一种抗癌药物帕博西林中间体的绿色制备方法与流程
1.本发明属于化药技术领域,具体涉及一种抗癌瘤药物帕博西林中间体的绿色制备方法。
背景技术:
2.帕博西林作为一种新型抗癌药物
‑
(cdk)4/6抑制剂,可以高选择性、可逆性抑制细胞周期蛋白依赖性激酶。2015年2月,美国食品与药品管理局(fda)批准ibrance(帕博西林(palbociclib))联合来曲唑作为内分泌治疗为基础的初始方案用于治疗er+/her2
‑
绝经后晚期乳腺癌。帕博西林(palbociclib)是首个获批的用于治疗乳腺癌的cdk4/6抑制剂。
[0003]2‑
硝基
‑5‑
溴吡啶是制备帕博西林原料药的一个关键中间体(济南大学硕士学位论文,2017年6月,帕博西林关键中间体的合成工艺研究),其由2
‑
氨基
‑5‑
溴吡啶氧化而成,反应式如scheme 1所示:
[0004][0005]
目前该氧化反应大多都是在酸性条件下采用h2o2或过氧酸作为氧化剂进行氧化反应,如cn106187867b中采用冰醋酸为溶剂、过氧乙酸为氧化剂,反应后蒸出部分醋酸,然后加碱性水溶液调节ph析晶得到目标产物,收率可达80%左右;但该反应后处理过程中需要大量碱进行中和,产生高盐废水。bioorganic and medicinal chemistry,1999,7(3):467
–
479中采用h2so4/h2o2体系进行氧化反应,收率仅为73%。org.process res.dev.2017,21,451
‑
459对该氧化反应做了详细的反应热及其反应过程研究,研究发现采用h2so4/h2o2体系氧化会产生显著量的3,5
‑
二溴
‑2‑
氨基吡啶和1,2
‑
双(5
‑
溴吡啶
‑2‑
基)偶氮氧化物等副产物;该论文重点研究了如何控制反应热使反应利于生产放大,但其最终收率48.6%。
技术实现要素:
[0006]
本发明的目的是克服2
‑
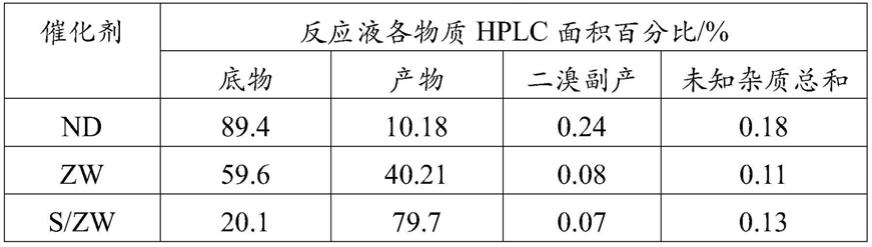
硝基
‑5‑
溴吡啶现有生产工艺废酸产量大、副反应多,收率低的缺陷,提供一种高收率绿色方法制备抗癌药物帕博西林中间体2
‑
硝基
‑5‑
溴吡啶。本发明采用自制的酸化的钨/锆双金属氧化物作为催化剂,与氧化剂形成活性氧化物种用于催化氧化2
‑
氨基
‑5‑
溴吡啶制备2
‑
硝基
‑5‑
溴吡啶,本发明催化氧化工艺绿色无污染,不会产生废酸;且氧化工艺较为温和,氧化过程中基本无3,5
‑
二溴
‑2‑
氨基吡啶副产的生成,所以粗品纯度和收率较高。
[0007]
根据本发明的一个方面,本发明提供了一种抗癌药物帕博西林中间体的绿色制备方法,在溶剂和催化剂的存在下,滴加氧化剂氧化2
‑
氨基
‑5‑
溴吡啶得2
‑
硝基
‑5‑
溴吡啶;
[0008]
所述催化剂为酸化的钨/锆双金属氧化物,其制备方法包括如下步骤:
[0009]
1)硝酸锆、偏钨酸铵、尿素溶于水中搅拌分散均匀,然后在ph=8.5
‑
9.0的条件下
加热回流反应12
‑
24h后降温至室温,过滤、水洗,收集滤饼在马弗炉中于600
‑
700℃下热处理后降温至室温得zr/w复合氧化物;按摩尔比计算,硝酸锆:偏钨酸铵:尿素=1:2
‑
5:20;本发明采用均相沉淀法制备出zr/w复合氧化物,其中尿素起到了沉淀剂的作用,使制备出的zr/w复合氧化物粒子更加均匀;
[0010]
2)将zr/w复合氧化物粉碎至粒径为200
‑
300目后浸渍于硫酸水溶液中,超声10
‑
30min后,过滤、水洗,收集滤饼在马弗炉中于600
‑
700℃下热处理后降温至室温得酸化的钨/锆双金属氧化物。
[0011]
优选的,所述在溶剂和催化剂的存在下,双氧水氧化2
‑
氨基
‑5‑
溴吡啶得2
‑
硝基
‑5‑
溴吡啶,具体步骤为:
[0012]
1)将2
‑
氨基
‑5‑
溴吡啶、催化剂采用溶剂在20
‑
30℃下搅拌均匀,然后将反应体系控温至10
‑
15℃,然后滴加氧化剂;通过滴加氧化剂速度来调节反应温度,避免反应剧烈导致局部过热产生3,5
‑
二溴
‑2‑
氨基吡啶等副产物;
[0013]
2)氧化剂滴加结束后,在10
‑
15℃保温反应1
‑
2h,然后以2℃/min的升温速率升温至30℃保温反应2h,最后以1℃/min的升温速率升温至40℃直至采用hplc检测底物2
‑
氨基
‑5‑
溴吡啶浓度不再下降;
[0014]
3)降温至室温,过滤去除催化剂,滤液中加入连二亚硫酸钠搅拌1
‑
2h;
[0015]
4)采用淀粉碘化钾试纸测试体系中无过量氧化剂后滴加碱水溶液调节体系ph至8.0,最后滴加反溶剂析晶,过滤,干燥得2
‑
硝基
‑5‑
溴吡啶。
[0016]
优选的,所述溶剂为甲醇、乙醇、丙酮、水或其任意比例混合液;进一步优选为丙酮与水的混合液,体积比为丙酮:水=5
‑
8:1;
[0017]
优选的,本发明所述氧化剂为双氧水或次氯酸钠,进一步优选为双氧水;所述氧化剂的用量与2
‑
氨基
‑5‑
溴吡啶的摩尔比为2
‑
5:1,进一步优选为3:1;
[0018]
优选的,所述催化剂重量为2
‑
氨基
‑5‑
溴吡啶重量的0.01
‑
0.4;进一步优选为,催化剂重量为2
‑
氨基
‑5‑
溴吡啶重量的0.1
‑
0.2;
[0019]
优选的,所述反溶剂为纯化水或纯化水与异丙醇的混合液;按照体积比纯化水:异丙醇=3
‑
5:1。
[0020]
本发明首次采用均相沉淀法制备出zr/w复合氧化物,并经硫酸浸渍制备出具有催化氧化性能的催化剂,酸化的钨/锆双金属氧化物中的钨和锆两种过渡金属可与氧化剂形成活性氧化物种,从而起到催化氧化效果;可用于催化氧化2
‑
氨基
‑5‑
溴吡啶制备抗癌药物帕博西林中间体2
‑
硝基
‑5‑
溴吡啶,与现有氧化工艺相比,本发明具有如下优点:
[0021]
1)本发明氧化过程中无需加入有机酸或者无机酸,杜绝了后处理过程中酸性废水的产生;
[0022]
2)本发明催化剂在催化氧化过程中,反应条件温和,基本无副产物生成,2
‑
硝基
‑5‑
溴吡啶纯度和收率大大提高,收率稳定在93%左右,粗品纯度可达99.8%以上,无需进一步纯化;
[0023]
3)本发明催化剂可通过过滤从反应体系中分离出来,且能够回收套用,生产成本大大降低;
[0024]
4)本发明催化剂在多次回收套用后可通过煅烧、酸浸渍的方法使催化剂活性复活。
具体实施方式
[0025]
为使本发明的目的、技术方案和优点更加清楚明了,下面结合具体实施方式,对本发明进一步详细说明。应该理解,这些描述只是示例性的,而并非要限制本发明的范围。
[0026]
hplc检测方法:仪器:agilent 1260;色谱柱:phenomenex luna phenyl
‑
hexyl柱,4.6
×
150mm,5μm;柱温:35℃;流动相a:ph=7.0的磷酸盐缓冲溶液,流动相b:甲醇;流速:1.0ml/min.检测波长:210nm;进样量:5μl;稀释液:流动相a/b=50/50。按表1进行梯度洗脱:
[0027]
表1流动相梯度洗脱表
[0028]
时间/min流动相a/v%流动相b/v%070302703020109022109022.17030267030
[0029]2‑
氨基
‑5‑
溴吡啶来自于山东西亚化学工业有限公司,订制纯度为99.6%以上,最大单杂小于0.1%;偏钨酸铵水合物来自于西格玛奥德里奇(上海)贸易有限公司,分子量以3060.46计算(无水形式);其余原料未做说明均为市售分析级试剂。
[0030]
实施例1
[0031]
按如下步骤制备催化剂:
[0032]
1)将10mmol zr(no3)4·
5h2o、30mmol偏钨酸铵水合物、200mmol尿素溶于500ml水中搅拌分散均匀,滴加氨水溶液调节ph至8.5
‑
9.0后回流反应12
‑
24h,降温至室温,过滤、水洗,收集滤饼在马弗炉中于600
‑
700℃下热处理1
‑
2h后降温至室温得zr/w复合氧化物(简写为zw);
[0033]
2)将10.0g zr/w复合氧化物粉碎至粒径为200
‑
300目后浸渍于100ml 2mol/l的硫酸水溶液中,超声10
‑
30min后,过滤、水洗,收集滤饼在马弗炉中于600
‑
700℃下热处理后降温至室温得酸化的钨/锆双金属氧化物(简写为s/zw)。
[0034]
实施例2
[0035]
采用实施例1制备的zr/w复合氧化物(简写为zw)和酸化的钨/锆双金属氧化物(简写为s/zw)作为添加剂,催化氧化2
‑
氨基
‑5‑
溴吡啶制备2
‑
硝基
‑5‑
溴吡啶,方法如下:
[0036]
反应瓶中加入底物2
‑
氨基
‑5‑
溴吡啶(1.73g,10mmol)、添加剂(0.30g,~17.3wt%)、15ml甲醇中搅拌均匀,然后将反应液温度控温至0
‑
5℃滴加浓度为30wt%h2o2(50mmol,5.0eq);滴加完毕后升温至20℃反应2h,30℃反应2h,然后升温至40℃直至前后2h取的反应液中2
‑
氨基
‑5‑
溴吡啶的面积百分比不再下降,加入连二亚硫酸钠(42mmol)去除反应液中过量的氧化剂,统计还原后的反应液中底物2
‑
氨基
‑5‑
溴吡啶、产物2
‑
硝基
‑5‑
溴吡啶、副产物3,5
‑
二溴
‑2‑
氨基吡啶(简称为二溴副产)及其其它未知杂质总和,结果如表2所示:
[0037]
表2催化剂催化氧化效果
[0038][0039]
注:nd是指未加入任何形式的添加剂作为催化剂。
[0040]
试验结果表明,不添加任何催化剂直接采用h2o2作为氧化剂,几乎不发生氧化反应,仅有10%左右的目标产物生成;采用本发明制备的zr/w复合氧化物(简写为zw)和酸化的钨/锆双金属氧化物(简写为s/zw)大大提高了底物的转化率,尤其是对zr/w复合氧化物,产品面积百分比可达到79.7%;硫酸浸渍后催化剂表面酸性改变导致其氧化还原能力得到改善,并且氧化过程中仅生成少量二溴副产,可通过后处理分离步骤顺带去除。
[0041]
实施例3
[0042]
为了进一步提高底物的转化率,本发明对氧化反应中的溶剂种类和氧化剂种类做了进一步优化,方法如下:
[0043]
反应瓶中加入底物2
‑
氨基
‑5‑
溴吡啶(1.73g,10mmol)、酸化的钨/锆双金属氧化物(简写为s/zw)(0.30g,~17.3wt%)、15ml溶剂中搅拌均匀,然后将反应液温度控温至0
‑
5℃滴加氧化剂(50mmol,5.0eq);滴加完毕后升温至20℃反应2h,30℃反应2h,然后升温至40℃直至前后2h取的反应液中2
‑
氨基
‑5‑
溴吡啶的面积百分比不再下降,加入连二亚硫酸钠(42mmol)去除反应液中过量的氧化剂,统计还原后的反应液中底物2
‑
氨基
‑5‑
溴吡啶、产物2
‑
硝基
‑5‑
溴吡啶、副产物3,5
‑
二溴
‑2‑
氨基吡啶(简称为二溴副产)及其其它未知杂质总和,结果如表3所示:
[0044]
表3溶剂和氧化剂种类对反应的影响
[0045]
[0046]
注:丙酮/水是指丙酮与水的混合液,体积比丙酮/水=7/1。
[0047]
试验结果表明,采用本发明制备的酸化的钨/锆双金属氧化物(简写为s/zw)作为催化剂,氧化反应过程中,溶剂具有一定的溶剂效应,其中以丙酮/h2o的混合液(体积比丙酮/水=7/1)作为溶剂反应效果最好;氧化剂中反应效果h2o2>naocl>acooh,催化剂与双氧水中的活性氧结合性强,所以最终确定h2o2作为氧化剂。
[0048]
实施例4
[0049]
本发明进一步优化了h2o2和催化剂酸化的钨/锆双金属氧化物(简写为s/zw)的用量,方法如下:
[0050]
1)将2
‑
氨基
‑5‑
溴吡啶(1.73g,10mmol)、催化剂s/zw(17
‑
865mg,1.0~50.0wt%)采用15ml丙酮/水混合液(体积比丙酮/水=7/1)在20
‑
30℃下搅拌均匀,然后将反应体系控温至10
‑
15℃,然后滴加30wt%h2o2(10
‑
80mmol,1.0
‑
8.0eq);
[0051]
2)氧化剂滴加结束后,在10
‑
15℃保温反应1
‑
2h,然后以2℃/min的升温速率升温至30℃保温反应2h,最后以1℃/min的升温速率升温至40℃直至采用hplc检测底物2
‑
氨基
‑5‑
溴吡啶浓度不再下降;
[0052]
3)降温至室温,过滤去除催化剂,滤液中加入连二亚硫酸钠搅拌1
‑
2h;
[0053]
4)采用淀粉碘化钾试纸测试体系中无过量氧化剂后升温至45
‑
55℃搅拌10
‑
30min,降温至室温,统计还原后的反应液中底物2
‑
氨基
‑5‑
溴吡啶、产物2
‑
硝基
‑5‑
溴吡啶、副产物3,5
‑
二溴
‑2‑
氨基吡啶(简称为二溴副产)及其其它未知杂质总和,结果如表4所示:
[0054]
表4催化剂和氧化剂用量对反应的影响
[0055][0056]
实验结果表明,氧化剂用量需为底物2
‑
氨基
‑5‑
溴吡啶摩尔量的2倍以上,为降低成本减少氧化剂用量以3.0eq为宜;催化剂用量需大于底物2
‑
氨基
‑5‑
溴吡啶重量的
5.0wt%,当大于30.0wt%时其它未知杂质增高,出现其它氧化副反应;所以催化剂用量以底物2
‑
氨基
‑5‑
溴吡啶重量的10.0
‑
20.0wt%为宜。
[0057]
实施例5
[0058]
采用上述优化工艺条件,本发明进行了实验室规模放大反应,具体方法如下:
[0059]
1)5l三口玻璃瓶中加入2
‑
氨基
‑5‑
溴吡啶(173g,1.0mol)、催化剂s/zw(26.0g,~15.0wt%)、1.5l丙酮/水混合液(体积比丙酮/水=7/1)在20
‑
30℃下采用机械搅拌器搅拌均匀,然后将反应体系控温至10
‑
15℃,采用恒压滴液漏斗滴加30wt%h2o2(3.5mol,3.0eq);
[0060]
2)h2o2滴加结束后,在10
‑
15℃保温反应1
‑
2h,然后以2℃/min的升温速率升温至30℃保温反应2h,最后以1℃/min的升温速率升温至40℃直至采用hplc检测底物2
‑
氨基
‑5‑
溴吡啶浓度不再下降;(40℃下反应4h,hplc检测物料面积百分比:底物0.18%、产物99.4%、二溴副产0.16%、其它未知杂质总和0.26%;与小试反应结果差别不大,说明本发明氧化反应按照现有工艺可以进行放大);
[0061]
3)降温至室温,过滤去除催化剂,滤液中加入连二亚硫酸钠搅拌1
‑
2h;
[0062]
4)采用淀粉碘化钾试纸测试体系中无过量氧化剂后升温至45
‑
55℃搅拌10
‑
30min,然后滴加碱水溶液调节体系ph至8.0,最后滴加反溶剂2.0l(体积比,纯化水/异丙醇=4/1)析晶,降温至室温过滤,干燥得2
‑
硝基
‑5‑
溴吡啶188.4g(收率为92.8%),hplc检测纯度为99.82%,最大单杂为二溴副产0.06%。1h
‑
nmr(dmso):8.83(1h,dd,),8.51(1h,dd),8.26(1h).lc/ms:202.9[m+h]
+
,204.8[m+h]
+
[0063]
对过滤所得催化剂采用丙酮超声洗涤后晾干,并在45℃下真空干燥至恒重得25.2g,回收率为96.9%,补加0.8g新鲜催化剂至26.0g;按照上述方法进行回收套用试验,统计反应结束后反应液中各物料的面积百分比,衡量催化剂使用寿命,结果如表5所示:
[0064]
表5催化剂套用结果
[0065][0066][0067]
注:w
‑
5是指对套用5次后的催化剂进行活化处理后的催化结果。
[0068]
回收套用结果表明,随着套用次数的增加,底物转化率呈现下降趋势,尤其是回收套用5次以后,底物剩余量达到5.96%;但套用第一次和第二次催化剂催化效果几乎没变,所以本发明制备的催化剂具有一定的稳定性;w
‑
5是对回收套用5次后的催化剂进行活化处
理,方法如下:
[0069]
对过滤分离所得催化剂采用丙酮超声洗涤1
‑
2h,然后在在马弗炉中于600
‑
700℃下热处理1
‑
2h,降温至室温后浸渍于2mol/l的硫酸水溶液中,超声10
‑
30min后,过滤、水洗,收集滤饼在马弗炉中于600℃下热处理后降温至室温得活化后的酸化的钨/锆双金属氧化物(简写为r
‑
s/zw)。
[0070]
对使用多次的催化剂,按照催化剂的热处理和酸浸渍方法进行活化处理可以解决催化剂多次使用后活性变低的缺点。
[0071]
尽管已经详细描述了本发明的实施方式,但是应该理解的是,在不偏离本发明的精神和范围的情况下,可以对本发明的实施方式做出各种改变、替换和变更。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1