金属有机配位化合物及其制备方法和应用
1.本发明属于有机光电材料技术领域,具体涉及金属有机配位化合物及其制备方法和应用。
背景技术:
2.配合物是一类具有特征化学结构的化合物,由中心原子(或离子,统称中心原子),以及围绕它的分子或离子(称为配位体/配体),完全或部分通过配位键结合,按一定的组成和空间构型所形成。研究配合物的化学分支称为配位化学。目前配位化学的研究对象和研究内容已经由简单的配合物拓展到复杂的配合物,由简单的配合物合成、结构研究拓展到更加注重配合物的应用方面的研究。
3.金属有机配位化合物是由金属离子和有机配体通过配位键、氢键等分子间作用力形成的高度有序、具有不同空间维度的配合物。金属有机配位化合物作为一种无极
‑
有机杂化材料,不但具有无机物的稳定性,同时还具有有机物的荧光量子效率高的特点,已成为最有应用前景的发光材料之一。相关技术中,金属有机配位化合物的研究主要集中在新型结构设计、新型材料制备及晶体结构分析方面。利用光谱手段能够对金属有机配位化合物进行结构及性质研究,分析确定其结构,为进一步探索新型光电材料的结构研究提供了帮助。
4.相关技术中,将配位化学引入到高分子设计中,延伸出了配位聚合物这一分支。这类聚合物是由有机配体和金属离子通过配位键、氢键或其他分子间作用力组装形成一维、二维或多维结构的聚合物。主要分为两类,一类是金属离子与高分子配体所形成的配合物;一类是低分子的配体与金属离子经配位作用而产生的高分子量化合物。配位聚合物中,高分子链部分在结构与功能上具有独特的作用,主要表现在两个方面:其一是通过对高分子链进行精确设计和制备,能够充分利用高分子链在金属离子附近形成微畴场作用,从而实现对配位聚合物的结构和稳定性、电子状态和氧化还原性质的调节;其二是配位聚合物具有高分子结构骨架,易于加工为各种形状,便于成膜增大表面积,从而提高了材料的实用性。
5.合成配位聚合物的关键在于金属有机配位化合的结构设计及制备,而相关技术中,可用于制备共轭聚合物的有机配位化合物普遍结构较复杂,成本较高,不利于大规模应用。
技术实现要素:
6.本发明旨在至少解决现有技术中存在的上述技术问题之一。为此,本发明提供了金属有机配位化合物,该金属有机配位化合物结构简单,成本低,利于大规模应用。
7.本发明还提供了上述金属有机配位化合物的制备方法。
8.本发明还提供了上述金属有机配位化合物的应用。
9.本发明的第一方面提供了金属有机配位化合物,具有以下结构:
[0010][0011]
其中r为具有环状共轭结构的基团。
[0012]
r为具有环状共轭结构的基团,可用于共聚形成导电聚合物。
[0013]
根据本发明的一些实施方式,所述r为
‑
c4h7s。
[0014]
根据本发明的一些实施方式,所述r的结构为:
[0015][0016]
r为
‑
c4h7s时,所述金属有机配位化合物为5
‑
(3
‑
噻吩甲酰胺)
‑
1,10
‑
菲罗啉(th
‑
phen)。
[0017]5‑
(3
‑
噻吩甲酰胺)
‑
1,10
‑
菲罗啉(th
‑
phen)可以用于制备配位聚合物。
[0018]
根据本发明的一些实施方式,所述r为
‑
c
13
h9。
[0019]
根据本发明的一些实施方式,所述r的结构为:
[0020][0021]
r为
‑
c
13
h9时,所述金属有机配位化合物为5
‑
(9
‑
芴甲酰胺)
‑
1,10
‑
菲罗啉(fl
‑
phen)。
[0022]5‑
(9
‑
芴甲酰胺)
‑
1,10
‑
菲罗啉(fl
‑
phen)可以用于制备配位聚合物。
[0023]
根据本发明的一些实施方式,5
‑
(3
‑
噻吩甲酰胺)
‑
1,10
‑
菲罗啉(th
‑
phen)的制备方法为:将3
‑
噻吩甲酰氯与1,10
‑
菲罗啉
‑5‑
氨基进行酰胺化反应,即得。
[0024]
根据本发明的一些实施方式,所述酰胺化反应,温度为25~45℃。
[0025]
根据本发明的一些实施方式,5
‑
(3
‑
噻吩甲酰胺)
‑
1,10
‑
菲罗啉(th
‑
phen)的制备方法为:
[0026]
s1:以氯化亚砜(socl2)为酰氯试剂,将3
‑
噻吩甲酸(tpa)酰氯化,制备3
‑
噻吩甲酰氯;
[0027]
s2:将3
‑
噻吩甲酰氯与1,10
‑
菲罗啉
‑5‑
氨基(nh2‑
phen)发生酰胺化反应,即得5
‑
(3
‑
噻吩甲酰胺)
‑
1,10
‑
菲罗啉(th
‑
phen)。
[0028]
根据本发明的一些实施方式,5
‑
(3
‑
噻吩甲酰胺)
‑
1,10
‑
菲罗啉(th
‑
phen)的合成路线为:
[0029][0030]
根据本发明的一些实施方式,步骤s1中,酰氯化反应的反应温度为75℃~85℃。
[0031]
温度过高反应速度过快,影响产物产率,过低影响进程。根据本发明的一些实施方式,步骤s1中,酰氯化反应的时间为1~4h。
[0032]
根据本发明的一些实施方式,步骤s1中,酰氯化反应在300rpm~600rpm的搅拌下进行。
[0033]
根据本发明的一些实施方式,步骤s1中,酰氯化反应的溶剂为四氯化碳(ccl4)。
[0034]
根据本发明的一些实施方式,步骤s1中,氯化亚砜(socl2)、3
‑
噻吩甲酸(tpa)与四氯化碳(ccl4)的配比为(0.01~0.02)g:(0.03~0.06)g:1ml。
[0035]
根据本发明的一些实施方式,步骤s1反应完成后,快速除去溶剂,所得固体产物真空干燥24h后备用,真空干燥温度为40℃。
[0036]
根据本发明的一些实施方式,步骤s2中,酰胺化反应的反应温度为25℃~45℃。
[0037]
根据本发明的一些实施方式,步骤s2中,酰胺化反应的时间为2~8h。
[0038]
根据本发明的一些实施方式,步骤s2中,酰胺化反应在300rpm~600rpm的搅拌下进行。
[0039]
根据本发明的一些实施方式,步骤s2中,酰胺化反应的溶剂为二氯甲烷(ch2cl2)。
[0040]
根据本发明的一些实施方式,步骤s2中,酰胺化反应添加吡啶(c5h5n)作为催化剂。
[0041]
根据本发明的一些实施方式,步骤s2中,3
‑
噻吩甲酰氯、1,10
‑
菲罗啉
‑5‑
氨基、吡啶(c5h5n)与二氯甲烷(ch2cl2)的配比为(0.004~0.008)g:(0.006~0.012)g:0.02ml:1ml。
[0042]
根据本发明的一些实施方式,5
‑
(9
‑
芴甲酰胺)
‑
1,10
‑
菲罗啉(fl
‑
phen)的制备方法为:将9
‑
芴甲酰氯与1,10
‑
菲罗啉
‑5‑
氨基发生酰胺化反应,即得。
[0043]
根据本发明的一些实施方式,所述酰胺化反应,温度为25~45℃。
[0044]
根据本发明的一些实施方式,5
‑
(9
‑
芴甲酰胺)
‑
1,10
‑
菲罗啉(fl
‑
phen)的制备方法为:
[0045]
(1):以氯化亚砜(socl2)为酰氯试剂,将9
‑
芴甲酸(flca)酰氯化,制备9
‑
芴甲酰氯;
[0046]
(2):将9
‑
芴甲酰氯与1,10
‑
菲罗啉
‑5‑
氨基(nh2‑
phen)发生酰胺化反应,即得5
‑
(9
‑
芴甲酰胺)
‑
1,10
‑
菲罗啉(fl
‑
phen)。
[0047]
根据本发明的一些实施方式,5
‑
(9
‑
芴甲酰胺)
‑
1,10
‑
菲罗啉(fl
‑
phen)的合成路线为:
[0048][0049]
根据本发明的一些实施方式,步骤(1)中,酰氯化反应的反应温度为75℃~85℃。
[0050]
温度过高反应速度过快,影响产物产率,过低影响进程。
[0051]
根据本发明的一些实施方式,步骤(1)中,酰氯化反应的时间为1~4h。
[0052]
根据本发明的一些实施方式,步骤(1)中,酰氯化反应在300rpm~600rpm的搅拌下进行。
[0053]
根据本发明的一些实施方式,步骤(1)中,酰氯化反应的溶剂为四氯化碳(ccl4)。
[0054]
根据本发明的一些实施方式,步骤(1)中,氯化亚砜(socl2)、9
‑
芴甲酸(flca)与四氯化碳(ccl4)的配比为(0.01~0.02)g:(0.02~0.06)g:1ml。
[0055]
根据本发明的一些实施方式,步骤(1)反应完成后,快速除去溶剂,所得固体产物真空干燥24h后备用,真空干燥温度为40℃。
[0056]
根据本发明的一些实施方式,步骤(2)中,酰胺化反应的反应温度为25℃~45℃。
[0057]
根据本发明的一些实施方式,步骤(2)中,酰胺化反应的时间为2~8h。
[0058]
根据本发明的一些实施方式,步骤(2)中,酰胺化反应在300rpm~600rpm的搅拌下进行。
[0059]
根据本发明的一些实施方式,步骤(2)中,酰胺化反应的溶剂为二氯甲烷(ch2cl2)。
[0060]
根据本发明的一些实施方式,步骤(2)中,酰胺化反应添加吡啶(c5h5n)作为催化剂。
[0061]
根据本发明的一些实施方式,步骤(2)中,9
‑
芴甲酰氯、1,10
‑
菲罗啉
‑5‑
氨基、吡啶(c5h5n)与二氯甲烷(ch2cl2)的配比为(0.005~0.008)g:(0.006~0.012)g:0.02ml:1ml。
[0062]
本发明结合酰氯化和酰胺化两个步骤来制备含噻吩基团或芴基团的金属有机配位化合物。具体而言,首先以氯化亚砜(socl2)为酰氯试剂,将带有特征基团的羧酸酰氯化,然后在吡啶催化下,与1,10
‑
菲罗啉
‑5‑
氨基(nh2‑
phen)发生酰胺化反应,得到带有特征基团的有机配位化合物。
[0063]
本发明的第四方面提供了上述的金属有机配位化合物在制备光电功能材料中的应用。
[0064]
本发明的金属有机配位化合物,至少具有以下有益效果:
[0065]
通过酰氯化和酰胺化两个步骤,即可制备结构简单的有机配位化合物,该有机配位化合物可用于制备导电共轭聚合物的有机配位化合物,实验方案简便,原料来源便宜;
[0066]
本发明合成金属有机配位化合物的方法,除了可以制备含有噻吩基团的配位化合物,还可以拓展至含有吡咯基团、芴基团等配位化合物的合成。
附图说明
[0067]
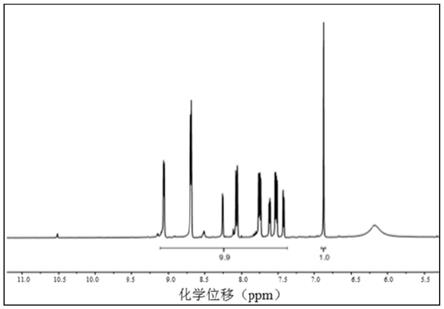
图1为5
‑
(3
‑
噻吩甲酰胺)
‑
1,10
‑
菲罗啉的核磁谱图。
[0068]
图2为5
‑
(3
‑
噻吩甲酰胺)
‑
1,10
‑
菲罗啉的紫外吸收谱图。
[0069]
图3为5
‑
(9
‑
芴甲酰胺)
‑
1,10
‑
菲罗啉(fl
‑
phen)的核磁谱图。
[0070]
图4为5
‑
(9
‑
芴甲酰胺)
‑
1,10
‑
菲罗啉(fl
‑
phen)的紫外吸收谱图。
具体实施方式
[0071]
以下是本发明的具体实施例,并结合实施例对本发明的技术方案作进一步的描述,但本发明并不限于这些实施例。
[0072]
实施例1
[0073]
本实施例制备了5
‑
(3
‑
噻吩甲酰胺)
‑
1,10
‑
菲罗啉(th
‑
phen)。具体步骤如下:
[0074]3‑
噻吩甲酰氯的合成:
[0075]
将0.32g 3
‑
噻吩甲酸,4ml二甲亚砜,20ml四氯化碳加入100ml单口烧瓶中,在300r/min的搅拌下溶解后,安装冷凝装置,然后置于75℃油浴中回流反应2h。反应结束后,将混合溶液快速旋蒸,除去溶剂和未反应的二甲亚砜,产物置于40℃下真空干燥24h得到3
‑
噻吩甲酰氯(约0.37g)。
[0076]5‑
(3
‑
噻吩甲酰胺)
‑
1,10
‑
菲罗啉(th
‑
phen)的合成:
[0077]
将0.37g 3
‑
噻吩甲酰氯,0.4875g 1,10
‑
菲罗啉
‑5‑
氨基(nh2‑
phen),2ml吡啶和100ml二氯甲烷加入250ml单口烧瓶中,在300r/min的搅拌下溶解后置于25℃油浴中反应6h。反应结束后,将混合溶液快速过滤,除去红色不溶物。旋蒸滤液,除去溶剂后将油状物溶液100ml水。过滤后,冷冻结晶,趁冷过滤,取滤液。高温旋蒸后,得到5
‑
(3
‑
噻吩甲酰胺)
‑
1,10
‑
菲罗啉。
[0078]
本实施例制备的5
‑
(3
‑
噻吩甲酰胺)
‑
1,10
‑
菲罗啉的核磁谱图如图1所示。
[0079]
本实施例制备的5
‑
(3
‑
噻吩甲酰胺)
‑
1,10
‑
菲罗啉的紫外吸收谱图如图2所示。
[0080]
实施例2
[0081]
本实施例制备了5
‑
(3
‑
噻吩甲酰胺)
‑
1,10
‑
菲罗啉(th
‑
phen)。具体步骤如下:
[0082]3‑
噻吩甲酰氯的合成:
[0083]
将0.29g 3
‑
噻吩甲酸,4ml二甲亚砜,20ml四氯化碳加入100ml单口烧瓶中,在300r/min的搅拌下溶解后,安装冷凝装置,然后置于80℃油浴中回流反应3h。反应结束后,将混合溶液快速旋蒸,除去溶剂和未反应的二甲亚砜,产物置于40℃下真空干燥24h得到3
‑
噻吩甲酰氯(约0.33g)。
[0084]5‑
(3
‑
噻吩甲酰胺)
‑
1,10
‑
菲罗啉(th
‑
phen)的合成:
[0085]
将0.33g 3
‑
噻吩甲酰氯,0.44g 1,10
‑
菲罗啉
‑5‑
氨基(nh2‑
phen),2ml吡啶和100ml二氯甲烷加入250ml单口烧瓶中,在300r/min的搅拌下溶解后置于35℃油浴中反应4h。反应结束后,将混合溶液快速过滤,除去红色不溶物。旋蒸滤液,除去溶剂后将油状物溶液100ml水。过滤后,冷冻结晶,趁冷过滤,取滤液。高温旋蒸后,得到5
‑
(3
‑
噻吩甲酰胺)
‑
1,10
‑
菲罗啉。
[0086]
实施例3
[0087]
本实施例制备了5
‑
(3
‑
噻吩甲酰胺)
‑
1,10
‑
菲罗啉(th
‑
phen)。具体步骤如下:
[0088]3‑
噻吩甲酰氯的合成:
[0089]
将0.35g 3
‑
噻吩甲酸,4ml二甲亚砜,20ml四氯化碳加入100ml单口烧瓶中,在
300r/min的搅拌下溶解后,安装冷凝装置,然后置于75℃油浴中回流反应4h。反应结束后,将混合溶液快速旋蒸,除去溶剂和未反应的二甲亚砜,产物置于40℃下真空干燥24h得到3
‑
噻吩甲酰氯(约0.37g)。
[0090]5‑
(3
‑
噻吩甲酰胺)
‑
1,10
‑
菲罗啉(th
‑
phen)的合成:
[0091]
将0.41g 3
‑
噻吩甲酰氯,0.54g 1,10
‑
菲罗啉
‑5‑
氨基(nh2‑
phen),2ml吡啶和100ml二氯甲烷加入250ml单口烧瓶中,在500r/min的搅拌下溶解后置于25℃油浴中反应8h。反应结束后,将混合溶液快速过滤,除去红色不溶物。旋蒸滤液,除去溶剂后将油状物溶液100ml水。过滤后,冷冻结晶,趁冷过滤,取滤液。高温旋蒸后,得到5
‑
(3
‑
噻吩甲酰胺)
‑
1,10
‑
菲罗啉。
[0092]
实施例4
[0093]
本实施例制备了5
‑
(3
‑
噻吩甲酰胺)
‑
1,10
‑
菲罗啉(th
‑
phen)。具体步骤如下:
[0094]3‑
噻吩甲酰氯的合成:
[0095]
将0.38g 3
‑
噻吩甲酸,4ml二甲亚砜,20ml四氯化碳加入100ml单口烧瓶中,在300r/min的搅拌下溶解后,安装冷凝装置,然后置于85℃油浴中回流反应2h。反应结束后,将混合溶液快速旋蒸,除去溶剂和未反应的二甲亚砜,产物置于40℃下真空干燥24h得到3
‑
噻吩甲酰氯(约0.344g)。
[0096]5‑
(3
‑
噻吩甲酰胺)
‑
1,10
‑
菲罗啉(th
‑
phen)的合成:
[0097]
将0.44g 3
‑
噻吩甲酰氯,0.585g 1,10
‑
菲罗啉
‑5‑
氨基(nh2‑
phen),2ml吡啶和100ml二氯甲烷加入250ml单口烧瓶中,在300r/min的搅拌下溶解后置于35℃油浴中反应6h。反应结束后,将混合溶液快速过滤,除去红色不溶物。旋蒸滤液,除去溶剂后将油状物溶液100ml水。过滤后,冷冻结晶,趁冷过滤,取滤液。高温旋蒸后,得到5
‑
(3
‑
噻吩甲酰胺)
‑
1,10
‑
菲罗啉。
[0098]
实施例5
[0099]
本实施例制备了5
‑
(3
‑
噻吩甲酰胺)
‑
1,10
‑
菲罗啉(th
‑
phen)。具体步骤如下:
[0100]3‑
噻吩甲酰氯的合成:
[0101]
将0.416g 3
‑
噻吩甲酸,4ml二甲亚砜,20ml四氯化碳加入100ml单口烧瓶中,在300r/min的搅拌下溶解后,安装冷凝装置,然后置于85℃油浴中回流反应4h。反应结束后,将混合溶液快速旋蒸,除去溶剂和未反应的二甲亚砜,产物置于40℃下真空干燥24h得到3
‑
噻吩甲酰氯(约0.481g)。
[0102]5‑
(3
‑
噻吩甲酰胺)
‑
1,10
‑
菲罗啉(th
‑
phen)的合成:
[0103]
将0.481g 3
‑
噻吩甲酰氯,0.634g 1,10
‑
菲罗啉
‑5‑
氨基(nh2‑
phen),2ml吡啶和100ml二氯甲烷加入250ml单口烧瓶中,在500r/min的搅拌下溶解后置于45℃油浴中反应4h。反应结束后,将混合溶液快速过滤,除去红色不溶物。旋蒸滤液,除去溶剂后将油状物溶液100ml水。过滤后,冷冻结晶,趁冷过滤,取滤液。高温旋蒸后,得到5
‑
(3
‑
噻吩甲酰胺)
‑
1,10
‑
菲罗啉。
[0104]
实施例6
[0105]
本实施例制备了5
‑
(9
‑
芴甲酰胺)
‑
1,10
‑
菲罗啉(fl
‑
phen)。具体步骤如下:
[0106]9‑
芴甲酰氯的合成:
[0107]
将0.525g 9
‑
芴甲酸,4ml二甲亚砜,20ml四氯化碳加入100ml单口烧瓶中,在300r/
min的搅拌下溶解后,安装冷凝装置,然后置于75℃油浴中回流反应2h。反应结束后,将混合溶液快速旋蒸,除去溶剂和未反应的二甲亚砜,产物置于40℃下真空干燥24h得到9
‑
芴甲酰氯(约0.57g)。
[0108]5‑
(9
‑
芴甲酰胺)
‑
1,10
‑
菲罗啉(fl
‑
phen)的合成:
[0109]
将0.57g 9
‑
芴甲酰氯,0.4875g 1,10
‑
菲罗啉
‑5‑
氨基(nh2‑
phen),2ml吡啶和100ml二氯甲烷加入250ml单口烧瓶中,在300r/min的搅拌下溶解后置于25℃油浴中反应6h。反应结束后,将混合溶液快速过滤,除去红色不溶物。旋蒸滤液,除去溶剂后将产物溶于100ml乙酸乙酯。过滤后,冷冻结晶,趁冷过滤,取滤液。旋蒸后,得到5
‑
(9
‑
芴甲酰胺)
‑
1,10
‑
菲罗啉。
[0110]
本实施例制备的5
‑
(9
‑
芴甲酰胺)
‑
1,10
‑
菲罗啉的核磁谱图如图3所示。
[0111]
本实施例制备的5
‑
(9
‑
芴甲酰胺)
‑
1,10
‑
菲罗啉的紫外吸收谱图如图4所示。
[0112]
实施例7
[0113]
本实施例制备了5
‑
(9
‑
芴甲酰胺)
‑
1,10
‑
菲罗啉(fl
‑
phen)。具体步骤如下:
[0114]9‑
芴甲酰氯的合成:
[0115]
将0.47g 9
‑
芴甲酸,4ml二甲亚砜,20ml四氯化碳加入100ml单口烧瓶中,在300r/min的搅拌下溶解后,安装冷凝装置,然后置于80℃油浴中回流反应3h。反应结束后,将混合溶液快速旋蒸,除去溶剂和未反应的二甲亚砜,产物置于40℃下真空干燥24h得到9
‑
芴甲酰氯(约0.51g)。
[0116]5‑
(9
‑
芴甲酰胺)
‑
1,10
‑
菲罗啉(fl
‑
phen)的合成:
[0117]
将0.51g 9
‑
芴甲酰氯,0.44g 1,10
‑
菲罗啉
‑5‑
氨基(nh2‑
phen),2ml吡啶和100ml二氯甲烷加入250ml单口烧瓶中,在300r/min的搅拌下溶解后置于35℃油浴中反应4h。反应结束后,将混合溶液快速过滤,除去红色不溶物。旋蒸滤液,除去溶剂后将产物溶于100ml乙酸乙酯。过滤后,冷冻结晶,趁冷过滤,取滤液。旋蒸后,得到5
‑
(9
‑
芴甲酰胺)
‑
1,10
‑
菲罗啉。
[0118]
实施例8
[0119]
本实施例制备了5
‑
(9
‑
芴甲酰胺)
‑
1,10
‑
菲罗啉(fl
‑
phen)。具体步骤如下:
[0120]9‑
芴甲酰氯的合成:
[0121]
将0.577g 9
‑
芴甲酸,4ml二甲亚砜,20ml四氯化碳加入100ml单口烧瓶中,在300r/min的搅拌下溶解后,安装冷凝装置,然后置于75℃油浴中回流反应4h。反应结束后,将混合溶液快速旋蒸,除去溶剂和未反应的二甲亚砜,产物置于40℃下真空干燥24h得到9
‑
芴甲酰氯(约0.57g)。
[0122]5‑
(9
‑
芴甲酰胺)
‑
1,10
‑
菲罗啉(fl
‑
phen)的合成:
[0123]
将0.627g 9
‑
芴甲酰氯,0.536g 1,10
‑
菲罗啉
‑5‑
氨基(nh2‑
phen),2ml吡啶和100ml二氯甲烷加入250ml单口烧瓶中,在500r/min的搅拌下溶解后置于25℃油浴中反应8h。反应结束后,将混合溶液快速过滤,除去红色不溶物。旋蒸滤液,除去溶剂后将产物溶于100ml乙酸乙酯。过滤后,冷冻结晶,趁冷过滤,取滤液。旋蒸后,得到5
‑
(9
‑
芴甲酰胺)
‑
1,10
‑
菲罗啉。
[0124]
实施例9
[0125]
本实施例制备了5
‑
(9
‑
芴甲酰胺)
‑
1,10
‑
菲罗啉(fl
‑
phen)。具体步骤如下:
[0126]9‑
芴甲酰氯的合成:
[0127]
将0.63g 9
‑
芴甲酸,4ml二甲亚砜,20ml四氯化碳加入100ml单口烧瓶中,在300r/min的搅拌下溶解后,安装冷凝装置,然后置于85℃油浴中回流反应2h。反应结束后,将混合溶液快速旋蒸,除去溶剂和未反应的二甲亚砜,产物置于40℃下真空干燥24h得到9
‑
芴甲酰氯(约0.68g)。
[0128]5‑
(9
‑
芴甲酰胺)
‑
1,10
‑
菲罗啉(fl
‑
phen)的合成:
[0129]
将0.68g 9
‑
芴甲酰氯,0.585g 1,10
‑
菲罗啉
‑5‑
氨基(nh2‑
phen),2ml吡啶和100ml二氯甲烷加入250ml单口烧瓶中,在500r/min的搅拌下溶解后置于35℃油浴中反应6h。反应结束后,将混合溶液快速过滤,除去红色不溶物。旋蒸滤液,除去溶剂后将产物溶于100ml乙酸乙酯。过滤后,冷冻结晶,趁冷过滤,取滤液。旋蒸后,得到5
‑
(9
‑
芴甲酰胺)
‑
1,10
‑
菲罗啉。
[0130]
实施例10
[0131]
本实施例制备了5
‑
(9
‑
芴甲酰胺)
‑
1,10
‑
菲罗啉(fl
‑
phen)。具体步骤如下:
[0132]9‑
芴甲酰氯的合成:
[0133]
将0.68g 9
‑
芴甲酸,4ml二甲亚砜,20ml四氯化碳加入100ml单口烧瓶中,在300r/min的搅拌下溶解后,安装冷凝装置,然后置于85℃油浴中回流反应4h。反应结束后,将混合溶液快速旋蒸,除去溶剂和未反应的二甲亚砜,产物置于40℃下真空干燥24h得到9
‑
芴甲酰氯(约0.74g)。
[0134]5‑
(9
‑
芴甲酰胺)
‑
1,10
‑
菲罗啉(fl
‑
phen)的合成:
[0135]
将0.74g 9
‑
芴甲酰氯,0.634g 1,10
‑
菲罗啉
‑5‑
氨基(nh2‑
phen),2ml吡啶和100ml二氯甲烷加入250ml单口烧瓶中,在500r/min的搅拌下溶解后置于45℃油浴中反应4h。反应结束后,将混合溶液快速过滤,除去红色不溶物。旋蒸滤液,除去溶剂后将产物溶于100ml乙酸乙酯。过滤后,冷冻结晶,趁冷过滤,取滤液。旋蒸后,得到5
‑
(9
‑
芴甲酰胺)
‑
1,10
‑
菲罗啉。
[0136]
上面结合实施例对本发明作了详细说明,但是本发明不限于上述实施例,在所属技术领域普通技术人员所具备的知识范围内,还可以在不脱离本发明宗旨的前提下作出各种变化。比如,5
‑
(3
‑
噻吩甲酰胺)
‑
1,10
‑
菲罗啉(th
‑
phen)与5
‑
(9
‑
芴甲酰胺)
‑
1,10
‑
菲罗啉(fl
‑
phen)各自都可以用于制备配位聚合物。
[0137]
本发明的金属有机配位化合物,可以应用于制备光电功能材料。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1