加120m l50%氢氧化钠水溶液。滴加完毕后,保温搅拌2小时,过滤,滤饼用纯化水洗涤。所得滤饼干燥后,得标题化合物6.95g,收率80.1%,hplc纯度97%以上。
[0083]1h nmr(400mhz,dmso)δ8.93(s,1h),8.30(dd,j=4.2,2.5hz,1h),8.00
–
7.83(m,2h),7.73(m,,1h),7.56(br,2h),2.79(d,j=4.6hz,3h).
[0084]
实施例3 2-氟-n-甲基-4-[7-[(喹啉-6-基)甲基]咪唑并[1,2-b]-[1,2,4]三嗪-2-基]苯甲酰胺(化合物6)的制备
[0085][0086]
方法a
[0087]
向反应瓶内加入4-(3-氨基-1,2,4-三嗪-6-基)-2-氟-n-甲基苯甲酰胺(化合物4)1.0g,2-氯-3-(喹啉-6-基)丙醛(化合物5-1)1.33g和10ml乙二醇。反应体系氮气置换后,升温至120℃搅拌反应2小时,当原料基本反应完全,反应体系降至室温,向反应体系滴加纯化水20ml。滴加完毕后,室温条件下搅拌0.5h后,过滤。再将滤饼置于反应瓶中,加入dmf/mtbe的混合溶剂(dmf:mtbe=1:5)20ml回流打浆1h后降至室温,过滤,所得滤饼用mtbe洗涤。干燥后,得标题化合物。
[0088]1h nmr(400mhz,dmso)δ9.23(s,1h),8.85(dd,j=4.2,1.7hz,1h),8.41(d,j=2.8hz,1h),8.31(dd,j=8.3,1.3hz,1h),8.06(s,1h),8.05
–
8.02(m,1h),8.01(s,1h),7.98(m,2h),7.84
–
7.77(m,2h),7.51(dd,j=8.3,4.2hz,1h),4.64(s,2h),2.81(d,j=4.6hz,3h).
[0089]
实施例4 2-氟-n-甲基-4-[7-[(喹啉-6-基)甲基]咪唑并[1,2-b]-[1,2,4]三嗪-2-基]苯甲酰胺(化合物6)的制备
[0090][0091]
方法b
[0092]
向反应瓶内加入4-(3-氨基-1,2,4-三嗪-6-基)-2-氟-n-甲基苯甲酰胺(化合物4)2.80g,1-(2-氯-1-羟基-3-(喹啉-6-基)丙基)吡咯烷-2,5-二酮(化合物5-2)4.31g和28ml乙二醇。反应体系氮气置换后,升温至120℃搅拌反应2小时,当
原料基本反应完全,将反应体系降至室温后,向反应体系滴加纯化水60ml。滴加完毕后,室温条件下保温搅拌0.5h,过滤。再将滤饼置于反应瓶中,加入dmf/mtbe的混合溶剂(dmf:mtbe=1:5)60ml回流打浆1h后降至室温,过滤,所得滤饼用mtbe洗涤。所得滤饼干燥后,得标题化合物2.97g,收率63.8%,hplc纯度97%以上。
[0093]
核磁数据与实施例3一致。
[0094]
实施例5 2-氟-n-甲基-4-[7-[(喹啉-6-基)甲基]咪唑并[1,2-b]-[1,2,4]三嗪-2-基]苯甲酰胺(化合物6)的制备
[0095][0096]
方法c
[0097]
向反应瓶内加入3-(喹啉-6-基)丙醛(化合物5-3)2.00g,再加入氯仿10ml,完全溶清后向反应体系依次加入l-脯氨酸0.2g,ncs 1.58g。反应体系在室温条件下反应2h后浓缩,向浓缩剩余物加入4-(3-氨基-1,2,4-三嗪-6-基)-2-氟-n-甲基苯甲酰胺(化合物4)2.14g以及乙二醇20ml。反应体系氮气置换后,升温至120℃搅拌反应2小时,当原料基本反应完全,将反应体系降至室温后,向反应体系滴加纯化水40ml。滴加完毕后,室温条件下保温搅拌0.5h,过滤。再将滤饼置于反应瓶中,加入dmf/mtbe的混合溶剂(dmf:mtbe=1:5)40ml回流打浆1h后降至室温,过滤,所得滤饼用mtbe洗涤。所得滤饼干燥后,得标题化合物。核磁数据与实施例3一致。
[0098]
实施例6 2-氟-n-甲基-4-(7-(喹啉-6-基甲基)咪唑并[1,2-b][1,2,4]三嗪-2-基)苯甲酰胺二盐酸盐(化合物1)的制备
[0099][0100]
向反应瓶内加入2-氟-n-甲基-4-[7-[(喹啉-6-基)甲基]咪唑并[1,2-b]-[1,2,4]三嗪-2-基]苯甲酰胺(化合物6)2.14g,再加入30ml无水甲醇,升温至55℃搅拌0.5小时后,再向反应体系加入2.1ml浓盐酸。搅拌0.5小时后,向反应体系滴加30m lmtbe,随后保温搅拌1小时。反应体系缓慢降温至室温后,过滤,并用mtbe洗涤。所得滤饼干燥后,得标题化合物2.41g,收率81.1%,hplc纯度99%以上。
[0101]1h nmr(400mhz,dmso)δ9.52(s,1h),9.26(d,j=5.2hz,1h),9.15(d,j=8.3hz,1h),8.52(m,2h),8.37(s,1h),8.30(s,1h),8.24(d,j=7.5hz,1h),8.12
–
8.02(m,3h),7.80(t,j=7.8hz,1h),4.80(s,1h),2.79(d,j=4.5hz,3h).
[0102]
实施例7 2-氯-3-(喹啉-6-基)丙醛(化合物5-1)的制备
[0103][0104]
(1)6-(3,3-二乙氧基丙基)喹啉的制备
[0105]
向反应瓶内加入9-bbn 100ml(0.5mol/l),氮气置换后,置于冰水浴中降温至5℃以下,再向反应体系滴加丙烯酸缩二乙醇6.51g。滴加完毕后,升至室温反应过夜。随后向反应体系依次加入6-溴喹啉5.95g,碳酸钾7.90g,三环己基膦0.80g以及乙酸钯0.60g。反应体系氮气置换后,升温至65℃回流反应3小时,期间取样tlc监测反应进程。当原料基本反应完全,将反应体系降至室温后,向反应体系加入乙酸乙酯50ml,搅拌均匀后,垫硅藻土过滤。滤液浓缩后,经快速柱层析得6-(3,3-二乙氧基丙基)喹啉6.25g,收率84.5%。([m+1]+(m/z 260))
[0106]
(2)3-(喹啉-6-基)丙醛(化合物5-3)的制备
[0107]
向反应瓶内加入6-(3,3-二乙氧基丙基)喹啉6.25g,乙酸乙酯94ml,置于冰水浴条件下降至5℃以下。随后向反应体系滴加6ml盐酸水溶液(2mol/l),滴加完毕后,保温搅拌2小时。期间取样tlc监测反应进程。当原料基本反应完全,向反应体系滴加25%氢氧化钠水溶液调节ph至8-9。用乙酸乙酯(50ml*3)萃取反应液,合并有机相后,无水硫酸钠干燥。过滤,滤液浓缩后,经快速柱层析得3-(喹啉-6-基)丙醛3.93g,收率88.3%。([m+1]
+
(m/z 186)),1h nmr(400mhz,cdcl3)δ9.83(s,1h),8.84(d,j=3.9hz,1h),8.07(d,j=8.2hz,1h),8.03(d,j=8.6hz,1h),7.59(s,1h),7.54(d,j=8.7hz,1h),7.36(dd,j=8.3,4.2hz,1h),3.12(t,j=7.4hz,2h),2.87(t,j=7.4hz,2h).
[0108]
(3)2-氯-3-(喹啉-6-基)丙醛(化合物5-1)的制备
[0109]
在0℃条件下,向反应瓶内加入3-(喹啉-6-基)丙醛3.30g,再加入氯仿30ml。随后向反应体系依次加入l-脯氨酸0.35g,ncs 2.11g,搅拌均匀后反应体系氮气置换后升至室温反应1h,期间tlc监测反应。反应结束后,浓缩溶剂。所得浓缩剩余物经快速柱层析后,得2-氯-3-(喹啉-6-基)丙醛。([m+1+h2o]
+
(m/z 238.0/239.9))
[0110]
实施例8
[0111]
1-(2-氯-1-羟基-3-(喹啉-6-基)丙基)吡咯烷-2,5-二酮(化合物5-2)的制备
[0112][0113]
向反应瓶内加入3-(喹啉-6-基)丙醛(化合物5-3)3.93g,再加入氯仿16ml,完全溶清后置于冰水浴中降温至5℃以下。随后向反应体系依次加入l-脯氨酸0.24g,ncs 2.98g,搅拌均匀后反应体系氮气置换后升至室温反应过夜后,将反应体系置于冰水浴中降
温至5℃以下,向反应体系滴加乙酸乙酯16ml,滴加完毕后,保温搅拌1h。过滤,滤饼用乙酸乙酯洗涤。所得滤饼干燥后,得1-(2-氯-1-羟基-3-(喹啉-6-基)丙基)吡咯烷-2,5-二酮4.47g,收率66.2%,hplc纯度99%以上。1h nmr(400mhz,cdcl3)δ9.83(s,1h),8.84(d,j=3.9hz,1h),8.07(d,j=8.2hz,1h),8.03(d,j=8.6hz,1h),7.59(s,1h),7.54(d,j=8.7hz,1h),7.36(dd,j=8.3,4.2hz,1h),3.12(t,j=7.4hz,2h),2.87(t,j=7.4hz,2h).
[0114]
以上所述仅为本发明的优选实施方式,并非因此限制本发明的专利范围,凡是在本发明的发明构思下,利用本发明说明书所作的等效结构变换,或直接/间接运用在其他相关的技术领域均包括在本发明的专利保护范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1