接枝高分子与包含其的复合材料的制作方法
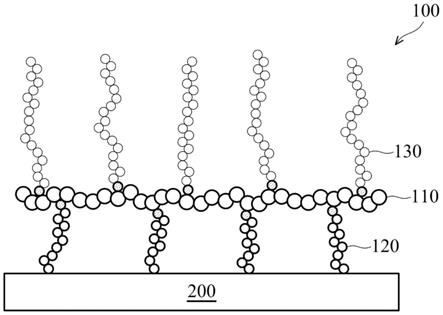
1.本揭露是关于接枝高分子,且特别是关于以接枝高分子包覆金属材料所形成的复合材料及其使用方法。
背景技术:
2.据统计,全球目前约超过三千万个病患受到严重下背部疼痛之苦,且进入高龄化社会,预期相关病患势必会更多。调研机构global market insights针对神经电刺激设备市场的报告即指出,相关产品于2017年的市场产值约是40亿美元,预估至2023年将成长至130亿美元,其中又以植入式脊髓电刺激器(spinal cord stimulation,scs)的产品市场潜力最为庞大,预估其产值可于2023年时迅速增长至超过90亿美元。
3.目前,植入式脊髓电刺激器在帕金森氏症、癫痫、疼痛、强迫症等疾病的临床治疗上皆有不错的疗效。然而,此类疗法需将神经刺激器的电极植入人体,而电极与血液及体液接触多日后往往会因蛋白质沉积吸附而提高电阻,进而导致止痛效果下降。虽然,藉由移除已沾粘蛋白质的电极再将新电极植入皮下可获得改善,但此举不仅耗费医疗成本更造成病人痛苦,并非理想的方案。
4.再者,以目前医疗产品电极表面抑制沾粘的处理技术而言,商业化涂布技术多采用表面亲水分子(peg/pvp),且处理基材多以高分子耗材为主,金属类较为罕见。并且,若表面处理(例如等离子体、臭氧、电晕等)复杂形状的金属电极,易产生不均匀以及成本高等问题。
5.因此,目前亟需针对此植入式电极的表面进行抗沾粘的改质研究。
技术实现要素:
6.本发明的目的在于提供一种能够抑制植入式电极的表面的沾粘问题的复合材料。
7.本揭露一实施例提供的接枝高分子,包括:具有多个羟基的高分子主链;保护基修饰的组胺酸,接枝至高分子主链的侧部;以及末端具有反应基的亲水性高分子,接枝至高分子主链的侧部。
8.在一些实施例中,高分子主链包括聚乙烯醇(polyvinyl alcohol,pva)、聚烯烃基二醇(polyalkylene glycol,pag)、聚醋酸乙烯酯(polyvinyl acetate,pvac)、乙烯醇
‑
醋酸乙烯酯共聚物(ethylene vinyl acetate copolymer,eva)、乙烯
‑
乙烯醇共聚物(ethylene vinyl alcohol copolymer,evoh)、透明质酸(hyaluronic acid,ha)、淀粉(starch)、纤维素(cellulose)、甲基纤维素(methyl cellulose,mc)、羟丙基甲基纤维素(hydroxypropyl methyl cellulose,hpmc)、氧化纤维素(oxycellulose)、葡聚醣(glucan)、硬葡聚醣(scleroglucan polysaccharide)、几丁质(chitin)、几丁聚醣(chitosan)、卡德兰胶(curdlan gum)、褐藻胶(algin)、角叉藻胶(carrageenan)、果胶(pectin)、阿拉伯树胶(arabic gum)、关华豆胶(guar gum)、结兰胶(gellan gum)、普路兰胶(pullulan)、软骨素(chondroitin)、肝素(heparin)、或硫酸角蛋白(keratin sulfate)。
9.在一些实施例中,高分子主链的重均分子量为500至100000。
10.在一些实施例中,末端具有反应基的亲水性高分子中,亲水性高分子包括单甲醚聚烷二醇(methoxypolyalkylene glycol)。
11.在一些实施例中,末端具有反应基的亲水性高分子中,反应基包括异氰酸酯基(isocyanate group)、羧酸基(carboxyl group)、酰卤基(acyl halide group)、或环氧基(epoxy group)。
12.在一些实施例中,末端具有反应基的亲水性高分子的重均分子量为500至20000。
13.在一些实施例中,保护基修饰的组胺酸中,保护基包括叔丁氧基羰基(tert
‑
butoxycarbonyl,boc)、苄氧羰基(carbobenzoxy,cbz)、芴甲氧羰基(fluorenylmethyloxycarbonyl,fmoc)、或乙酰基(acetyl group)。
14.在一些实施例中,高分子主链与保护基修饰的组胺酸的重量比为100:0.05至100:350,而高分子主链与末端具有反应基的亲水性高分子的重量比为100:0.1至100:1050。
15.本揭露一实施例提供的复合材料,包括:金属材料;以及接枝高分子,包覆金属材料的表面,其中接枝高分子包括:具有多个羟基的高分子主链;保护基修饰的组胺酸,接枝至高分子主链的侧部;以及末端具有反应基的亲水性高分子,接枝至高分子主链的侧部,其中,接枝高分子以保护基修饰的组胺酸吸附至金属材料的表面。
16.在一些实施例中,金属材料包括铂、金、铱、钯、或上述的合金。
17.在一些实施例中,高分子主链包括聚乙烯醇、聚烯烃基二醇、聚醋酸乙烯酯、乙烯醇
‑
醋酸乙烯酯共聚物、乙烯
‑
乙烯醇共聚物、透明质酸、淀粉、纤维素、甲基纤维素、羟丙基甲基纤维素、氧化纤维素、葡聚醣、硬葡聚醣、几丁质、几丁聚醣、卡德兰胶、褐藻胶、角叉藻胶、果胶、阿拉伯树胶、关华豆胶、结兰胶、普路兰、软骨素、肝素、或硫酸角蛋白。
18.在一些实施例中,高分子主链的重均分子量为500至100000。
19.在一些实施例中,末端具有反应基的亲水性高分子中,亲水性高分子包括单甲醚聚烷二醇。
20.在一些实施例中,末端具有反应基的亲水性高分子中,反应基包括异氰酸酯基、羧酸基、酰卤基、或环氧基。
21.在一些实施例中,末端具有反应基的亲水性高分子的重均分子量为500至20000。
22.在一些实施例中,保护基修饰的组胺酸中,保护基包括叔丁氧基羰基、苄氧羰基、或芴甲氧羰基、或乙酰基。
23.在一些实施例中,高分子主链与保护基修饰的组胺酸的重量比为100:0.05至100:350,而高分子主链与末端具有反应基的亲水性高分子的重量比为100:0.1至100:1050。
24.与现有技术相比,本发明的有益效果在于:通过将本发明的接枝高分子螯合至金属材料的表面,可有效降低蛋白质的吸附量,即具抗蛋白质吸附的效果,因此该复合材料植入生物体中可以避免沾粘问题。
附图说明
25.图1是本发明一实施例中,复合材料的示意图;
26.图2是本发明一实施例中,复合材料浸泡pbs溶液前后的ir图谱;
27.图3a、3b、3c、及3d是本发明一实施例中,复合材料浸泡pbs溶液前后的ir图谱;
28.图4a、4b、及4c是本发明一实施例中,复合材料浸泡pbs溶液前后的ir图谱;
29.其中,符号说明:
30.100接枝高分子
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
110高分子主链
31.120保护基修饰的组胺酸
ꢀꢀꢀꢀ
130亲水性高分子
32.200金属材料。
具体实施方式
33.本发明一实施例提供的接枝高分子如图1所示。在图1中,接枝高分子100包括高分子主链110。保护基修饰的组胺酸120,接枝至高分子主链110的侧部。末端具有反应基的亲水性高分子130,接枝至高分子主链110的侧部。由于高分子主链110的侧部原本具有多个羟基,保护基修饰的组胺酸120与末端具有反应基的亲水性高分子130可与高分子主链110的羟基反应,以接枝至高分子主链110的侧部。
34.可以理解的是,图1中的接枝高分子100仅为示意。举例来说,图1中保护基修饰的组胺酸120与末端具有反应基的亲水性高分子130可为规则的交错排列,或可为不规则排列,且两者的比例可非1:1。
35.在一实施例中,高分子主链110包括聚乙烯醇(polyvinyl alcohol,pva)、聚烯烃基二醇(polyalkylene glycol,pag)、聚醋酸乙烯酯(polyvinyl acetate,pvac)、乙烯醇
‑
醋酸乙烯酯共聚物(ethylene vinyl acetate copolymer,eva)、乙烯
‑
乙烯醇共聚物(ethylene vinyl alcohol copolymer,evoh)、透明质酸(hyaluronic acid,ha)、淀粉(starch)、纤维素(cellulose)、甲基纤维素(methyl cellulose,mc)、羟丙基甲基纤维素(hydroxypropyl methyl cellulose,hpmc)、氧化纤维素(oxycellulose)、葡聚醣(glucan)、硬葡聚醣(scleroglucan polysaccharide)、几丁质(chitin)、几丁聚醣(chitosan)、卡德兰胶(curdlan gum)、褐藻胶(algin)、角叉藻胶(carrageenan)、果胶(pectin)、阿拉伯树胶(arabic gum)、关华豆胶(guar gum)、结兰胶(gellan gum)、普路兰胶(pullulan)、软骨素(chondroitin)、肝素(heparin)、或硫酸角蛋白(keratin sulfate)。
36.在一实施例中,高分子主链110的重均分子量约为500至200000,例如可为约500至5000、约5000至10000、约10000至30000、约30000至50000、约50000至80000、约80000至100000、约100000至150000、约150000至200000、约1000至180000、约5000至150000、约8000至120000、约10000至160000、约20000至120000、或约30000至100000等,但不限于此。
37.在一实施例中,末端具有反应基的亲水性高分子130中,亲水性高分子130包括单甲醚聚烷二醇,例如可为单甲醚聚乙二醇(methoxypolyethylene glycol,mpeg)、单甲醚聚丙二醇(methoxypolypropylene glycol,mppg)、单甲醚聚丁二醇(methoxypolybutylene glycol,mpbg)、或其他合适的单甲醚聚烷二醇,但不限于此。在一实施例中,末端具有反应基的亲水性高分子130中,反应基包括异氰酸酯基、羧酸基、酰卤基、或环氧基。以单甲醚聚乙二醇(mpeg)为例,可取过当量的二异氰酸酯(diisocyanate)如甲苯二异氰酸酯(toluene diisocyanate,tdi)、异佛尔酮二异氰酸酯(isophorone diisocyanate,ipdi)、二苯基甲烷二异氰酸酯(methylene diphenyl diisocyanate,mdi)、二环己基甲烷二异氰酸酯(methylene
‑
bis(4
‑
cyclohexylisocyanate),hmdi)、赖氨酸二异氰酸酯(lysine diisocyanate,ldi)、或前述的类似物与mpeg反应,使mpeg末端的羟基与二异氰酸酯的一个
异氰酸酯基反应,并保留另一个异氰酸酯于亲水性高分子的末端。另一方面,可氧化mpeg末端的羟基,以形成羧酸基。上述羧酸基可与卤化亚砜(如socl2)反应,以形成酰卤基。此外,亦可取过量的卤化环氧基化合物如环氧氯丙烷与mpeg反应,使mpeg的羟基与卤化环氧基化合物的卤基进行取代反应,并保留环氧基于亲水性高分子的末端。如此一来,高分子主链110上的羟基可与亲水性高分子130的末端的反应基如异氰酸酯基、羧酸基、酰卤基、或环氧基反应,使亲水性高分子130接枝至高分子主链110的侧部。
38.在一实施例中,末端具有反应基的亲水性高分子130的重均分子量为500至20000,例如可为约500至1000、约1000至5000、约5000至8000、约8000至12000、约12000至15000、约15000至20000、约800至15000、约1200至12000、约1500至15000、或约2000至20000等,但不限于此。若亲水性高分子130的分子量过低,则可能降低亲水性高分子130的亲水性,而使抗沾粘效果下降。若亲水性高分子130的分子量过高,则可能产生立体障碍而影响接枝至主链的效果。
39.在一实施例中,保护基修饰的组胺酸120中,保护基包括叔丁氧基羰基(tert
‑
butoxycarbonyl,boc)、苄氧羰基(carbobenzyloxy,cbz)、或芴甲氧羰基(fluorenylmethoxycarbonyl,fmoc)、或乙酰基(acetyl group,ac)。举例来说,保护基可为boc。值得注意的是,若直接取组胺酸(无保护基保护)与高分子主链110进行反应,则一组胺酸的胺基可能会与另一组胺酸的羧酸基进行反应而形成聚组胺酸,而无法形成本发明实施例所需的接枝高分子100。一般而言,高分子主链110的羟基可与保护基修饰的组胺酸120的羧酸基进行酯化反应,使保护基修饰的组胺酸120接枝至高分子主链110的侧部。
40.在一实施例中,高分子主链110与保护基修饰的组胺酸120的重量比为100:0.05至100:350。若保护基修饰的组胺酸120用量过低,则接枝高分子100无法有效螯合至金属材料200。若保护基修饰的组胺酸120用量过高,则亲水性高分子130的用量可能不足。另一方面,高分子主链110与末端具有反应基的亲水性高分子130的重量比为100:0.1至100:1050。若亲水性高分子130的用量过低,则无法避免后述的复合材料在植入生物体之后的沾粘问题。若亲水性高分子130的用量过高,则保护基修饰的组胺酸120的用量可能不足。
41.本发明一实施例提供的复合材料如图1所示,包括:金属材料200;以及接枝高分子100,包覆金属材料200的表面。接枝高分子100中的保护基修饰的组胺酸120可螯合至金属材料200的表面。boc保护基修饰的组胺酸的结构如下:
[0042][0043]
在上述结构中,boc保护基修饰的组胺酸左侧的咪唑可螯合至金属材料200。举例来说,金属材料200可包括铂、金、铱、钯、或上述的合金。在一实施例中,金属材料200可为片状、棒状、块状、管状、网状、或其他合适构形,端视应用而定。不论金属材料200的构形为何,其表面均包覆接枝高分子100。
[0044]
在一些实施例中,复合材料可作为组织替代物、导管、血管通路装置、血液透析装
置、血管支架、胆道支架、或其他支撑物。另一方面,复合材料可植入生物体(如人或动物)内。举例来说,复合材料可作为电极,其连接至电线,而电线可连接至提供电源的装置如电刺激器、心脏节律器、植入式心脏去颤器、电子耳蜗、或连续式血糖侦测器。
[0045]
为让本揭露的上述内容和其他目的、特征、和优点能更明显易懂,下文特举出较佳实施例,并配合所附附图,作详细说明如下:
[0046]
【实施例】
[0047]
实施例1
‑
1:接枝高分子产物1070326 pvabp的制备
[0048]
首先,将nα
‑
(叔丁氧基羰基)
‑
l
‑
组氨酸(bochis,3.54g,13.8mmol)和4
‑
二甲氨基吡啶(dmap,1.54g,12.5mmol)加入二甲基乙酰胺(dmac,28ml)后,取1
‑
乙基
‑3‑
(3
‑
二甲基氨基丙基)碳酰二亚胺(edc,2.41g,12.5mmol)快速倒入一反应瓶中,于60℃反应3小时以活化bochis。将聚乙烯醇(pva,分子量为9,000
‑
10,000,2.2g,41.7mmol,80%水解)加入dmac(22ml)中,于80℃搅拌2小时至完全溶解成pva溶液。将活化后的bochis溶液快速加入上述pva溶液,并于90℃持续反应24小时以获得粗产物溶液。其中bochis的
‑
cooh基与pva上的
‑
oh基进行酯化反应,使bochis接枝至pva上。接着,取单甲氧基聚乙二醇琥珀酸(mpeg
‑
sa,分子量为2,000,10g,5mmol)置入双颈瓶,加入dmac(50ml)于50℃搅拌至均匀溶解,再降温至30℃,并依序加入dmap(0.513g,4.2mmol)与edc(0.805g,4.2mmol),于60℃反应3小时以活化mpeg
‑
sa。将活化完成的mpeg
‑
sa溶液加入接枝bochis的pva,反应后的粗产物溶液于90℃继续反应24小时,其中mpeg
‑
sa的
‑
cooh基与pva上的
‑
oh基进行酯化反应,使mpeg
‑
sa接枝至pva上。反应完成后将粗产物溶液使用切向流过滤法纯化(tff,mwco:10k da),纯化后的粗产物体积增加18~20倍,之后以回旋浓缩机(rotary evaporator)对纯化溶液进行浓缩直至完全干燥,得到接枝高分子固体产物1070326pvabp。经1h
‑
nmr实测,可得1070326pvabp的bochis接枝率8%,且mpeg
‑
sa接枝率5.5%。
[0049]
实施例1
‑
2:接枝高分子产物1070507pvabp的制备
[0050]
以实施例1
‑
1的方式制备pvabp,差别仅在于bochis的用量改为17.7g(69.3mmol),dmap的用量改为7.69g(63mmol),而edc的用量改为12.0g(63mmol)。至于mpeg
‑
sa与pva的用量、反应温度与时间、与纯化粗产物溶液的方法,则与实施例1
‑
1类似,于此不再赘述。经1h
‑
nmr实测,可得1070507pvabp接枝高分子的bochis接枝率25%,且mpeg
‑
sa接枝率1.5%。
[0051]
实施例1
‑
3:接枝高分子产物1070521 pvabp的制备
[0052]
以实施例1
‑
1的方式制备pvabp,差别在于bochis与mpeg
‑
sa的使用量。于配制bochis时,将其用量改为10.6g(41.6mmol),dmap的用量改为4.6g(37.8mmol),edc的用量改为7.2g(37.8mmol)。而于配制mpeg
‑
sa时,将其用量改为20g(10mmol),dmap的用量改为1.0g(8.4mmol),edc的用量改为1.6g(8.4mmol)。至于pva的用量、反应温度与时间、与纯化粗产物溶液的方法,则与实施例1
‑
1类似,于此不再赘述。经1h
‑
nmr实测,可得1070521pvabp接枝高分子的bochis接枝率18%,且mpeg
‑
sa接枝率1.0%。
[0053]
实施例1
‑
4:接枝高分子产物1070716 pvabp的制备
[0054]
以实施例1
‑
3的方式制备pvabp,差别在于接枝的顺序。先活化mpeg
‑
sa,将其接枝于pva上后,再活化bochis并接枝于pva上。其中,mpeg
‑
sa、bochis与pva的用量、反应温度与时间、与纯化粗产物溶液的方法,则与实施例1
‑
3类似,于此不再赘述。经1h
‑
nmr实测,可得1070716 pvabp接枝高分子的bochis接枝率29%,且mpeg
‑
sa接枝率3.0%。
[0055]
实施例1
‑
5:接枝高分子产物1080114 pvabp的制备
[0056]
以实施例1
‑
1的方式制备pvabp,差别在于接枝的顺序及bochis与mpeg
‑
sa的使用量。其中,先活化mpeg
‑
sa,将其接枝于pva上后,再活化bochis并接枝于pva上。于配制mpeg
‑
sa时,mpeg
‑
sa的分子量为2,100,用量改为6.35g(3.02mmol),dmap的用量改为0.31g(2.52mmol),edc的用量改为0.48g(2.52mmol)。而于配制bochis时,bochis的用量改为10.6g(41.6mmol),dmap的用量改为4.6g(37.8mmol),edc的用量改为7.2g(37.8mmol)。至于pva的用量、反应温度与时间、与纯化粗产物溶液的方法,则与实施例1
‑
1类似,于此不再赘述。经1h
‑
nmr实测,可得1080114pvabp接枝高分子的bochis接枝率28%,且mpeg
‑
sa接枝率5%。
[0057]
实施例1
‑
6:接枝高分子产物1080121 pvabp的制备
[0058]
以实施例1
‑
1的方式制备pvabp,差别在于接枝的顺序及bochis、mpeg
‑
sa与pva的使用量。其中,先活化mpeg
‑
sa,将其接枝于pva上后,再活化bochis并接枝于pva上。于配制mpeg
‑
sa时,mpeg
‑
sa的分子量为2,100,用量改为6.93g(3.3mmol),dmap的用量改为0.34g(2.75mmol),edc的用量改为0.53g(2.75mmol)。而于配制bochis时,bochis的用量改为19.3g(75.6mmol),dmap的用量改为8.4g(68.7mmol),edc的用量改为13.2g(68.7mmol)。另外,pva的用量改为4g(76.3mmol)。至于反应温度与时间、与纯化粗产物溶液的方法,则与实施例1
‑
1类似,于此不再赘述。经1h
‑
nmr实测,可得1080121 pvabp接枝高分子的bochis接枝率32%,且mpeg
‑
sa接枝率3.5%。
[0059]
实施例1
‑
7:接枝高分子产物1080325 pvabp的制备
[0060]
以实施例1
‑
1的方式制备pvabp,差别在于接枝的顺序及bochis、mpeg
‑
sa与pva的使用量。其中,先活化mpeg
‑
sa,将其接枝于pva上后,再活化bochis并接枝于pva上。于配制mpeg
‑
sa时,mpeg
‑
sa的分子量为2,100,用量改为5.39g(2.6mmol),dmap的用量改为0.26g(2.14mmol),edc的用量改为0.41g(2.14mmol)。而于配制bochis时,bochis的用量改为7.72g(30.2mmol),dmap的用量改为3.36g(27.5mmol),edc的用量改为5.27g(27.5mmol)。另外,pva的用量改为4g(76.3mmol)。至于反应温度与时间、与纯化粗产物溶液的方法,则与实施例1
‑
1类似,于此不再赘述。经1h
‑
nmr实测,可得1080325pvabp接枝高分子的bochis接枝率18%,且mpeg
‑
sa接枝率2.5%。
[0061]
实施例1
‑
8:接枝高分子产物1080422 pvabp的制备
[0062]
以实施例1
‑
1的方式制备pvabp,差别在于接枝的顺序及bochis、mpeg
‑
sa与pva的使用量。其中,先活化mpeg
‑
sa,将其接枝于pva上后,再活化bochis并接枝于pva上。于配制mpeg
‑
sa时,mpeg
‑
sa的分子量为2,100,用量改为6.93g(3.3mmol),dmap的用量改为0.34g(2.75mmol),edc的用量改为0.53g(2.75mmol)。而于配制bochis时,bochis的用量改为12.9g(50.4mmol),dmap的用量改为8.8g(45.8mmol),edc的用量改为5.6g(45.8mmol)。另外,pva的用量改为4g(76.3mmol)。至于反应温度与时间、与纯化粗产物溶液的方法,则与实施例1
‑
1类似,于此不再赘述。经1h
‑
nmr实测,可得1080422 pvabp接枝高分子的bochis接枝率23%,且mpeg
‑
sa接枝率2.8%。
[0063]
实施例1
‑
9:接枝高分子产物1080520 pvabp的制备
[0064]
以实施例1
‑
8的方式制备pvabp,差别仅在于bochis于配制前先以真空干燥进行预处理。此真空干燥预处理方式为于室温下(约20
‑
25℃)抽真空10分钟,之后回填氮气。经1h
‑
nmr实测,可得1080520 pvabp接枝高分子bochis接枝率28%、mpeg
‑
sa接枝率3.5%。
[0065]
实施例1
‑
10:接枝高分子产物1081104 pvabp的制备
[0066]
以实施例1
‑
1的方式制备pvabp,差别在于接枝的顺序及bochis、mpeg
‑
sa与pva的使用量。其中,先活化mpeg
‑
sa,将其接枝于pva上后,再活化bochis并接枝于pva上。于配制mpeg
‑
sa时,mpeg
‑
sa的分子量为2,100,用量改为3.46g(1.65mmol),dmap的用量改为0.17g(1.37mmol),edc的用量改为0.26g(1.37mmol)。而于配制bochis时,bochis的用量改为13.9g(54.6mmol),dmap的用量改为6.06g(49.6mmol),edc的用量改为9.51g(49.6mmol)。另外,pva的用量改为2g(38.2mmol)。至于反应温度与时间、与纯化粗产物溶液的方法,则与实施例1
‑
1类似,于此不再赘述。经1h
‑
nmr实测,可得1081104pvabp接枝高分子的bochis接枝率62%,且mpeg
‑
sa接枝率1%。
[0067]
实施例1
‑
11:接枝高分子产物1081118 pvabp的制备
[0068]
以实施例1
‑
1的方式制备pvabp,差别在于接枝的顺序及bochis、mpeg
‑
sa与pva的使用量。其中,先活化mpeg
‑
sa,将其接枝于pva上后,再活化bochis并接枝于pva上。于配制mpeg
‑
sa时,mpeg
‑
sa的分子量为650,用量改为6.85g(10.5mmol),dmap的用量改为1.07g(8.8mmol),edc的用量改为1.68g(8.8mmol)。而于配制bochis时,bochis的用量改为6.43g(25.2mmol),dmap的用量改为2.8g(22.9mmol),edc的用量改为4.4g(22.9mmol)。另外,pva的用量改为2g(38.2mmol)。至于反应温度与时间、与纯化粗产物溶液的方法,则与实施例1
‑
1类似,于此不再赘述。经1h
‑
nmr实测,可得1081118pvabp接枝高分子的bochis接枝率5%,且mpeg
‑
sa接枝率22%。
[0069]
实施例2
‑
1:接枝高分子产物1070521 pvabp的附着力测试
[0070]
取上述实施例1
‑
3的接枝高分子产物1070521 pvabp涂布于pt箔上,浸泡至磷酸盐缓冲盐水(pbs)中24小时,之后风干并进行百格测试(量测标准为iso2409)。结果显示其剥落面积仅为约5%至15%。由上述可知,接枝高分子可附着于pt箔的表面上,而不会因浸泡pbs而大量甚至完全剥落。
[0071]
实施例2
‑
2:接枝高分子产物1070716 pvabp的附着力测试
[0072]
取实施例1
‑
4的接枝高分子产物1070716 pvabp涂布于pt箔上,浸泡至pbs中24小时,之后风干并进行百格测试(量测标准为iso2409)。结果显示其完全无剥落。由上述可知,接枝高分子可稳定附着于pt箔的表面上,而不会因浸泡pbs而剥落。
[0073]
实施例3:蛋白质吸附抑制测试(抗沾粘试验)
[0074]
取实施例1
‑
4的接枝高分子产物1070716 pvabp涂布于pt箔上,浸泡至1%的bsa的pbs溶液中24小时后风干。另外取pt箔浸泡至1%的bsa的pbs溶液中24小时后风干。以压电石英晶体微天平(quartz crystal microbalance,qcm,美国ch instruments公司chi 400c)量测上述样品的振动频率,换算为蛋白质的吸附质量(使用7.995mhz的石英晶体时,吸附质量每增加1.34ng/0.196cm2,则振荡频率下降1hz),结果显示如表1。由表1可知,涂布有接枝高分子产物1070716pvabp的pt箔的蛋白质吸附量为40.76ng/cm2,而pt箔的蛋白质吸附量为1450ng/cm2,显见接枝高分子可有效降低蛋白质吸附量,即具抗沾粘效果。
[0075]
表1
[0076]
检测样品24小时后的蛋白质吸附量(ng/cm2)1070716 pvabp/pt箔40.76
pt箔1450
[0077]
实施例4:细胞贴附抑制测试(抗沾粘试验)
[0078]
取实施例1
‑
4的接枝高分子产物1070716 pvabp涂布于pt箔上(1x1 cm2面积大小),放入12well培养盘中。取200μl hsf细胞溶液(human skin fibroblasts,约5x 105cell/ml)均匀种植于样品表面,以达细胞密度约为1x 105cells/样品。待细胞贴附于材料上,每个well分别加入1.5ml细胞培养液(90%dmem+10%fetal bovine serum),再将培养盘移入37℃细胞培养箱进行培养。培养1
‑
4天后,各well内加入1ml neutral red solution(购自sigma的n2889),再将培养盘移入细胞培养箱(astec.co.,ltd.sca
‑
165d)培养0.5
‑
1小时。活细胞核染色后,移去neutral red solution,再以collagenase/trypsin酵素溶液取下测试样品内粘附的细胞,各取溶解后溶液200μl放置于96孔微量分析盘(96well micro
‑
plate)孔槽内。以elisa reader测量其ex/em 535/615荧光值,并依此荧光值计算样品内含有的细胞数量。
[0079]
另外重复上述步骤,差别在于样品为pt箔而无涂布接枝高分子。若将pt箔的hsf细胞吸附量设定为100%,则上述1070716 pvabp/pt箔的hsf细胞吸附量不到10%。显见本发明实施例的接枝高分子可有效降低hsf细胞吸附量,即具抗沾粘的效果。
[0080]
实施例5:阻抗(electrical impedance)测试
[0081]
取pt箔浸入电阻液(1%bsa in pbs)中,量测其阻抗(约10ω)。将实施例1
‑
4的接枝高分子产物1070716 pvabp涂布于pt箔上,并以此结构浸入电阻液(1%bsa in pbs)中,量测其阻抗(约50ω)。一般而言,阻抗小于或等于100ω即可作为电极。显示本发明实施例的接枝高分子可用于电极。
[0082]
实施例6
‑
1:pt箔于仿体的电阻变化测试
[0083]
取pt箔浸入1%牛血清蛋白(bsa)的pbs溶液中,量测其电阻(13.8ω)。将pt箔浸入1%bsa的pbs溶液中14天后,再量测其电阻(25.9ω),即电阻增加约87%。电阻增加的原因应来自于pt箔吸附牛血清蛋白,即所谓的沾粘现象。
[0084]
实施例6
‑
2:涂布有接枝高分子产物的pt箔于仿体的电阻变化测试
[0085]
将实施例1
‑
4的接枝高分子产物1070716 pvabp涂布于pt箔上,取上述结构浸入1%bsa的pbs溶液中,量测其电阻(11.6ω)。将上述结构浸入1%牛血清蛋白的pbs溶液中14天后,再量测其电阻(13.9ω),即电阻仅增加约19.8%。与实施例6
‑
1相较,接枝高分子的电阻增加率明显较低,显示其可大幅降低沾粘的问题。
[0086]
实施例7
‑
1:依美国专利us8198364所制备的接枝高分子的粘着性测试
[0087]
依美国专利us8198364所示方法,将peg接枝至pva上以形成接枝高分子。将此接枝高分子涂布于pt箔上,并以衰减式全反射(attenuated total reflectance,atr)红外线光谱仪量测其红外线光谱(infrared spectroscopy,ir),如图2所示。接着将上述结构浸入0.1m的pbs溶液两天后,再量测其ir光谱,如图2所示。由图2的比较可知,依美国专利us8198364所制备的接枝高分子,于浸泡后已量测不到接枝高分子的讯号,显见此接枝高分子对pt箔的粘着性差。
[0088]
实施例7
‑
2:本发明所制备的接枝高分子的粘着性测试
[0089]
分别将上述实施例1
‑
5的接枝高分子产物1080114 pvabp及实施例1
‑
7的接枝高分子产物1080325 pvabp涂布于pt箔上,并以衰减式全反射红外线光谱仪量测其ir光谱,结果
分别如图3a(实施例1
‑
5)及图3c(实施例1
‑
7)所示。接着将上述结构浸入0.1m的pbs溶液60天后,再量测其ir光谱,结果分别如图3b(实施例1
‑
5)及图3d(实施例1
‑
7)所示。由图3a与3b的比较以及图3c与3d的比较可知,本发明所制备的接枝高分子于长时间浸泡后的ir图谱与浸泡前类似,显见本发明实施例的接枝高分子对pt箔的粘着性极佳。
[0090]
实施例8:本发明所制备的接枝高分子的抗蛋白质吸附测试
[0091]
分别将实施例1
‑
1的接枝高分子产物1070326 pvabp涂布于pt箔上与实施例1
‑
3的接枝高分子产物1070521 pvabp涂布于另一pt箔上,接着将上述两结构分别浸泡于蛋白质溶液(50%fbs)中,并量测上述两结构所吸附的蛋白质量。同时,也将pt箔浸泡于蛋白质溶液中以进行比较,并量测pt箔所吸附的蛋白质量。上述结构所吸附的蛋白质量皆是以lowry protein assay法量测。将pt箔所吸附的蛋白质量设定为100%,则1070326 pvabp/pt箔与1070521 pvabp/pt箔的蛋白质吸附量分别为61.14%与51.57%。显示本发明实施例的接枝高分子可有效降低蛋白质的吸附量,即具抗蛋白质吸附的效果。
[0092]
实施例9:本发明所制备的接枝高分子的毒性测试
[0093]
将pt箔涂布于pet基材上作为对照组。将pt箔涂布于pet基材上,并取实施例1
‑
4的接枝高分子产物1070716 pvabp涂布于pt箔上,使pt箔夹设于pet与1070716 pvabp之间。接着,参考astm f895
‑
84(2006)所述流程并以iso10993
‑
5:2009标准测试方法测试上述两种结构的agar毒性,结果显示两者在样品放置区域之内与之外均没有细胞受到影响而死亡,显示本发明实施例的接枝高分子不具细胞毒性。
[0094]
比较例1:本发明所制备的接枝高分子与接枝他种胺基酸的接枝高分子的稳定性比较测试
[0095]
接枝高分子产物pvapp的制备
[0096]
首先,将nα
‑
(叔丁氧基羰基)
‑
l
‑
苯胺酸(boc
‑
l
‑
phenylalanine,bocphe,13.4g,50.4mmol)和4
‑
二甲氨基吡啶(dmap,5.6g,45.8mmol)加入二甲基乙酰胺(dmac,101ml)后,取1
‑
乙基
‑3‑
(3
‑
二甲基氨基丙基)碳酰二亚胺(edc,8.8g,45.8mmol)快速倒入一反应瓶中,于60℃反应3小时以活化bocphe。将聚乙烯醇(pva,分子量为9,000
‑
10,000,4g,76.3mmol,80%水解)加入dmac(40ml)中,于80℃搅拌2小时至完全溶解成pva溶液。将活化后的bocphe溶液快速加入上述pva溶液,并于90℃持续反应24小时以获得粗产物溶液。其中bocphe的
‑
cooh基与pva上的
‑
oh基进行酯化反应,使bocphe接枝至pva上。接着,取单甲氧基聚乙二醇琥珀酸(mpeg
‑
sa,分子量为2,100,6.93g,3.3mmol)置入双颈瓶,加入dmac(33ml)于50℃搅拌至均匀溶解,再降温至30℃,并依序加入dmap(0.34g,2.75mmol)与edc(0.53g,2.75mmol),于60℃反应3小时以活化mpeg
‑
sa。将活化完成的mpeg
‑
sa溶液加入接枝bocphe的pva,反应后的粗产物溶液于90℃继续反应24小时,其中mpeg
‑
sa的
‑
cooh基与pva上的
‑
oh基进行酯化反应,使mpeg
‑
sa接枝至pva上。反应完成后将粗产物溶液使用切向流过滤法纯化(tff,mwco:10k da),纯化后的粗产物体积增加18~20倍,之后以回旋浓缩机对纯化溶液进行浓缩直至完全干燥,得到接枝高分子固体产物pvapp。经1h
‑
nmr实测,可得pvapp的bocphe接枝率10%,且mpeg
‑
sa接枝率3%。将上述接枝高分子产物pvapp涂布于pt箔上,并以衰减式全反射红外线光谱仪量测其ir光谱,结果如图4a所示。接着将上述结构浸入0.1m的pbs溶液,分别量测其浸入pbs溶液1天后及2天后的ir光谱,结果如图4b及4c所示,于浸入pbs溶液1天后即测不到任何材料讯号。相较于接枝高分子产物pvapp,上述实施例7
‑
2所示
的本发明所制备的接枝高分子于长时间浸泡后的ir图谱与浸泡前类似,稳定性较佳。由比较例1可知,组氨酸以外的其他胺基酸(如苯胺酸)所形成的接枝高分子并不适用于包覆电极。
[0097]
虽然本揭露已以数个较佳实施例揭露如上,然其并非用以限定本揭露,任何所属技术领域中具有通常知识者,在不脱离本揭露的精神和范围内,当可作任意的更动与润饰,因此本揭露的保护范围当视后附的申请专利范围所界定者为准。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1