基于萘酰亚胺衍生物的荧光应力响应材料及其制备与应用
1.本发明属于精细化工领域,具体涉及一种基于萘酰亚胺衍生物的荧光应力响应材料及其制备与应用。
背景技术:
2.发光功能材料由于其具有在受到外部刺激下能表现出发光变化这一特性,已经广泛应用于生物探针、化学传感器、电致发光器件等诸多领域。其中,荧光应力响应材料具有对外力刺激产生明显的荧光信号变化的特性,这种材料的荧光发射变化是通过外力刺激其有序晶体结构和无序无定型结构之间的转变而实现的。
3.荧光应力响应材料不仅在物质结构的基础研究中具有重要意义,而且在传感器、电致发光器件、光学数据存储、安全油墨等应用领域也引起了人们的广泛关注。这种材料大致可以分为:有机小分子、配合物、有机高分子、配位聚合物、超分子材料等。其中有机小分子具有结构易于修饰、种类多、功能化策略齐全、合成技术成熟、结构重复性好等优势,特别是随着聚集诱导发光现象的发现,具有聚集诱导发光性质的有机小分子克服了聚集态下荧光猝灭的缺点。近年来,基于有机小分子的荧光应力响应材料得到了快速的发展,出现了一系列以四苯基乙烯(q.qi,et al.,adv.funct.mater.,2015,25,4005
‑
4010;s.
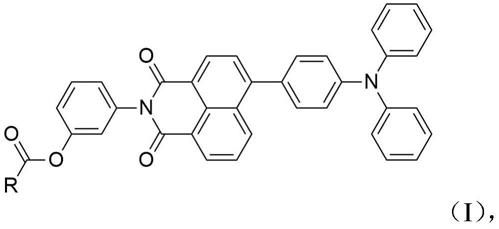
‑
s.zhao,et al.,chem.commun.,2017,53,7048
‑
7051;b.xu,et al.,chem.sci.,2015,6,3236
‑
3241),吩噻嗪(b.huang,et al.,angew.chem.int.ed.,2018,57,12473
‑
12477;y.chen,et al.,dyes.pigments.,2019,161,44
‑
50),氟硼荧(g.zhang,et al.,j.am.chem.soc.,2010,132,2160
‑
2162),蒽(d.tu,et al.,chem.commun.,2016,52,12494
‑
12497)等荧光分子构建的小分子应力响应材料。上述材料中,多通过将四苯基乙烯引入其它分子结构中构建出新的聚集诱导发光分子,而性能优异的单一荧光团小分子则相对较少。
4.近来,一些报道指出通过合适的修饰,萘酰亚胺衍生物可以实现单一分子的聚集诱导发光效应,并进一步制备出相应的应力响应荧光功能材料(y.yin,et al.,acs.omega.,2019,4,14324
‑
14332)。但是,现有报道的萘酰亚胺类材料的开发仍集中于黄绿光区域(500
‑
550nm),而在黄绿光区域发光的材料具有信号通道单一、信号分辨率低等缺点。少数较长波段的萘酰亚胺类材料也难以达到红光区域,且这类材料结构复杂、合成难度大、荧光量子效率很低,开发价值不高。因此,开发出量子效率高的多通道长波荧光应力响应材料对进一步的实用化荧光传感器、光学信息安全保密等具有重要的现实意义。
技术实现要素:
5.本发明的目的就是提供一种基于萘酰亚胺衍生物的荧光应力响应材料及其制备与应用,提供的应力响应材料具有不同量子效率的聚集态发光性质,不同的自组装结构可产生荧光差异,实现voc气体检测和数据存储。
6.本发明的目的通过以下技术方案实现:
7.一种基于萘酰亚胺衍生物的荧光应力响应材料,通过链长的调控,可以制备出一
系列微观形貌为针状或层状的晶体,晶体长度在1~5微米范围内,层状晶体厚度在200~300纳米范围内,且具有100~500纳米不等之孔隙。所述材料均具有式(ⅰ)所示的结构式:
8.其中,r为烷基。
9.优选地,r为c1~c7烷基。
10.本发明制备的荧光应力响应材料均在390nm处激发,在自组装状态(即原始状态)下具有553~566nm发射峰,在无定型状态下具有568~610nm发射峰,荧光应力响应材料在未受外力刺激的自组装状态的量子效率为23.9~92.2%,在受到外力刺激下形成无定型态的量子效率为43.7~59.2%。具体地,nitpac1在390nm激发,在自组装状态下具有560nm发射峰,量子效率为92.2%,在无定型状态下具有610nm发射峰,量子效率为59.2%;nitpac3在390nm激发,在自组装状态下具有566nm发射峰,量子效率为23.9%,在无定型状态下具有602nm发射峰,量子效率为43.7%;nitpac5在390nm激发,在自组装状态下具有557nm发射峰,量子效率为43.9%,在无定型状态下具有581nm发射峰,量子效率为58.0%;nitpac7在390nm激发,在自组装状态下具有553nm发射峰,量子效率为41.7%,在无定型状态下具有568nm发射峰,量子效率为46.1%。
11.一种如上述所述的基于萘酰亚胺衍生物的荧光应力响应材料的制备方法,该合成路线如下式(ⅱ)所示:
12.13.所述制备方法具体包括以下步骤:
14.(a)取间氨基苯酚和4
‑
溴
‑
1,8
‑
萘二甲酸酐(命名为ni)溶于乙酸中并进行加热反应,经第一次后处理得到n
‑
间苯酚
‑4‑
溴
‑
1,8
‑
萘酰亚胺(命名为nip);
15.(b)取步骤(a)得到的n
‑
间苯酚
‑4‑
溴
‑
1,8
‑
萘酰亚胺、三苯胺
‑4‑
硼酸和四(三苯基膦)钯(别名为pd(pph3)4)溶于n,n
‑
二甲基甲酰胺中,再加入碳酸钾溶液进行加热反应,经第二次后处理得到4
‑
位含三苯胺单元的1,8
‑
萘酰亚胺
‑
n
‑
间苯酚(命名为nitpa
‑
phenol);
16.(c)取步骤(b)得到的4
‑
位含三苯胺单元的1,8
‑
萘酰亚胺
‑
n
‑
间苯酚、4
‑
二甲氨基吡啶和n,n
′‑
二环己基碳二亚胺溶于四氢呋喃中,再加入脂肪酸进行反应,经第三次后处理得到4
‑
位含三苯胺单元的1,8
‑
萘酰亚胺
‑
n
‑
间苯酚烷基酯(命名为nitpac)。
17.步骤(a)中,所述的4
‑
溴
‑
1,8
‑
萘二甲酸酐、间氨基苯酚和乙酸的摩尔体积比为(1.0~1.2)mmol:(1.95~2.3)mmol:(15~30)ml,优选为1.1mmol:2mmol:15ml。
18.步骤(a)中,加热反应的温度为110~130℃,优选为120℃,加热反应的时间为4~6h。
19.步骤(a)中,进行反应时采用氮气鼓泡脱除氧气,间氨基酚会被氧化成醌,脱除氧气可预防出现过多的副产物。
20.步骤(a)中,第一次后处理具体为:待反应结束之后,冷却至室温,再加入水抽滤,得到的滤饼真空干燥后用无水乙醇重结晶。
21.步骤(b)中,所述的n
‑
间苯酚
‑4‑
溴
‑
1,8
‑
萘酰亚胺、三苯胺
‑4‑
硼酸、四(三苯基膦)钯和n,n
‑
二甲基甲酰胺(别名为dmf)的摩尔体积比为(1.0~1.2)mmol:(1.1~1.2)mmol:(0.026~0.028)mmol:(10~15)ml,优选为1.1mmol:1.1mmol:0.026mmol:10ml所述碳酸钾溶液的浓度为(1.5~1.62)mol/l,优选为1.62mol/l。
22.步骤(b)中,加热反应的温度为140~160℃,优选为150℃,加热反应的时间为8~12h。
23.步骤(b)中,进行反应时采用氮气鼓泡脱除氧气。
24.步骤(b)中,第二次后处理具体为:待反应结束后,冷却至室温,依次加入二氯甲烷、饱和食盐水和无水na2so4分别进行萃取、洗涤和干燥,最后用100
‑
200目硅胶层析柱提纯。
25.硅胶层析柱的洗脱剂为二氯甲烷。
26.步骤(c)中,所述的n
‑
间苯酚
‑4‑
三苯胺基
‑
1,8
‑
萘酰亚胺、4
‑
二甲氨基吡啶(别名为dmap)、n,n
′‑
二环己基碳二亚胺(别名为dcc)和四氢呋喃(别名为thf)的摩尔体积比为(1.0~1.2)mmol:(1.25~1.40)mmol:(1.63~1.80)mmol:(10~12)ml。
27.步骤(c)中,所述的n
‑
间苯酚
‑4‑
三苯胺基
‑
1,8
‑
萘酰亚胺和脂肪酸的反应投料摩尔比为(1.0~1.2):(2.5~4)。
28.步骤(c)中,在冰浴条件下加入脂肪酸,后升温至室温进行反应,反应的时间为2~4h。考虑到有不饱和键的脂肪酸或具有更长链的脂肪酸需要较低温度和较长反应时间,因此在冰浴条件下加入脂肪酸,并适当延长了反应时间。
29.步骤(c)中,进行反应时采用氮气鼓泡脱除氧气。
30.步骤(c)中,第三次后处理具体为:待反应结束后,减压浓缩四氢呋喃,再加入饱和碳酸氢钠溶液抽滤,得到的滤饼真空干燥后过100
‑
200目硅胶层析柱提纯。
31.硅胶层析柱的洗脱剂为二氯甲烷和石油醚的混合溶液,所述二氯甲烷和石油醚的体积比为3:1。
32.步骤(a)和步骤(c)中,在反应时均进行搅拌。
33.一种如上述所述的基于萘酰亚胺衍生物的荧光应力响应材料的应用,所述应用包括检测多种voc气体和光学信息安全保密。
34.基于萘酰亚胺衍生物的荧光应力响应材料在固相状态下检测voc气体。在检测voc气体过程中,基于萘酰亚胺衍生物的荧光应力响应材料固态的微观结构由自组装结构转变为块状无定型结构(可通过场发射扫描电镜予以甄别)。荧光应力响应材料可以在粉末、压片、浸泡试纸、旋涂膜、聚合物掺杂膜中与voc气体作用,作用后可实现荧光发光颜色和强度的变化,通过imagej软件,可对荧光应力响应材料产生的绿光、红光通道进行数字信号提取,并进一步处理成单、双通道检测信号,从而对voc进行定性和定量检测,该材料可作为一类优良的比色比率荧光探针。
35.光学信息安全保密过程中,采用基于萘酰亚胺衍生物的荧光应力响应材料涂画的信号在自然光条件下难以肉眼识别,但可在365nm紫外辐射下清晰显示。
36.本发明以萘酰亚胺为母体和电子受体,引入三苯胺作为电子给体,构建强的分子内电荷转移,以实现不同聚集状态下分子内电荷分布的调制,再通过引入不同长度的烷基链作为自组装结构的调节单元。1,8
‑
萘酰亚胺衍生物(即含有结构式为的一类化合物),由于其自身具有很高的荧光量子效率、强固态荧光、stokes位移大、荧光聚集性能好、紫外光区摩尔消光系数大、光稳定性好、成膜性好等优势,可用作荧光探针、激光染料、染料敏化太阳能电池敏化剂以及电致发光材料等。在电子给体芳胺(即三苯胺)和电子受体萘酰亚胺(即结构式为萘酰亚胺单元)之间插入一个可旋的刚性苯环,构建了具有一定二面角的共轭分子,通过不同聚集状态下二面角的变化可以调节分子内电荷转移的幅度,进而实现不同聚集态下荧光波长的变化。在本发明中,自组装结构的有序性堆积会使得分子趋向平面化,从而降低分子内的二面角角度,使得荧光波长变短,而研磨后产生的无定形态将进一步增大分子内二面角的角度,使得分子内扭曲增大,荧光波长向红移动。
37.本发明提供的应力响应材料具有不同量子效率的聚集态发光性质,解决了现有技术中应力响应材料结构复杂、合成难度大、成本高、长波段应力响应材料稀缺、量子效率较低等技术难题,并利用不同自组装结构下的荧光差异实现voc气体检测和光学信息安全保密。
附图说明
38.图1为实施例1制备的nitpac1在未受外力刺激时的场发射扫描电镜图;
39.图2为实施例1制备的nitpac1在受外力刺激时的场发射扫描电镜图;
40.图3为实施例3制备的nitpac3在未受外力刺激时的场发射扫描电镜图;
41.图4为实施例3制备的nitpac3在受外力刺激时的场发射扫描电镜图;
42.图5为实施例5制备的nitpac5在未受外力刺激时的场发射扫描电镜图;
43.图6为实施例5制备的nitpac5在受外力刺激时的场发射扫描电镜图;
44.图7为实施例7制备的nitpac7在未受外力刺激时的场发射扫描电镜图;
45.图8为实施例7制备的nitpac7在受外力刺激时的场发射扫描电镜图;
46.图9为实施例1制备的nitpac1在未受外力刺激时和受外力刺激时的固体荧光发射光谱比较图;
47.图10为实施例3制备的nitpac3未受外力刺激时和受外力刺激时的固体荧光发射光谱比较图;
48.图11为实施例5制备的nitpac5未受外力刺激时和受外力刺激时的固体荧光发射光谱比较图;
49.图12为实施例7制备的nitpac7未受外力刺激时和受外力刺激时的固体荧光发射光谱比较图;
50.图13为实施例1制备的nitpac1对voc检测在不同响应时间下的荧光变化图;
51.图14为实施例1制备的nitpac1对voc检测的单一通道数字信号图;
52.图15为实施例1制备的nitpac1对voc检测的双通道比率信号图;
53.图16为实施例1制备的nitpac1在应用于光学信息安全保密时的加密和可视化图;
54.图17为实施例1制备的nitpac1的核磁氢谱图;
55.图18为实施例1制备的nitpac1的核磁碳谱图;
56.图19为实施例1制备的nitpac1的高分辨质谱图;
57.图20为实施例3制备的nitpac3的核磁氢谱图;
58.图21为实施例3制备的nitpac3的核磁碳谱图;
59.图22为实施例3制备的nitpac3的高分辨质谱图;
60.图23为实施例5制备的nitpac5的核磁氢谱图;
61.图24为实施例5制备的nitpac5的核磁碳谱图;
62.图25为实施例5制备的nitpac5的高分辨质谱图;
63.图26为实施例7制备的nitpac7的核磁氢谱图;
64.图27为实施例7制备的nitpac7的核磁碳谱图;
65.图28为实施例7制备的nitpac7的高分辨质谱图;
66.图29为将nitpac1进行研磨后经蒸汽熏恢复原结构状态的示意图;
67.图30为图29中nitpac1三种状态下的固体荧光发射光谱比较图;
68.图31为nitpac1、nitpac3、nitpac5和nitpac7研磨前后的荧光变化图;
69.图32为nitpac1的发射可逆循环次数图。
具体实施方式
70.下面结合附图和具体实施例对本发明进行详细说明。
71.一种基于萘酰亚胺衍生物的荧光应力响应材料,具有式(ⅰ)所示的结构式:
72.其中,r为烷基。
73.一种如上述所述的基于萘酰亚胺衍生物的荧光应力响应材料的制备方法,所述制备方法具体包括以下步骤:
74.(a)取间氨基苯酚和4
‑
溴
‑
1,8
‑
萘二甲酸酐溶于乙酸中并在110~130℃下进行加热反应4~6h,进行反应时采用氮气鼓泡脱除氧气,待反应结束之后,冷却至室温,再加入水抽滤,得到的滤饼真空干燥后用无水乙醇重结晶,得到n
‑
间苯酚
‑4‑
溴
‑
1,8
‑
萘酰亚胺,其中4
‑
溴
‑
1,8
‑
萘二甲酸酐、间氨基苯酚和乙酸的摩尔体积比为(1.0~1.2)mmol:(1.95~2.3)mmol:(15~30)ml;
75.(b)取步骤(a)得到的n
‑
间苯酚
‑4‑
溴
‑
1,8
‑
萘酰亚胺、三苯胺
‑4‑
硼酸和四(三苯基膦)钯溶于n,n
‑
二甲基甲酰胺中,再加入碳酸钾溶液在140~160℃下进行加热反应8~12h,进行反应时采用氮气鼓泡脱除氧气,待反应结束后,冷却至室温,依次加入二氯甲烷、饱和食盐水和无水na2so4分别进行萃取、洗涤和干燥,最后用100
‑
200目硅胶层析柱提纯,得到4
‑
位含三苯胺单元的1,8
‑
萘酰亚胺
‑
n
‑
间苯酚,其中n
‑
间苯酚
‑4‑
溴
‑
1,8
‑
萘酰亚胺、三苯胺
‑4‑
硼酸、四(三苯基膦)钯催化剂和n,n
‑
二甲基甲酰胺的摩尔体积比为(1.0~1.2)mmol:(1.1~1.2)mmol:(0.026~0.028)mmol:(10~15)ml,所述碳酸钾溶液的浓度为(1.35~1.62)mol/l;
76.(c)取步骤(b)得到的4
‑
位含三苯胺单元的1,8
‑
萘酰亚胺
‑
n
‑
间苯酚、4
‑
二甲氨基吡啶和n,n
′‑
二环己基碳二亚胺溶于四氢呋喃中,再在冰浴条件下加入脂肪酸,后升温至室温进行反应2~4h,进行反应时采用氮气鼓泡脱除氧气,待反应结束后,减压浓缩四氢呋喃,再加入饱和碳酸氢钠溶液抽滤,得到的滤饼真空干燥后过100
‑
200目硅胶层析柱提纯,得到4
‑
位含三苯胺单元的1,8
‑
萘酰亚胺
‑
n
‑
间苯酚烷基酯,其中所述的n
‑
间苯酚
‑4‑
三苯胺基
‑
1,8
‑
萘酰亚胺、4
‑
二甲氨基吡啶、n,n
′‑
二环己基碳酰亚胺和四氢呋喃的摩尔体积比为(1.0~1.2)mmol:(1.25~1.40)mmol:(1.63~1.80)mmol:(10~12)ml,所述的n
‑
间苯酚
‑4‑
三苯胺基
‑
1,8
‑
萘酰亚胺和脂肪酸的反应投料摩尔比为(1.0~1.2):(2.5~4)。本发明采用的4
‑
溴
‑
1,8
‑
萘二甲酸酐和三苯胺
‑4‑
硼酸为市售化合物,也可采用文献常规方法进行制备,本发明不再赘述。
77.一种如上述所述的基于萘酰亚胺衍生物的荧光应力响应材料的应用,包括检测多种voc气体和光学信息安全保密。
78.下面通过实施例对本发明作进一步阐述,其目的仅在于更好地理解本发明的内容,因此所举之例并不限制本发明的保护范围。
79.实施例1
80.一种基于萘酰亚胺衍生物的荧光应力响应材料,具有式(ⅰ)所示的结构式:
81.其中,r为ch3,采用以下步骤制备得到:
82.(1)nip的合成
[0083][0084]
在一个配有磁性搅拌子的100ml三口烧瓶中,依次加入4
‑
溴
‑
1,8
‑
萘二甲酸酐(ni)(340mg,1.1mmol)、间氨基苯酚(218mg,2mmol)以及15ml乙酸,氮气鼓泡脱除氧气,升温至120℃,搅拌回流6h。自然冷却至室温,然后加入20ml水,静置抽滤,滤饼真空干燥,用无水乙醇重结晶,得到292mg灰黄色粉末,化学名为n
‑
间苯酚
‑4‑
溴
‑
1,8
‑
萘酰亚胺(nip),产率为79%。1h nmr(400mhz,dmso
‑
d6)δppm:9.67(s,1h),8.58(s,1h),8.61(dd,2h),8.34
‑
8.36(d,j=7.48,1h),8.26(d,j=8.23,1h),8.02
‑
8.05(m,1h),7.29
‑
7.32(m,1h),6.85
‑
6.87(m,1h),6.79(m,2h).
[0085]
(2)nitpa
‑
phenol的合成
[0086][0087]
室温条件下,将nip(405mg,1.1mmol)、4
‑
硼酸三苯胺(318mg,1.1mmol)、四(三苯基膦)钯(30mg,0.026mmol)以及10ml n,n
‑
二甲基甲酰胺置于100ml反应管中。然后加入1.12g k2co3/5ml h2o溶液(即k2co3溶液的浓度为1.62mol/l),使固体化合物完全溶解,得到澄清透明溶液,鼓泡脱除氧气,氮气保护,逐渐升温至150℃,反应8h。tlc监测反应,反应完全后,停止加热,让其自然冷却至室温,加入二氯甲烷(20ml
×
2)萃取,饱和食盐水(20ml
×
2)洗涤,
无水na2so4干燥,用100
‑
200目硅胶层析柱提纯(洗脱剂为二氯甲烷),得到523mg橙红色纯品,化学名为4
‑
位含三苯胺单元的1,8
‑
萘酰亚胺
‑
n
‑
间苯酚(nitpa
‑
phenol),产率为89%。1h nmr(400mhz,dmso
‑
d6)δppm:9.67(s,1h),8.57
‑
8.51(m,2h),8.45(d,j=8.5hz,1h),7.95
‑
7.88(m,1h),7.84(d,j=7.6hz,1h),7.52(d,j=8.5hz,2h),7.39(t,j=7.9hz,4h),7.31(t,j=8.0hz,1h),7.15(dd,j=16.1,7.7hz,8h),6.87(d,j=7.5hz,1h),6.80(d,j=6.9hz,2h).
13
c nmr(100mhz,dmso
‑
d6)δppm:164.06,163.84,158.30,148.22,147.26,131.94,131.49,130.91,130.23,129.88,125.24,124.27,122.48.hrms(esi)calcd.for c
36
h
25
n2o3[m+h]
+
533.1865,found 533.1864.
[0088]
(3)化合物nitpac1的合成
[0089][0090]
将nitpa
‑
phenol(532mg,1.0mmol)、n,n
′‑
二环己基碳二亚胺(336mg,1.63mmol)和4
‑
二甲氨基吡啶(153mg,1.25mmol)加入到50ml烧瓶中,加入10ml四氢呋喃溶解。然后在冰浴条件下,缓慢加入(150mg,2.5mmol)乙酸,脱除氧气,移至室温下反应2小时。待反应结束后,减压浓缩溶剂,加入饱和碳酸氢钠溶液,过滤得到固体粗产物,最后通过100
‑
200目硅胶层析柱(洗脱剂体积比二氯甲烷:石油醚=3:1)纯化得到黄绿色目标产物390mg,黄绿色目标产物的化学名为4
‑
位含三苯胺单元的1,8
‑
萘酰亚胺
‑
n
‑
间苯酚烷基酯,记为nitpac1,nitpac1为针状形晶体,平均长度大约为2~3微米,厚度约为200~300nm,孔隙大约为200~300nm,该材料的氢谱如下:1h nmr(400mhz,cdcl3)δ8.69(d,j=7.6hz,2h),8.50(d,j=9.4hz,1h),7.81
–
7.75(m,2h),7.58(t,j=8.1hz,1h),7.43(d,j=8.6hz,2h),7.39
–
7.33(m,4h),7.31(s,1h),7.25(dd,j=8.0,2.5hz,7h),7.20(t,j=2.0hz,1h),7.13(t,j=7.3hz,2h),2.32(s,3h).13c nmr(100mhz,cdcl3)δ168.69,164.22,164.00,151.25,148.48,147.33,147.25,136.21,133.26,131.81,131.62,131.36,130.79,130.23,129.69,129.49,129.26,127.77,126.74,126.12,125.09,123.69,122.92,122.46,122.21,121.79,121.23,77.32,77.00,76.68,21.15.hr
‑
ms(esi)calcd.for c
38
h
26
n2o4[m+na]+597.1790,found 597.1788.
[0091]
取80mg目标产物nitpac1于玛瑙研钵中,紫外照射下发射出黄绿色荧光(绝对量子效率:92.2%),实物图如图31所示,该材料未受外力刺激时的场发射扫描电镜图具体如图1所示,当用研杵对目标产物进行研磨3分钟后,其荧光发射为红色(绝对量子效率:59.2%),实物图如图31所示,受外力刺激(外力刺激在本发明中即为研磨,下同)时的场发射扫描电
镜图具体如图2所示(先进行研磨后进行扫描,下同),从图1中可以看到nitpac1发生了明显的自组装行为(即nitpac1被制备出来还未被研磨时本身就是一种自组装),对比nitpac1、nitpac3、nitpac5和nitpac7原始状态下的量子效率以及研磨状态下的效率,可发现nitpac1的量子效率经研磨后降低,而其他三种的量子效率经研磨后均提高,这种现象可以从以下方面进行解释:无定形态的量子效率与侧链长度关系不大,故研磨后四种产物的量子效率趋同,但自组装状态的量子效率受侧链长度影响大,而nitpac1的链长最短,自组装后平面化程度最大,形成的类似晶态的结构具有强荧光,因此,只有nitpac1的量子效率经研磨后是降低的。
[0092]
图9为nitpac1的固体荧光发射光谱比较图(图中,横坐标为发射波长,单位为纳米;纵坐标为归一化荧光发射强度,下同),从图9中可以看到nitpac1受到外力刺激前后发生约50nm的光谱位移,nitpac1在390nm激发,在自组装状态下具有560nm发射峰,在无定型状态下具有610nm发射峰。
[0093]
nitpac1的核磁氢谱图如图17所示,nitpac1的核磁碳谱图如图18所示,nitpac1的高分辨质谱图如图19所示,此三幅图均是在nitpac1原始状态下做的检测,下同,原始状态和研磨状态这两种状态下材料的检测结果是一样的,因为研磨只是改变了宏观状态,并未改变化学结构。
[0094]
采用上述所得的nitpac1化合物对voc进行定性和定量检测,自然环境条件下,将洁净的圆形小滤纸片在nitpac1染料的二氯甲烷溶液中浸泡15s后取出,电吹风吹干溶剂。在365nm紫外光辐射下,对其荧光图像进行拍摄。然后将5ml不同voc溶剂(以二氯甲烷为例)分别置于10ml称量瓶中,使其达到饱和蒸汽压。将附载nitpac1染料的圆形小滤纸片粘贴在称量瓶盖内侧,然后盖在称量瓶上,放置相同时间后,在365nm紫外光辐射下,进行荧光拍摄,分别测试nitpac1制成膜识别voc前和识别voc后的红绿通道数字信号,并进行比率计算,然后以响应时间为横坐标,数字信号强度或数字信号比率为纵坐标,分别绘制工作曲线,具体如图13、14、15所示(图13中横坐标为不同voc检测,纵坐标为不同响应时间,单位为秒,图14中横坐标为响应时间,单位为秒,纵坐标为数字信号强度(图14中的阴影部分指信号转化时间,即响应时间),图15中横坐标为响应时间,单位为秒,纵坐标为数字比率信号(图15中的阴影部分也指信号转化时间,即响应时间)。图13~15表明这种基于萘酰亚胺衍生物的荧光应力响应材料在检测voc的过程中,转变速度较快(响应速度可达20~40s),灵敏度较高(数字比率信号增益高达400%),对比图14和图15,可得到:nitpac1这类荧光应力响应材料在使用两个通道信号的比率值作为检测信号(图15)的响应时间时比单独使用某一通道信号作为检测信号(图14)时的响应时间更短。
[0095]
进一步地,将上述所得的nitpac1化合物应用在光学信息安全保密中。通过用含有低浓度nitpac1染料的二甲基亚砜溶液(1mm)在滤纸片上写上“sues”字样,然后自然风干。自然光条件下很难通过肉眼识别出滤纸片上的信号,但是在365nm紫外辐射下,清晰地显现出“sues”字样,具体如图16所示,图16中左边为加密后的区域自然光照图片,右边为可视化解密后的区域365nm紫外辐射图片,由此可见,本发明的基于萘酰亚胺衍生物的荧光应力响应材料对同一区域的信号进行加密与可视化可实现光学信息的安全保密。
[0096]
图29为将nitpac1进行研磨后经蒸汽熏恢复原结构状态示意图,恢复原状态可再经研磨,图30为nitpac1在原始状态下、研磨状态下以及经蒸汽熏状态下三种情况下的固体
荧光发射光谱比较图,可看到代表蒸汽熏的线和代表原始状态的线重合度很高,说明nitpac1经蒸汽熏基本能恢复到原始状态。
[0097]
实施例2
[0098]
一种基于萘酰亚胺衍生物的荧光应力响应材料,除了将nitpa
‑
phenol(639mg,1.2mmol)、n,n
′‑
二环己基碳二亚胺(371mg,1.8mmol)和4
‑
二甲氨基吡啶(171mg,1.4mmol)加入到50ml烧瓶中,加入12ml四氢呋喃溶解。然后在冰浴条件下,缓慢加入(240mg,4.0mmol)乙酸之外,其余均与实施例1相同,最终得到结构与实施例1相同的黄绿色目标产物440mg。该材料的氢谱如下:1h nmr(400mhz,cdcl3)δ8.69(d,j=7.6hz,2h),8.50(d,j=9.4hz,1h),7.81
–
7.75(m,2h),7.58(t,j=8.1hz,1h),7.43(d,j=8.6hz,2h),7.39
–
7.33(m,4h),7.31(s,1h),7.25(dd,j=8.0,2.5hz,7h),7.20(t,j=2.0hz,1h),7.13(t,j=7.3hz,2h),2.32(s,3h).13c nmr(100mhz,cdcl3)δ168.69,164.22,164.00,151.25,148.48,147.33,147.25,136.21,133.26,131.81,131.62,131.36,130.79,130.23,129.69,129.49,129.26,127.77,126.74,126.12,125.09,123.69,122.92,122.46,122.21,121.79,121.23,77.32,77.00,76.68,21.15.hr
‑
ms(esi)calcd.for c
38
h
26
n2o4[m+na]+597.1790,found 597.1788.
[0099]
实施例3
[0100]
一种基于萘酰亚胺衍生物的荧光应力响应材料,具有式(ⅰ)所示的结构式:
[0101]
其中,r为c3h7,采用以下步骤制
[0102]
备得到,其中前两步和实施例1相同,第三步如下:
[0103]
(3)化合物nitpac3的合成
[0104][0105]
将nitpa
‑
phenol(532mg,1.0mmol)、n,n
′‑
二环己基碳二亚胺(336mg,1.63mmol)和4
‑
二甲氨基吡啶(153mg,1.25mmol)加入到50ml烧瓶中,用10ml四氢呋喃溶解。然后在冰浴条件下,缓慢加入(220mg,2.5mmol)丁酸,脱除氧气,移至室温下反应2小时。待反应结束后,
减压浓缩溶剂,加入饱和碳酸氢钠溶液,过滤得到固体粗产物,最后通过100
‑
200目硅胶层析柱(洗脱剂体积比二氯甲烷:石油醚=3:1)纯化得到黄色目标产物443mg,黄绿色目标产物的化学名为4
‑
位含三苯胺单元的1,8
‑
萘酰亚胺
‑
n
‑
间苯酚烷基酯,记为nitpac3,nitpac3为层状形晶体,平均长度大约为3~5微米,厚度约为200~300nm,孔隙大约为200~300nm,该材料的氢谱如下:1h nmr(400mhz,cdcl3)δ8.70(s,2h),8.50(s,1h),7.78(s,2h),7.59(s,1h),7.30(q,j=46.2,35.9hz,18h),2.57(s,2h),1.82(s,2h),1.69(s,2h),1.07(s,3h).
13
c nmr(101mhz,cdcl3)δ171.41,164.25,151.34,148.48,147.33,136.20,133.27,131.81,131.62,131.37,130.81,130.21,129.70,129.49,129.26,127.77,126.75,126.02,125.09,123.69,122.92,122.47,122.24,121.84,121.23,77.34,36.29,18.39,13.65.hr
‑
ms(esi)calcd.for c
40
h
30
n2o4[m+h]
+
603.2284,found 603.2264.
[0106]
取80mg目标产物nitpac3于玛瑙研钵中,紫外照射下发射出黄绿色荧光(绝对量子效率:23.9%),实物图如图31所示,该材料未受外力刺激时的场发射扫描电镜图具体如图3所示,当用研杵对目标产物进行研磨3分钟后,其荧光发射为橙黄色(绝对量子效率:43.7%),实物图如图31所示,受外力刺激时的场发射扫描电镜图具体如图4所示,从图3中可以看到nitpac3发生了明显的自组装行为。图10为nitpac3的固体荧光发射光谱比较图,从图10中可以看到nitpac3受到外力刺激前后发生约36nm的光谱位移,nitpac3在390nm激发,在自组装状态下具有566nm发射峰,在无定型状态下具有602nm发射峰。nitpac3的核磁氢谱图如图20所示,nitpac3的核磁碳谱图如图21所示,nitpac3的高分辨质谱图如图22所示。
[0107]
实施例4
[0108]
一种基于萘酰亚胺衍生物的荧光应力响应材料,除了将nitpa
‑
phenol(639mg,1.2mmol)、n,n
′‑
二环己基碳二亚胺(371mg,1.8mmol)和4
‑
二甲氨基吡啶(171mg,1.4mmol)加入到50ml烧瓶中,加入12ml四氢呋喃溶解。然后在冰浴条件下,缓慢加入(352mg,4mmol)丁酸之外,其余均与实施例3相同,最终得到结构与实施例3相同的黄绿色目标产物460mg。该材料的氢谱如下:1h nmr(400mhz,cdcl3)δ8.70(s,2h),8.50(s,1h),7.78(s,2h),7.59(s,1h),7.30(q,j=46.2,35.9hz,18h),2.57(s,2h),1.82(s,2h),1.69(s,2h),1.07(s,3h).
13
c nmr(101mhz,cdcl3)δ171.41,164.25,151.34,148.48,147.33,136.20,133.27,131.81,131.62,131.37,130.81,130.21,129.70,129.49,129.26,127.77,126.75,126.02,125.09,123.69,122.92,122.47,122.24,121.84,121.23,77.34,36.29,18.39,13.65.hr
‑
ms(esi)calcd.for c
40
h
30
n2o4[m+h]+603.2284,found 603.2264.
[0109]
实施例5
[0110]
一种基于萘酰亚胺衍生物的荧光应力响应材料,具有式(ⅰ)所示的结构式:其中,r为c5h
11
,采用以下步骤制
[0111]
备得到,其中前两步和实施例1相同,第三步如下:
[0112]
(3)化合物nitpac5的合成
[0113][0114]
将nitpa
‑
phenol(532mg,1.0mmol)、n,n
′‑
二环己基碳二亚胺(336mg,1.63mmol)和4
‑
二甲氨基吡啶(153mg,1.25mmol)加入到50ml烧瓶中,用10ml四氢呋喃溶解。然后在冰浴条件下,缓慢加入(290mg,2.5mmol)己酸,脱除氧气,移至室温下反应2小时。待反应结束后,减压浓缩溶剂,加入饱和碳酸氢钠溶液,过滤得到固体粗产物,最后通过100
‑
200目硅胶层析柱(洗脱剂体积比二氯甲烷:石油醚=3:1)纯化得到黄色目标产物363mg,黄绿色目标产物的化学名为4
‑
位含三苯胺单元的1,8
‑
萘酰亚胺
‑
n
‑
间苯酚烷基酯,记为nitpac5,nitpac5为层状形晶体,平均长度大约为1~3微米,厚度约为200~300nm,孔隙大约为100~200nm,该材料的氢谱如下:1h nmr(400mhz,cdcl3)δ8.69(d,j=7.6hz,2h),8.50(d,j=8.4hz,1h),7.78(t,j=8.2hz,2h),7.58(t,j=8.1hz,1h),7.43(d,j=8.4hz,2h),7.36(t,j=7.7hz,4h),7.31(s,1h),7.24(d,j=7.8hz,7h),7.19(s,1h),7.13(t,j=7.2hz,2h),2.58(t,j=7.5hz,2h),1.78(p,j=7.3hz,2h),1.47
–
1.34(m,4h),0.94(t,j=6.8hz,3h).
13
c nmr(101mhz,cdcl3)δ171.57,164.25,164.03,151.36,148.48,147.33,147.24,136.19,133.26,131.82,131.61,131.36,130.22,129.69,129.48,129.26,127.77,126.74,125.99,125.09,123.68,122.93,122.47,122.22,121.82,121.24,77.31,34.41,31.25,24.56,22.30,13.86.hr
‑
ms(esi)calcd.for c
42
h
34
n2o4[m+h]
+
631.2596,found 631.2597.
[0115]
取80mg目标产物nitpac5于玛瑙研钵中,紫外照射下发射出黄绿色荧光(绝对量子效率:43.9%),实物图如图31所示,该材料未受外力刺激时的场发射扫描电镜图具体如图5所示,当用研杵对目标产物进行研磨3分钟后,其荧光发射为黄色(绝对量子效率:58.0%),实物图如图31所示,受外力刺激时的场发射扫描电镜图具体如图6所示,从图5中可以看出,nitpac5发生了明显的自组装行为。图11为nitpac5的固体荧光发射光谱比较图,从图11中可以看到nitpac5受到外力刺激前后发生约24nm的光谱位移,nitpac5在390nm激发,在自组装状态下具有557nm发射峰,在无定型状态下具有581nm发射峰。nitpac5的核磁氢谱图如图23所示,nitpac5的核磁碳谱图如图24所示,nitpac5的高分辨质谱图如图25所示。
[0116]
实施例6
[0117]
一种基于萘酰亚胺衍生物的荧光应力响应材料,除了将nitpa
‑
phenol(639mg,1.2mmol)、n,n
′‑
二环己基碳二亚胺(371mg,1.8mmol)和4
‑
二甲氨基吡啶(171mg,1.4mmol)
加入到50ml烧瓶中,加入12ml四氢呋喃溶解。然后在冰浴条件下,缓慢加入(464mg,4mmol)己酸之外,其余均与实施例5相同,最终得到结构与实施例5相同的黄绿色目标产物460mg。该材料的氢谱如下:1h nmr(400mhz,cdcl3)δ8.69(d,j=7.6hz,2h),8.50(d,j=8.4hz,1h),7.78(t,j=8.2hz,2h),7.58(t,j=8.1hz,1h),7.43(d,j=8.4hz,2h),7.36(t,j=7.7hz,4h),7.31(s,1h),7.24(d,j=7.8hz,7h),7.19(s,1h),7.13(t,j=7.2hz,2h),2.58(t,j=7.5hz,2h),1.78(p,j=7.3hz,2h),1.47
–
1.34(m,4h),0.94(t,j=6.8hz,3h).
13
c nmr(101mhz,cdcl3)δ171.57,164.25,164.03,151.36,148.48,147.33,147.24,136.19,133.26,131.82,131.61,131.36,130.22,129.69,129.48,129.26,127.77,126.74,125.99,125.09,123.68,122.93,122.47,122.22,121.82,121.24,77.31,34.41,31.25,24.56,22.30,13.86.hr
‑
ms(esi)calcd.for c
42
h
34
n2o4[m+h]
+
631.2596,found 631.2597.
[0118]
实施例7
[0119]
一种基于萘酰亚胺衍生物的荧光应力响应材料,具有式(ⅰ)所示的结构式:
[0120]
其中,r为c7h
15
,采用以下步骤制备得到,其中前两步和实施例1相同,第三步如下:
[0121]
(3)化合物nitpac7的合成
[0122][0123]
将nitpa
‑
phenol(532mg,1.0mmol)、n,n
′‑
二环己基碳二亚胺(336mg,1.63mmol)和4
‑
二甲氨基吡啶(153mg,1.25mmol)加入到50ml烧瓶中,用10ml四氢呋喃溶解。然后在冰浴条件下,缓慢加入(360mg,2.5mmol)辛酸,脱除氧气,移至室温下反应2小时。待反应结束后,减压浓缩溶剂,加入饱和碳酸氢钠溶液,过滤得到固体粗产物,最后通过100
‑
200目硅胶层析柱(洗脱剂体积比二氯甲烷:石油醚=3:1)纯化得到黄绿色目标产物430mg,黄绿色目标产物的化学名为4
‑
位含三苯胺单元的1,8
‑
萘酰亚胺
‑
n
‑
间苯酚烷基酯,记为nitpac7,nitpac7为层状形晶体,平均长度大约为1~3微米,厚度约为200~300nm,孔隙大约为200~
500nm,该材料的氢谱如下:1h nmr(400mhz,cdcl3)δ8.70(dd,j=7.4,1.9hz,2h),8.50(d,j=8.4hz,1h),7.78(t,j=8.1hz,2h),7.58(t,j=8.1hz,1h),7.43(d,j=8.5hz,2h),7.36(t,j=7.9hz,4h),7.31(s,1h),7.25(dd,j=8.1,2.5hz,7h),7.19(s,1h),7.13(t,j=7.3hz,2h),2.58(t,j=7.5hz,2h),1.77(p,j=7.4hz,2h),1.48
–
1.28(m,8h),0.91(t,j=6.8hz,3h).
13
cnmr(101mhz,cdcl3)δ171.55,164.23,164.01,151.37,148.49,147.34,147.23,136.19,133.24,131.83,131.60,131.35,130.79,130.23,129.66,129.48,129.26,127.76,126.73,125.99,125.09,123.68,122.94,122.47,122.22,121.80,121.26,77.31,76.99,76.68,34.45,31.62,29.05,28.89,24.88,22.57,14.01.hr
‑
ms(esi)calcd.for c
44
h
38
n2o4[m+h]
+
659.2910,found 659.2955.
[0124]
取80mg目标产物nitpac7于玛瑙研钵中,紫外照射下发射出黄绿色荧光(绝对量子效率:41.7%),实物图如图31所示,该材料未受外力刺激时的场发射扫描电镜图具体如图7所示,当用研杵对目标产物进行研磨3分钟后,其荧光发射为黄色(绝对量子效率:46.1%),实物图如图31所示,受外力刺激时的场发射扫描电镜图具体如图8所示,从图7可以看出nitpac7发生了明显的自组装行为。图12为nitpac7的固体荧光发射光谱比较图,从图12中可以看到nitpac7受到外力刺激前后发生约15nm的光谱位移,nitpac7在390nm激发,在自组装状态下具有553nm发射峰,在无定型状态下具有568nm发射峰。nitpac7的核磁氢谱图如图26所示,nitpac7的核磁碳谱图如27所示,nitpac7的高分辨质谱图如图28所示。
[0125]
实施例8
[0126]
一种基于萘酰亚胺衍生物的荧光应力响应材料,除了将nitpa
‑
phenol(639mg,1.2mmol)、n,n
′‑
二环己基碳二亚胺(371mg,1.8mmol)和(171mg,1.4mmol)4
‑
二甲氨基吡啶加入到50ml烧瓶中,加入12ml四氢呋喃溶解。然后在冰浴条件下,缓慢加入(576mg,,4.0mmol)辛酸之外,其余均与实施例7相同,最终得到结构与实施例7相同的黄绿色目标产物485mg。该材料的氢谱如下:1h nmr(400mhz,cdcl3)δ8.70(dd,j=7.4,1.9hz,2h),8.50(d,j=8.4hz,1h),7.78(t,j=8.1hz,2h),7.58(t,j=8.1hz,1h),7.43(d,j=8.5hz,2h),7.36(t,j=7.9hz,4h),7.31(s,1h),7.25(dd,j=8.1,2.5hz,7h),7.19(s,1h),7.13(t,j=7.3hz,2h),2.58(t,j=7.5hz,2h),1.77(p,j=7.4hz,2h),1.48
–
1.28(m,8h),0.91(t,j=6.8hz,3h).
13
cnmr(101mhz,cdcl3)δ171.55,164.23,164.01,151.37,148.49,147.34,147.23,136.19,133.24,131.83,131.60,131.35,130.79,130.23,129.66,129.48,129.26,127.76,126.73,125.99,125.09,123.68,122.94,122.47,122.22,121.80,121.26,77.31,76.99,76.68,34.45,31.62,29.05,28.89,24.88,22.57,14.01.hr
‑
ms(esi)calcd.for c
44
h
38
n2o4[m+h]
+
659.2910,found 659.2955.
[0127]
以上结果证明,nitpac1、nitpac3、nitpac5、nitpac7都表现出荧光应力响应。其中碳链最短的nitpac1表现出高对比的应力响应荧光变色效果,它在原始状态下可以发射黄绿色荧光,但是经过机械力研磨之后,呈现出红色荧光,这种高对比的发射状态可以通过挥发性溶剂蒸汽熏和研磨进行可逆切换(即发射峰值可以经蒸汽熏和研磨两种操作在566nm和610nm之间来回切换),并且可以重复使用(重复使用指的是荧光应力响应材料可根据应用的场景和要求来选择研磨或蒸汽熏来改变材料的宏观状态,并且性能不变)。碳链的长度不同,分子在聚集状态下的自组装行为也不同。这种对应力、挥发性有机溶剂具有显著响应,且响应信号位于人眼敏感的红绿色区的基于萘酰亚胺衍生物的荧光应力响应材料在光
学数据记录、数据安全、数据保护等领域具有潜在的应用前景。
[0128]
实施例9
[0129]
本实施例提供荧光应力响应材料的可逆切换实验,具体为:(1)取80mg的自组装状态下的nitpac1进行荧光光谱测试,激发波长为390nm,获得荧光发射峰峰值处的发射波长为566nm。(2)将样品置于玛瑙研钵手动研磨两分钟,刮取样品(此为无定型状态样品),测试其荧光发射,激发波长为:390nm,获得荧光发射峰峰值处发射波长为:610nm。(3)将(2)中的样品置于一小培养皿中,取10毫升二氯甲烷(作为挥发性溶剂进行蒸汽熏操作)置于一大培养皿中,用烧杯将两个培养皿罩住,在大培养皿底部40度加热一分钟。然后收集小培养皿中的样品,测试其荧光光谱,激发波长为:390nm,获得荧光发射峰峰值为:610nm。然后按照上述(1)
‑
(2)
‑
(3)顺序循环多次实验,测试该样品的荧光发射峰峰值可逆切换次数,以评定其荧光信号重复使用性能,具体如图32所示。从图32可以看到,这种发射波长的信号切换在循环30次时仍可以保持荧光发射峰峰值信号在10nm以内,说明本发明的荧光应力响应材料的可逆切换性能优越。
[0130]
上述的对实施例的描述是为便于该技术领域的普通技术人员能理解和使用发明。熟悉本领域技术的人员显然可以容易地对这些实施例做出各种修改,并把在此说明的一般原理应用到其他实施例中而不必经过创造性的劳动。因此,本发明不限于上述实施例,本领域技术人员根据本发明的揭示,不脱离本发明范畴所做出的改进和修改都应该在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1