一种吩嗪衍生物类光引发剂及其制备方法和用途与流程
1.本发明属于光引发剂领域,涉及一种吩嗪衍生物类光引发剂及其制备方法和用途。
背景技术:
[0002]2‑
甲基
‑1‑
(4
‑
甲硫基苯基)
‑2‑
吗啉
‑1‑
丙酮(光引发剂907)是一种自由基(ⅰ)型光引发剂,主要用于对相应的树脂的uv聚合固化;其分子式如下所示:
[0003][0004]
目前商用的紫外发光二级管(uv
‑
led)发射的主要是单波长的不可见光,波长包括385nm、395nm、405nm等,而光引发剂907的紫外吸收波长集中在231nm、307nm,其与uv
‑
led的发射波长匹配性差,造成光吸收效率低;且光引发剂907存在使用过程中还存在迁移的风险,限制了其应用范围;
[0005]
吩嗪,别名夹二氮杂蒽,主要用于染料、医药、有机合成中间体及生化研究;现有技术公开了一种苯并吩嗪光敏剂,可用于自由基和阳离子聚合,但是其对光源要求较为苛刻,仅能在高强度氙灯照射下进行光固化反应,限制了其应用。
[0006]
因此,开发一种能与uv
‑
led具有更高的匹配度、且固化效率高,迁移率低的光引发剂仍具有重要意义。
技术实现要素:
[0007]
本发明的目的在于提供一种吩嗪衍生物类光引发剂及其制备方法和用途,所述吩嗪衍生物类光引发剂包含苯并吩嗪基团及连接在其苯环上的2
‑
甲基
‑2‑
吗啉基
‑
丙酮基团,相较于光引发剂907,其吸收波长红移,因而与uv
‑
led的发射波长具有更高的匹配度,使得光吸收效率明显提升;且本发明所述吩嗪衍生物类光引发剂中苯并吩嗪基团为富电子基团,其与2
‑
甲基
‑2‑
吗啉基
‑
丙酮基团的相互作用使得其具有更高的固化效率,且使用过程中的迁移率明显降低,使得其应用范围明显拓宽。
[0008]
为达到此发明目的,本发明采用以下技术方案:
[0009]
第一方面,本发明提供了一种吩嗪衍生物类光引发剂,所述吩嗪衍生物类光引发剂的分子式如下式(ⅰ)所示;
[0010][0011]
其中,r1、r2、r3、r4各自独立的选自h、c1~c4的烷基及c1~c4的烷氧基中的至少一种;
[0012]
本发明中,吩嗪衍生物类光引发剂采用上述结构,其中富电子基团苯并吩嗪与连接在其苯环上的2
‑
甲基
‑2‑
吗啉基
‑
丙酮基团相互作用,提升了光引发剂的吸收波长,使得所得光引发剂与uv
‑
led的匹配性明显改善,光吸收效率明显提升,同时,其还具备了高的固化效率、低的迁移率。
[0013]
优选地,所述吩嗪衍生物类光引发剂选自如下所示结构中的至少一种;
[0014][0015]
优选地,所述吩嗪衍生物类光引发剂选自如下式(ⅱ)所示;
[0016][0017]
本发明中,如式(ⅱ)所示的吩嗪衍生物类光引发剂的吸收波长与uv
‑
led具有良好
的匹配性,使得在uv
‑
led下其具有高的光吸收效率;相较于光引发剂907,其固化效率也明显提升,且具有低迁移率的特点。
[0018]
第二方面,本发明提供了如第一方面所述的吩嗪衍生物类光引发剂的制备方法,所述制备方法包括以下步骤:
[0019]
(1)将1,2
‑
萘醌、异丁酰氯及催化剂分散在溶剂中,经傅克酰基化反应,得到式(ⅲ)所示化合物;
[0020][0021]
(2)将步骤(1)得到的式(ⅲ)所示化合物与式(ⅳ)所示化合物在溶剂中混合,进行反应,得到如式(
ⅴ
)所示化合物;
[0022][0023][0024]
其中,r1、r2、r3、r4各自独立的选自h、c1~c4的烷基及c1~c4的烷氧基中的至少一种;
[0025]
(3)将步骤(2)得到的式(
ⅴ
)所示化合物与卤素单质在溶剂中反应得到式(
ⅵ
)所示化合物;
[0026][0027]
其中,x选自cl和/或br;
[0028]
(4)将步骤(3)得到的式(
ⅵ
)所示化合物与甲醇钠混合进行环氧化反应,得到式(
ⅶ
)所示化合物;
[0029][0030]
(5)将步骤(4)得到的式(
ⅶ
)所示化合物与吗啉混合进行吗啉取代反应,得到式(ⅰ)所示化合物。
[0031]
本发明中,上述制备方法的反应方程式如下所示:
[0032][0033]
其中,r1、r2、r3、r4各自独立的选自h、c1~c4的烷基及c1~c4的烷氧基中的至少一种;
[0034]
x选自br或cl。
[0035]
优选地,步骤(1)所述溶剂选自二氯乙烷、二氯甲烷及氯苯中的至少一种。
[0036]
优选地,所述催化剂选自三氯化铝。
[0037]
优选地,所述傅克酰基化反应的温度为0~35℃,例如5℃、10℃、15℃、20℃、25℃或30℃等,优选为5~15℃。
[0038]
优选地,步骤(1)中将1,2
‑
萘醌、异丁酰氯及催化剂分散在溶剂中的方法包括将1,2
‑
萘醌分散在溶剂中,之后加入异丁酰氯。
[0039]
优选地,所述加入异丁酰氯采用滴加方式。
[0040]
优选地,进行傅克酰基化反应的时间为1~24h,例如3h、5h、7h、9h、11h、13h、15h、17h、19h、21h或23h等。
[0041]
优选地,所述傅克酰基化反应还包括将反应结束后的反应液与盐酸混合水解,分液、脱溶,得到式(ⅲ)所示化合物。
[0042]
优选地,步骤(2)中溶剂选自乙醇和/或乙酸。
[0043]
此处步骤(2)中采用乙醇作为溶剂时,可加入硫酸作为助剂催化反应进行。
[0044]
优选地,步骤(2)中反应的温度为50~85℃,例如55℃、60℃、65℃、70℃、75℃或80℃等。
[0045]
优选地,步骤(2)中反应的时间为1~24h,例如3h、5h、7h、9h、11h、13h、15h、17h、
19h、21h或23h等。
[0046]
优选地,步骤(2)中反应的过程中伴随搅拌。
[0047]
优选地,步骤(2)中反应结束后还包括脱溶。
[0048]
优选地,步骤(3)所述溶剂选自二氯乙烷、二氯甲烷及氯苯中的至少一种。
[0049]
优选地,步骤(3)中反应温度为20~60℃,例如25℃、30℃、35℃、40℃、45℃、50℃或55℃等。
[0050]
优选地,步骤(3)中还包括加入助剂。
[0051]
优选地,所述助剂包括第一组分和第二组分;第一组分选自盐酸、磷酸、硫酸、甲酸及乙酸中的至少一种;所述第二组分选自双氧水和/或过氧乙酸。
[0052]
优选地,所述第一组分选自15wt%~31wt%(例如20wt%、25wt%或30wt%等)盐酸、60wt%~98wt%(65wt%、70wt%、75wt%、80wt%、85wt%、90wt%或95wt%等)硫酸及50wt%~80wt%(55wt%、60wt%、65wt%、70wt%或75wt%等)磷酸中的至少一种。
[0053]
优选地,所述第一组分与卤素单质的摩尔比为0.8~2.2:1,例如0.9:1、1.2:1、1.5:1、1.8:1或2:1等。
[0054]
优选地,所述第二组分与卤素单质的摩尔比为0.7~1.2:1,例如0.8:1、0.9:1、1:1或1.1:1等。
[0055]
在上述步骤(3)中卤素取代的反应中,加入上述助剂,其有利于促进卤素取代反应的进行,尤其适用于br的取代反应。
[0056]
优选地,步骤(3)中反应结束后还包括水洗、脱溶,得到式(
ⅵ
)所示化合物。
[0057]
优选地,步骤(4)所述将步骤(3)得到的式(
ⅵ
)所示化合物与甲醇钠混合进行环氧化反应的方法包括将式(
ⅵ
)所示化合物和甲醇钠分散在甲醇中,控温反应,升温蒸馏甲醇。
[0058]
优选地,所述控温反应的温度为25~60℃,例如30℃、35℃、40℃、45℃、50℃或55℃等。
[0059]
优选地,步骤(5)中吗啉取代反应的方法包括向步骤(4)的反应产物中加入吗啉和助剂,控温反应。
[0060]
优选地,所述助剂选自水、强酸弱碱盐水溶液及强碱弱酸盐水溶液中的至少一种。
[0061]
本发明中,吗啉取代反应过程中,加入上述助剂,其有利于开环反应的进行,进而提升吗啉取代反应的反应效率,本发明中上述助剂加入反应体系中,其能起到缓冲溶液ph的作用,进而有利于促进反应的进行。相较于传统吗啉取代反应中加入白土等助剂作为催化剂,其避免了后续催化剂的过滤工艺,降低了工艺操作的难度,同时避免细小白土进入产品中,影响产品澄清度的问题。本发明所述制备过程中采用上述助剂,其所得产品的澄清度较高,进而有利于得到纯度较高的吩嗪衍生物类光引发剂产品。
[0062]
优选地,所述助剂选自水,加入的吗啉的质量与助剂的体积之比为1:(0.06~0.11)kg/l,例如1:0.07kg/l、1:0.08kg/l、1:0.09kg/l或1:0.1kg/l等。
[0063]
本发明所述制备方法中,助剂水的加入量过低时,开环反应进行缓慢,进而使得吗啉取代反应效率低,本发明中控制水的加入量在上述范围内,其有利于促进吗啉取代反应的进行,同时避免产生过多废液。
[0064]
优选地,所述强碱弱酸盐水溶液的溶质选自碳酸钠和/或碳酸氢钠。
[0065]
优选地,所述强酸弱碱盐水溶液的溶质选自碱土金属盐,优选为氯化钙和/或氯化
镁。
[0066]
优选地,所述强碱弱酸盐水溶液的浓度为0.01~0.15mol/l,例如0.05mol/l或0.1mol/l等。
[0067]
优选地,所述强酸弱碱盐水溶液的浓度为0.001~0.01mol/l;例如0.002mol/l、0.005mol/l、0.008mol/l或0.01mol/l等。
[0068]
优选地,所述助剂选自强碱弱酸盐水溶液,加入吗啉的质量与助剂的体积之比为1:(0.002~0.02)kg/l,例如1:0.005kg/l、1:0.01kg/l或1:0.015kg/l等。
[0069]
优选地,所述助剂选自强酸弱碱盐水溶液,中加入吗啉的质量与助剂的体积之比为1:(0.002~0.02)kg/l,例如1:0.005kg/l、1:0.01kg/l或1:0.015kg/l等。
[0070]
优选地,吗啉取代反应中控温反应的温度为102~110℃,例如105℃或108℃等。
[0071]
本发明中,在吗啉取代反应中加入上述强碱弱酸盐水溶液或强酸弱碱盐水溶液,其相较于单纯加入水,其溶液加入量明显减少,且对吗啉取代反应的促进作用明显,有利于减少废液的产生。
[0072]
优选地,步骤(5)中待所述吗啉取代反应结束后,还包括减压蒸馏吗啉,之后加入非极性溶剂,水洗,分液,真空浓缩、重结晶,得到式(ⅰ)所示化合物。
[0073]
优选地,所述非极性溶剂包括甲苯。
[0074]
优选地,所述重结晶溶剂选自低分子醇,优选为甲醇。
[0075]
作为本发明优选的技术方案,所述吩嗪衍生物类光引发剂的制备方法包括以下步骤:
[0076]
(1)将1,2
‑
萘醌及催化剂分散在溶剂中,之后滴加异丁酰氯,在5~15℃条件下经傅克酰基化反应,之后与盐酸混合水解,分液,脱溶,得到式(ⅲ)所示化合物;
[0077][0078]
(2)将步骤(1)得到的式(ⅲ)所示化合物与式(ⅳ)所示化合物在溶剂中混合,搅拌作用下,50~85℃下进行反应,脱溶,得到如式(
ⅴ
)所示化合物;
[0079][0080]
其中,r1、r2、r3、r4各自独立的选自h、c1~c4的烷基及c1~c4的烷氧基中的至少一种;
[0081]
(3)将步骤(2)得到的式(
ⅴ
)所示化合物与卤素单质分散在溶剂中,20~60℃下进行反应,脱溶,得到式(
ⅵ
)所示化合物;
[0082][0083]
其中,x选自cl和/或br;
[0084]
(4)将步骤(3)得到的式(
ⅵ
)所示化合物和甲醇钠分散在甲醇中,25~60℃控温反应,升温蒸馏甲醇,得到式(
ⅶ
)所示化合物;
[0085][0086]
(5)将步骤(4)得到的式(
ⅶ
)所示化合物、吗啉及助剂混合,102~110℃下进行吗啉取代反应,减压蒸馏吗啉,之后加入非极性溶剂,水洗,分液,真空浓缩、重结晶,得到式(ⅰ)所示化合物。
[0087]
第三方面,本发明提供了如第一方面所述的吩嗪衍生物类光引发剂的用途,所述吩嗪衍生物类光引发剂用于油墨、涂料或电子材料。
[0088]
相对于现有技术,本发明具有以下有益效果:
[0089]
(1)本发明所述吩嗪衍生物类光引发剂包含富电子的苯并吩嗪基团及连接在苯环上的2
‑
甲基
‑2‑
吗啉基
‑
丙酮基团,相较于光引发剂907,其吸收波长红移,且与uv
‑
led的发射波长具有更高的匹配度,使得其光吸收效率明显提升;
[0090]
(2)本发明所述吩嗪衍生物类光引发剂相较于光引发剂907,在uv
‑
led照射下,其具有更高的光固化效率,且后续迁移率均明显降低。
附图说明
[0091]
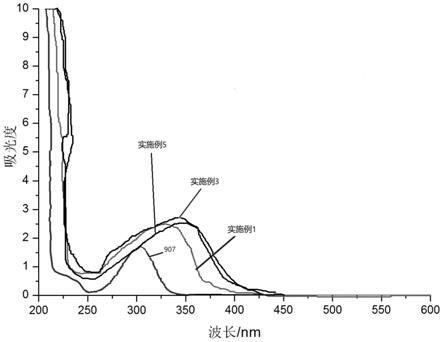
图1是本发明实施例1、3、5中吩嗪衍生物类光引发剂与光引发剂907的紫外吸收谱图;
具体实施方式
[0092]
下面通过具体实施方式来进一步说明本发明的技术方案。本领域技术人员应该明了,所述实施例仅仅是帮助理解本发明,不应视为对本发明的具体限制。
[0093]
实施例1
[0094]
本实施例中吩嗪衍生物类光引发剂的分子式如下式(ⅱ)所示:
[0095][0096]
本实施例提供了如上所示吩嗪衍生物类光引发剂的制备方法,所述方法包括以下步骤:
[0097]
(1)将1mol 1,2
‑
萘醌及1.4mol三氯化铝分散在500ml二氯乙烷中,之后滴加1.2mol异丁酰氯,在13℃条件下进行傅克酰基化反应2h,之后与800ml 2.5wt%的盐酸混合水解,分液,水洗,脱溶,得到式(ⅲ)所示化合物;
[0098][0099]
(2)将步骤(1)得到的式(ⅲ)所示化合物与邻苯二胺按照摩尔比为1:1的比例在乙酸中混合溶解,搅拌作用下,65℃下进行反应1.5h,脱溶,得到如下式所示化合物;
[0100][0101]
(3)将步骤(2)产物分散在二氯乙烷中,通入氯气,50℃下进行反应,脱溶,得到如下式所示反应产物;
[0102][0103]
(4)将步骤(3)产物和1.2mol甲醇钠分散在甲醇中,30℃下控温反应,升温蒸馏甲醇,得到如下式所示反应产物;
[0104][0105]
(5)将步骤(4)得到产物、吗啉按照摩尔比1:8的比例混合,加入水作为助剂,其中,吗啉的质量与水的体积之比为1:0.1kg/l,105℃下进行吗啉取代反应,减压蒸馏吗啉,之后加入甲苯,水洗,分液,真空浓缩、甲醇重结晶,得到式(ⅱ)所示化合物。
[0106]
本实施例中产物收率为83%,所得产物纯度为98.0%。
[0107]
本实施例所述光引发剂的质谱分析结果如下所示;
[0108]
ms:m/z[m+1]+=386.18(mw=385.46)。
[0109]
本实施例中光引发剂的h
‑
nmr分析结果如下所示;
[0110]
1h
‑
nmr(400mhz,cdcl3):δ8.18~8.12(m,3h),7.80
‑
7.60(m,5h),7.7.30~7.20(m,1h),3.65(t,4h),2.5(t,4h),1.45(s,6h)。
[0111]
实施例2
[0112]
本实施例与实施例1的区别仅在于,步骤(2)中将邻苯二胺等摩尔量的替换为4
‑
甲基邻苯二胺,分子式如下所示;
[0113][0114]
其他参数和条件与实施例1中完全相同。
[0115]
本实施例所得光引发剂中包含以下两种结构;
[0116][0117]
二者hplc含量为1:1;
[0118]
本实施例所述光引发剂的质谱分析结果如下所示;
[0119]
ms:m/z[m+1]+=400.19(mw=399.48)。
[0120]
本实施例中光引发剂的h
‑
nmr分析结果如下所示;
[0121]
1h
‑
nmr(400mhz,cdcl3):δ8.18~8.10(m,1h),7.95
‑
7.80(m,2h),7.75~7.67(m,3h),7.55(m,1h),7.30~7.20(m,1h)3.65(t,4h),2.40~2.30(m,7h),1.45(s,6h)。
[0122]
实施例3
[0123]
本实施例与实施例1的区别仅在于,步骤(2)中将邻苯二胺等摩尔量的替换为3,4
‑
二甲基邻苯二胺,分子式如下所示;
[0124][0125]
其他参数和条件与实施例1中完全相同。
[0126]
本实施例所得光引发剂中包含以下两种结构;
[0127]
二者hplc含量为1:1;
[0128]
本实施例所述光引发剂的质谱分析结果如下所示;
[0129]
ms:m/z[m+1]+=423.21(mw=413.51)。
[0130]
本实施例中光引发剂的h
‑
nmr分析结果如下所示;1h
‑
nmr(400mhz,cdcl3):δ8.08(d,1h),7.85~7.67(m,4h),7.36(d,1h),7.28(t,1h),3.68(t,4h),2.5~2.40(m,10h),1.45(s,6h)。
[0131]
实施例4
[0132]
本实施例与实施例1的区别仅在于,步骤(2)中将邻苯二胺等摩尔量的替换为3,4
‑
二氨基苯甲醚,分子式如下所示;
[0133][0134]
其他参数和条件与实施例1中完全相同。
[0135]
本实施例所得光引发剂中包含以下两种结构;
[0136]
二者hplc含量为1:1;
[0137]
本实施例所述光引发剂的质谱分析结果如下所示;
[0138]
ms:m/z[m+1]+=425.19(mw=415.48)。
[0139]
本实施例中光引发剂的h
‑
nmr分析结果如下所示;
[0140]
1h
‑
nmr(400mhz,cdcl3):δ8.10(d,1h),7.98(d,1h),7.82~7.50(m,4h),7.30~7.21(m,2h),3.83(s,3h),3.68(t,4h),2.50(t,4h),1.45(s,6h)。
[0141]
实施例5
[0142]
本实施例与实施例1的区别仅在于,步骤(2)中将邻苯二胺等摩尔量的替换为4,5
‑
二甲氧基
‑
1,2
‑
苯二胺,分子式如下所示;
[0143][0144]
其他参数和条件与实施例1中完全相同。
[0145]
本实施例所得光引发剂的结构如下所示;
[0146][0147]
本实施例所述光引发剂的质谱分析结果如下所示;
[0148]
ms:m/z[m+1]+=446.2(mw=445.51)。
[0149]
本实施例中光引发剂的h
‑
nmr分析结果如下所示;
[0150]
1h
‑
nmr(400mhz,cdcl3):δ8.07(d,1h),7.82~7.55(m,3h),7.47(s,2h),7.23(t,1h),3.80(s,6h),3.65(t,4h),2.51(t,4h),1.45(s,6h)。
[0151]
实施例6
[0152]
本实施例与实施例1的区别在于,步骤(5)中将助剂由水替换为浓度为0.15mol/l的碳酸钠水溶液,加入吗啉的质量与助剂的体积之比1:(0.018)kg/l,其他参数和条件与实
施例1中完全相同。
[0153]
本实施例中产物收率为87%,所得产物纯度为98.5%。
[0154]
实施例7
[0155]
本实施例与实施例1的区别在于,步骤(5)中将助剂由水替换为浓度为0.01mol/l的氯化钙水溶液,加入吗啉的质量与助剂的体积之比1:(0.015)kg/l,其他参数和条件与实施例1中完全相同。
[0156]
本实施例中产物收率为88%,所得产物纯度为98.5%。
[0157]
对比实施例1、6
‑
7可以看出,本发明所述光引发剂的制备过程中,步骤(5)中采用强酸弱碱盐或强碱弱酸盐作为助剂,其对步骤(5)中开环反应具有明显的促进作用,进而促进反应收率的提升。
[0158]
对比例1
[0159]
本对比例中采用光引发剂907,作为对照;
[0160]
对比例2
[0161]
本对比例光引发剂的结构如下所示;
[0162][0163]
本对比例所述光引发剂的结构质谱分析结果如下所示;
[0164]
ms:m/z[m+1]+=476.15(mw=475.45)。
[0165]
本对比例中光引发剂的h
‑
nmr分析结果如下所示;
[0166]
1h
‑
nmr(400mhz,cdcl3):δ9.26(s,2h),8.07(d,1h),7.80~7.60(m,3h),7.24(t,1h),3.65(t,4h),2.35(t,4h),1.35(s,6h)。
[0167]
对比例3
[0168]
本对比例光引发剂的结构如下所示;
[0169][0170]
本对比例所述光引发剂的结构质谱分析结果如下所示;
[0171]
ms:m/z[m+1]+=336.16(mw=335.4)。
[0172]
本对比例中光引发剂的h
‑
nmr分析结果如下所示;
[0173]
1h
‑
nmr(400mhz,cdcl3):δ8.70(d,1h),8.31(dd,1h),7.8~7.67(m,5h),3.65(t,4h),2.5(t,4h),1.45(s,6h)。
[0174]
性能测试:
[0175]
吸收波长:以实施例1、3、5和对比例1为例,得到的光引发剂紫外吸收谱图如下图1所示;可以看出,本发明所述光引发剂的吸收波长红移现象比较明显。
[0176]
固化性能测试:
[0177]
对实施例1
‑
5和对比例1
‑
3中得到的光引发剂进行光引发活性、迁移率测试,测试条件分别如下所示;
[0178]
光引发剂活性测试:
[0179]
试验配方及工作条件
[0180]
配方:
[0181][0182][0183]
工作条件与评价
[0184]
将上述混合物用涂刷器在玻璃板上涂膜,膜的固化用标准汞汽灯照射和led灯(360w,395nm,5s)分别照射。标准汞汽灯照射时,玻璃板以100米/分的速度通过灯照后,发现膜层擦拭坚固。记录表面彻底固化所需要的灯下通过次数。led灯照射时,照射时长为5s;结果如表1所示。
[0185]
对实施例1
‑
5和对比例1
‑
3中得到的光引发剂的迁移率进行测试,测试条件如下所示。
[0186]
迁移率测试的光固化组合物配方同上述光引发活性测试配方;用涂刷器在涂布上涂膜,膜的固化用标准汞汽灯照射。取(15
×
15cm2)固化后的涂布样品与直径为10cm的滤纸,放置于两块不锈钢片中间,在五吨压力下保持72小时,滤纸用thf提取加热回流三小时,用hplc测定样品和对比例的含量;
[0187]
上述测试结果如下表1所示;
[0188]
表1
[0189][0190][0191]
由图1可知,本发明所述吩嗪衍生物类光引发剂的吸收波长相较于光引发剂907明显红移,从而使得其对uv
‑
led的光吸收效率明显提升;对比实施例1
‑
5、对比例1
‑
2可知,相较于光引发剂907和硝基取代苯并吩嗪光引发剂,本发明所述吩嗪衍生物类光引发剂的固化速率明显提升,且迁移率低。
[0192]
由实施例1、实施例2
‑
5可以看出,在吩嗪基团上不与吗啉基团相邻的一侧苯环上增设烷基或烷氧基,其所得光引发剂仍具有高的光引发效率和低的迁移率。
[0193]
对比实施例1和对比例3可以看出,本发明所述光引发剂相较于对比例3中光引发剂,其固化速率明显提升,迁移率也明显降低。
[0194]
申请人声明,以上所述仅为本发明的具体实施方式,但本发明的保护范围并不局限于此,所属技术领域的技术人员应该明了,任何属于本技术领域的技术人员在本发明揭露的技术范围内,可轻易想到的变化或替换,均落在本发明的保护范围和公开范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1