一种拉司米地坦的制备方法与流程
1.本发明属于药物合成技术领域,具体涉及一种拉司米地坦的制备方法。
背景技术:
2.拉司米地坦(lasmiditan),化学名为2,4,6-三氟-n-[6-[(1-甲基-4-哌啶基)羰基]-2-吡啶基]苯甲酰胺,是elililly研发的一种口服处方药,临床上用其琥珀酸盐,用于成人伴或不伴先兆症状偏头痛的急性治疗,于2019年10月获得美国fda批准(商品名),是20多年来fda批准的首个新一类的急性偏头痛治疗药物。拉司米地坦是一种口服、中枢神经系统渗透性、选择性、5-羟色胺1f(5-ht
1f
)激动剂,在结构上和机制上不同于目前已获批的偏头痛药物,而且不存在血管收缩活性。其化学结构式如下:
[0003][0004]
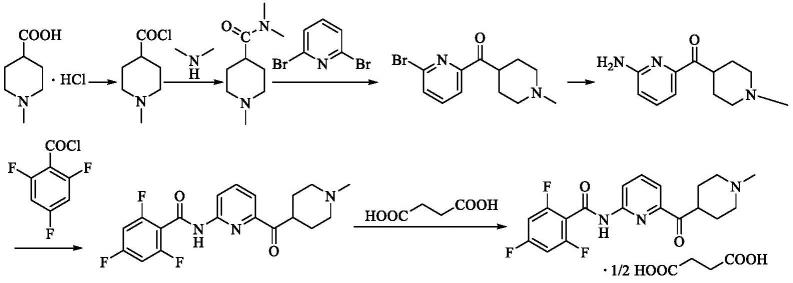
目前lasmiditan的合成工艺已经在多篇专利,如wo2011123654a1、us2019233393a1、cn03807363、us8697876b2、wo2011123654、cn110386918a中公开。但其思路与原研专利wo03084949大体一致,其公开的合成方法为:n-甲基-4-哌啶甲酸盐酸盐与dmf/草酰氯反应得到n-甲基-4-哌啶酰氯的盐酸盐,然后滴加二甲胺的thf溶液和三乙胺,经后处理得到n,n'-二甲基-n-甲基哌啶-4-甲酰胺;然后与2,6-二溴吡啶与正丁基锂在-70℃左右反应得到2-溴-6-(1-甲基哌啶-4-基酰基)-吡啶;之后在密封的高压釜中,乙二醇作溶剂高温加压与氨气反应20h后得到2-氨基-6-(1-甲基哌啶-4-基酰基)-吡啶的盐酸盐,然后调碱后游离得到2-氨基-6-(1-甲基哌啶-4-基酰基)-吡啶;之后氮气保护下,2-氨基-6-(1-甲基哌啶-4-基酰基)-吡啶与2,4,6-三氟苯甲酰氯在无水thf中反应得到2,4,6-三氟-n-[6-(l-甲基-哌啶-4-基酰基)-吡啶-2-基]-苯甲酰胺;最后,在丙酮中与琥珀酸成盐得到目标产品。
[0005][0006]
但该方法存在以下不足:
①
制备2-溴-6-(1-甲基哌啶-4-基酰基)-吡啶时,需要用到对水敏感,反应条件苛刻的正丁基锂,不利于工业化放大生产;
②
氨化制备2-氨基-6-(1-甲基哌啶-4-基酰基)-吡啶时,需要用到氨气加压约50psi(345kpa)反应,对设备要求高;并且2-氨基-6-(1-甲基哌啶-4-基酰基)-吡啶需反复调酸调碱进行纯化,不但操作复杂而且
收率(63%)较低;
③
制备拉司米地坦时,需要氮气保护,并且所用溶剂thf需要严格无水处理,同时后处理也需要反复调酸调碱进行提纯,操作繁琐。
[0007]
综上,目前拉司米地坦的制备方法在工艺安全、操作繁琐,收率不高,生产成本较高等方面存在许多不足,因此,研究寻找一条反应条件温和,操作过程简便,产品收率高、纯度高的适合工业化生产拉司米地坦的反应路线仍是目前需要解决的问题。
技术实现要素:
[0008]
针对目前现有拉司米地坦制备技术存在的问题,本发明提供了一种新的拉司米地坦的制备方法。
[0009]
本发明的具体技术方案如下:
[0010]
通过化合物sm-1和化合物sm-2反应得到化合物i,具体路线如下:
[0011][0012]
一种拉司米地坦的制备方法,具体包括以下步骤:
[0013]
惰性气体保护下,将催化剂、配体、碱和溶剂a加入密闭装置中,控温反应,反应完毕,反应液降至室温,加入化合物sm-1、化合物sm-2,升温反应,反应结束,经后处理制得化合物i。
[0014]
优选地,所述的催化剂为三苯基膦氯化铑、二聚醋酸铑、二氯(五甲基环戊二烯基)合铑(iii)二聚体、二羰基乙酰丙酮铑(i)、(1,5-环辛二烯)氯铑(i)二聚体中的一种或其组合,进一步优选三苯基膦氯化铑。
[0015]
优选地,所述的配体为1,10-菲罗啉、2,2'-联吡啶、3,8-二(噻吩-2-基)-1,10-菲罗啉、2,2'-联吡啶-4,4'-二甲醛、3-溴-1,10-菲罗啉的一种或其组合,进一步优选为1,10-菲罗啉。
[0016]
优选地,所述的碱为碳酸钾、碳酸氢钠、叔丁醇钠、叔丁醇钾、氢化钠中的一种或其组合,进一步优选为叔丁醇钠。
[0017]
优选地,所述的溶剂a为甲苯、二甲苯、n,n-二甲基甲酰胺、n-甲基吡咯烷酮的一种或其组合,进一步优选为甲苯。
[0018]
优选的,所述控温反应与升温反应,可将密封装置置于温度为100~120℃加热设备中,所述加热装置可选用油浴加热,电热套,蒸汽加热,电炉等加热设备;所述密封设备可选用密封玻璃管,密封性能好的不锈钢反应釜,密封schlenk装置等设备,本发明优选schlenk装置进行验证。
[0019]
优选地,所述的sm-1与sm-2、催化剂、配体、碱的投料摩尔比为1:1.05~1.3:0.06~0.10:0.06~0.10:0.1~0.5,进一步优选为1:1.1:0.08:0.08:0.3。
[0020]
优选地,所述的控温反应温度为100~120℃。
[0021]
优选地,所述的升温反应温度为100~120℃。
[0022]
优选地,所述的后处理步骤为:将反应液过滤,滤液减压浓缩至干后,用盐酸溶解,过滤,滤液用溶剂b洗涤,氢氧化钠溶液调节ph,溶剂c萃取,合并有机相,纯化水洗涤,无水
硫酸钠干燥,过滤,所得滤液减压浓缩至干,得化合物i。
[0023]
优选地,所述的盐酸溶液浓度为1~5mol/l。
[0024]
优选地,所述的溶剂b为乙酸乙酯、二氯甲烷、氯仿、甲基叔丁基醚中的一种或其组合,进一步优选为二氯甲烷。
[0025]
优选地,所述的氢氧化钠溶液浓度为4~7.5mol/l。
[0026]
优选地,所述的调节ph范围为12~14,优选ph为13。
[0027]
优选地,所述的溶剂c为乙酸乙酯、二氯甲烷、氯仿、甲基叔丁基醚中的一种或其组合,进一步优选为甲基叔丁基醚。
[0028]
本发明中,所述的惰性气体通常选择氮气、氩气,进一步优选为氩气。
[0029]
本发明取得的有益效果:
[0030]
1.本发明提供了一种新的拉司米地坦的制备方法。
[0031]
2.本发明制备工艺路线简便,所得化合物纯度高,收率高,适合工业化生产。
具体实施方式
[0032]
下面通过实施例来进一步说明本发明,应该正确理解的是:本发明的实施例仅仅是用于说明本发明,而不是对本发明的限制,所以,在本发明的方法前提下对本发明的简单改进均属于本发明要求保护的范围。
[0033]
对本发明得到的拉司米地坦化合物结构确证如下:
[0034][0035]
esi-hrms(m/z):378.1441[m+h]
+
;
[0036]1hnmr(400mhz,dmso-d6)δ:8.16(s,1h),7.91~7.78(m,1h),7.66~7.52(m,1h),7.50~7.39(m,1h),6.84~6.66(m,2h),2.98~2.83(m,1h),2.79~2.61(m,2h),2.28(s,3h),2.25~2.11(m,2h),1.99~1.87(m,2h),1.75~1.58(m,2h);
[0037]
13
cnmr(100mhz,dmso-d6)δ199.62,165.31,162.59,162.00,160.34,155.53,151.19,144.25,125.64,119.18,109.24,101.16,101.16,53.77,46.05,43.90,28.03;
[0038]
本发明采用hplc测定拉司米地坦的纯度,色谱条件如下:
[0039]
色谱柱:welchultimatexb-c
18
(4.6mm
×
250mm,5μm)或效能相当的色谱柱;
[0040]
流动相:流动相a:0.02mol/l磷酸二氢铵+2.1ml三乙胺,用磷酸调节ph至7.0,流动相b:乙腈,梯度洗脱(0min:a85%,25min:a65%,35min:50%,60min:85%);
[0041]
柱温:35℃;
[0042]
检测波长:223nm;
[0043]
流速:1.0ml/min;
[0044]
进样量:10μl;
[0045]
其中拉司米地坦的保留时间约在26.3min左右。
[0046]
以下各实施例中,未详细描述的各种过程和方法是本领域中公知的常规方法。
[0047]
实施例1
[0048]
氩气保护,将三苯基膦氯化铑(7.61g,8.0mmol)、1,10-菲罗啉(1.44g,8.0mmol)、叔丁醇钠(2.88g,0.03mol)、甲苯(250ml)加入schlenk装置中,控温105~110℃反应,反应完毕,反应液降至室温,加入sm-1(21.93g,0.1mol)、sm-2(19.61g,0.11mol),继续控温105~110℃反应,反应完毕,将反应液过滤,滤液减压浓缩至干后,用盐酸(2mol/l,200ml)溶解,过滤,滤液用二氯甲烷(50ml
×
2)洗涤,氢氧化钠(5mol/l)溶液调节ph至约13,甲基叔丁基醚(50ml
×
3)萃取,合并有机相,纯化水(50ml
×
2)洗涤,无水硫酸钠干燥,过滤,所得滤液减压浓缩至干,化合物i(36.49g),收率96.5%,纯度99.92%。
[0049]
实施例2
[0050]
氩气保护,将二聚醋酸铑(2.65g,6.0mmol)、2,2'-联吡啶(0.94g,6.0mmol)、叔丁醇钾(3.36g,0.03mol)、n-甲基吡咯烷酮(250ml)加入schlenk装置中,控温105~110℃,反应完毕,反应液降至室温,加入sm-1(21.93g,0.1mol)、sm-2(17.02g,0.105mol),继续控温105~110℃反应,反应完毕,过滤,滤液减压浓缩至干后,用盐酸(1mol/l,300ml)溶解,过滤,滤液二氯甲烷(70ml
×
2)洗涤,氢氧化钠(7.5mol/l)溶液调节ph至约13,甲基叔丁基醚(40ml
×
3)萃取,合并有机相,纯化水(40ml
×
2)洗涤,无水硫酸钠干燥,过滤,所得滤液减压浓缩至干,得化合物i(35.66g),收率94.3%,纯度99.85%。
[0051]
实施例3
[0052]
氩气保护,将二氯(五甲基环戊二烯基)合铑(iii)二聚体(4.95g,8.0mmol)、3,8-二(噻吩-2-基)-1,10-菲罗啉(2.76g,8.0mmol)、碳酸钾(4.14g,0.03mol)、n,n-二甲基甲酰胺(250ml)加入schlenk装置中,控温105~110℃,反应完毕,反应液降至室温,加入sm-1(21.93g,0.1mol)、sm-2(16.21g,0.1mol),继续控温105~110℃反应,反应完毕后,过滤,滤液减压浓缩至干后,用盐酸(2mol/l,200ml)溶解,过滤,滤液乙酸乙酯(50ml
×
2)洗涤,氢氧化钠(5mol/l)溶液调节ph至约12,甲基叔丁基醚(50ml
×
3)萃取,合并有机相,纯化水(50ml
×
2)洗涤,无水硫酸钠干燥,过滤,所得滤液减压浓缩至干,得化合物i(34.83g),收率92.1%,纯度99.83%。
[0053]
实施例4
[0054]
氩气保护,将二羰基乙酰丙酮铑(i)(2.07g,8.0mmol)、2,2'-联吡啶-4,4'-二甲醛1.69g,8.0mmol)、氢化钠(2.24g,0.03mol)、甲苯(250ml)加入schlenk装置中,控温105~110℃,反应完毕,反应液降至室温,加入sm-1(21.93g,0.1mol)、sm-2(21.07g,0.13mol),继续控温105~110℃反应,反应完毕后,过滤,滤液减压浓缩至干后,用盐酸(5mol/l,100ml)溶解,过滤,滤液二氯甲烷(30ml
×
2)洗涤,氢氧化钠(4mol/l)溶液调节ph至约13,甲基叔丁基醚(30ml
×
3)萃取,合并有机相,纯化水(30ml
×
2)洗涤,无水硫酸钠干燥,过滤,所得滤液减压浓缩至干,得化合物i(35.74g),收率94.5%,纯度99.79%。
[0055]
实施例5
[0056]
氩气保护,将(1,5-环辛二烯)氯铑(i)二聚体(3.95g,8.0mmol)、3-溴-1,10-菲罗啉(2.07g,8.0mmol)、碳酸氢钠(2.52g,0.03mol)、二甲苯(250ml)加入schlenk装置中,控温105~110℃,反应完毕,反应液降至室温,加入sm-1(21.93g,0.1mol)、sm-2(22.70g,0.14mol),继续控温105~110℃反应,反应完毕后,过滤,滤液减压浓缩至干后,用盐酸(1mol/l,200ml)溶解,过滤,滤液氯仿(50ml
×
2)洗涤,氢氧化钠(5mol/l)溶液调节ph至约13,乙酸乙酯(50ml
×
3)萃取,合并有机相,纯化水(50ml
×
2)洗涤,无水硫酸钠干燥,过滤,
所得滤液减压浓缩至干,得化合物i(34.64g),收率91.6%,纯度99.78%。
[0057]
实施例6
[0058]
氩气保护,将三苯基膦氯化铑(5.56g,6.0mmol)、1,10-菲罗啉(1.08g,6.0mmol)、叔丁醇钠(2.88g,0.03mol)、n-甲基吡咯烷酮(250ml)加入schlenk装置中,控温105~110℃,反应完毕,反应液降至室温,加入sm-1(21.93g,0.1mol)、sm-2(19.61g,0.11mol),继续控温105~110℃反应,反应完毕后,过滤,滤液减压浓缩至干后,用盐酸(2mol/l,200ml)溶解,过滤,滤液甲基叔丁基醚(50ml
×
2)洗涤,氢氧化钠(5mol/l)溶液调节ph至约13,二氯甲烷(50ml
×
3)萃取,合并有机相,纯化水(50ml
×
2)洗涤,无水硫酸钠干燥,过滤,所得滤液减压浓缩至干,得化合物i(34.98g),收率92.5%,纯度99.79%。
[0059]
实施例7
[0060]
氩气保护,将三苯基膦氯化铑(4.63g,5.0mmol)、1,10-菲罗啉(0.9g,5.0mmol)、叔丁醇钠(2.88g,0.03mol)、n,n-二甲基甲酰胺(250ml)加入schlenk装置中,控温105~110℃,反应完毕,反应液降至室温,加入sm-1(21.93g,0.1mol)、sm-2(19.61g,0.11mol),继续控温105~110℃反应,反应完毕后,过滤,滤液减压浓缩至干后,用盐酸(2mol/l,200ml)溶解,过滤,滤液乙酸乙酯(50ml
×
2)洗涤,氢氧化钠(5mol/l)溶液调节ph至约13,氯仿(50ml
×
3)萃取,合并有机相,纯化水(50ml
×
2)洗涤,无水硫酸钠干燥,过滤,所得滤液减压浓缩至干,得化合物i(34.34g),收率90.8%,纯度99.77%。
[0061]
实施例8
[0062]
氩气保护,将三苯基膦氯化铑(9.25g,10.0mmol)、1,10-菲罗啉(1.8g,10.0mmol)、叔丁醇钠(2.88g,0.03mol)、二甲苯(250ml)加入schlenk装置中,控温105~110℃,反应完毕,反应液降至室温,加入sm-1(21.93g,0.1mol)、sm-2(19.61g,0.11mol),继续控温105~110℃反应,反应完毕后,过滤,滤液减压浓缩至干后,用盐酸(2mol/l,200ml)溶解,过滤,滤液二氯甲烷(50ml
×
2)洗涤,氢氧化钠(5mol/l)溶液调节ph至约14,甲基叔丁基醚(50ml
×
3)萃取,合并有机相,纯化水(50ml
×
2)洗涤,无水硫酸钠干燥,过滤,所得滤液减压浓缩至干,得化合物i(35.09g),收率92.8%,纯度99.73%。
[0063]
实施例9
[0064]
氩气保护,将三苯基膦氯化铑(9.16g,11.0mmol)、1,10-菲罗啉(1.78g,11.0mmol)、叔丁醇钠(2.88g,0.03mol)、二甲苯(250ml)加入schlenk装置中,控温95~100℃,反应完毕,反应液降至室温,加入sm-1(21.93g,0.1mol)、sm-2(19.61g,0.11mol),继续控温105~110℃反应,反应完毕后,过滤,滤液减压浓缩至干后,用盐酸(2mol/l,200ml)溶解,过滤,滤液二氯甲烷(50ml
×
2)洗涤,氢氧化钠(5mol/l)溶液调节ph至约14,二氯甲烷(50ml
×
3)萃取,合并有机相,纯化水(50ml
×
2)洗涤,无水硫酸钠干燥,过滤,所得滤液减压浓缩至干,得化合物i(34.30g),收率90.7%,纯度98.73%。
[0065]
实施例10
[0066]
氩气保护,将三苯基膦氯化铑(5.71g,6.0mmol)、1,10-菲罗啉(1.08g,6.0mmol)、叔丁醇钠(0.96g,0.01mol)、n,n-二甲基甲酰胺(250ml)加入schlenk装置中,控温100~105℃,反应完毕,反应液降至室温,加入sm-1(21.93g,0.1mol)、sm-2(19.61g,0.11mol),继续控温105~110℃反应,反应完毕后,过滤,滤液减压浓缩至干后,用盐酸(2mol/l,200ml)溶解,过滤,滤液甲基叔丁基醚(50ml
×
2)洗涤,氢氧化钠(5mol/l)溶液调节ph至约13,甲基叔
丁基醚(50ml
×
3)萃取,合并有机相,纯化水(50ml
×
2)洗涤,无水硫酸钠干燥,过滤,所得滤液减压浓缩至干,得化合物i(35.47g),收率93.8%,纯度99.74%。
[0067]
实施例11
[0068]
氮气保护,将三苯基膦氯化铑(5.71g,6.0mmol)、1,10-菲罗啉(1.08g,6.0mmol)、叔丁醇钠(4.8g,0.05mol)、n,n-二甲基甲酰胺(250ml)加入schlenk装置中,控温110~120℃,反应完毕,反应液降至室温,加入sm-1(21.93g,0.1mol)、sm-2(19.61g,0.11mol),继续控温105~110℃反应,反应完毕后,过滤,滤液减压浓缩至干后,用盐酸(2mol/l,200ml)溶解,过滤,滤液二氯甲烷(50ml
×
2)洗涤,氢氧化钠(5mol/l)溶液调节ph至约13,甲基叔丁基醚(50ml
×
3)萃取,合并有机相,纯化水(50ml
×
2)洗涤,无水硫酸钠干燥,过滤,所得滤液减压浓缩至干,得化合物i(35.43g),收率93.7%,纯度99.72%。
[0069]
实施例12
[0070]
氮气保护,将三苯基膦氯化铑(5.71g,6.0mmol)、1,10-菲罗啉(1.08g,6.0mmol)、叔丁醇钠(0.48g,0.005mol)、二甲苯(250ml)加入schlenk装置中,控温120~125℃,反应完毕,反应液降至室温,加入sm-1(21.93g,0.1mol)、sm-2(19.61g,0.11mol),继续控温105~110℃反应,反应完毕后,过滤,滤液减压浓缩至干后,用盐酸(2mol/l,200ml)溶解,过滤,滤液二氯甲烷(50ml
×
2)洗涤,氢氧化钠(5mol/l)溶液调节ph至约13,甲基叔丁基醚(50ml
×
3)萃取,合并有机相,纯化水(50ml
×
2)洗涤,无水硫酸钠干燥,过滤,所得滤液减压浓缩至干,得化合物i(34.30g),收率90.7%,纯度99.70%。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1