倍他米松磷酸酯二水合物及其制备方法和应用与流程
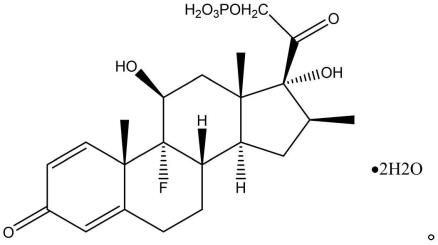
1.本发明涉及医药技术领域,尤其涉及一种倍他米松磷酸酯二水合物及其制备方法和应用。
背景技术:
2.倍他米松磷酸酯,英文名称:betamethasone phosphate,其结构式如下:
[0003][0004]
倍他米松磷酸酯是倍他米松系列药物制备的重要原料,其成盐后得到的倍他米松磷酸钠具有抗炎、抗过敏、抑制免疫、增强应激反应、抗毒素和抗休克等药理作用;可制成快速起效的注射剂型。
[0005]
国家对制备此类药物制剂有较高的质量要求。例如,对于倍他米松磷酸钠,中国药典2015版不仅规定了有关物质的限度(单个杂质含量不大于1%,总杂质含量不大于3%),而且新增规定了溶液澄清度与颜色,即:将原料药用新沸过的冷水溶解后,溶液应澄清无色,如显色,与黄色或橙黄色2号标准比色液比较,不得更深。因此倍他米松磷酸酯作为倍他米松磷酸钠的上游产品,控制其有关物质对下游制剂产品的质量具有重要意义。
[0006]
目前关于倍他米松磷酸酯纯化的报道不多,具体如下:
[0007]
cn106432390a中以倍他米松磷酸酯为原料,先将其溶于碱溶液,再加入酸溶液酸化,得到纯化物。该方法在纯化过程中使用了大量的强酸、强碱,操作繁琐,不利于工业化。
[0008]
cn104744543a中以倍他米松磷酸钠为原料,在有机溶剂-水混合体系中加入酸溶液得到纯化后的倍他米松磷酸酯,有机溶剂使用酯类溶剂、低级醚类溶剂、芳烃类溶剂、环烷烃类溶剂或氯代烃类溶剂等与水不互溶的有机溶剂。该方法在纯化过程中使用了大量的强酸,操作繁琐,不利于工业化。
[0009]
文献(倍他米松磷酸钠合成新工艺[j].医药工业,1983(09):3-4.)以倍他米松磷酸钠为原料,将其用醋酸哌嗪盐制成倍他米松磷酸哌嗪盐,然后用732h型阳离子交换树脂处理得到倍他米松磷酸酯,未对其晶型及纯度进行分析。该方法操作繁琐,成本较高,不利于工业化。
[0010]
cn110964075a中将倍他米松磷酸酯在混合有机溶剂(醚类和烷烃混合溶剂)中重结晶得到纯化物。该方法得到的纯化物纯度在95.9%-99.9%,质量不稳定。
[0011]
上述文献中报道的方法得到的倍他米松磷酸酯经x射线粉末衍射测定,均为无水物,其结晶性差,易包夹一定量的杂质,不利于作为高质量的原料参与下游产品的生产。
[0012]
有鉴于此,特提出本发明。
技术实现要素:
[0013]
本发明的主要目的在于提供一种倍他米松磷酸酯二水合物及其制备方法和应用,以期至少部分地解决上述技术问题中的至少之一。
[0014]
作为本发明的第一个方面,本发明提供了一种倍他米松磷酸酯二水合物,结构式如下:
[0015][0016]
进一步的,所述倍他米松磷酸酯二水合物的单晶属于正交晶系,空间群为p212121,晶胞参数:晶胞参数:α=90
°
,β=90
°
,γ=90
°
,晶胞体积胞体积晶胞内分子数z=4,单位晶格的独立区域内含有1个分子。
[0017]
进一步的,其x射线粉末衍射在衍射角2θ=11.5
°±
0.2
°
、15.1
°±
0.2
°
、16.2
°±
0.2
°
、19.1
°±
0.2
°
、21.7
°±
0.2
°
处有特征峰。
[0018]
进一步的,其x射线粉末衍射在衍射角2θ=11.5
°±
0.2
°
、13.8
°±
0.2
°
、14.2
°±
0.2
°
、14.8
°±
0.2
°
、15.1
°±
0.2
°
、16.2
°±
0.2
°
、17.8
°±
0.2
°
、19.1
°±
0.2
°
、21.7
°±
0.2
°
、27.7
°±
0.2
°
有特征峰。
[0019]
需要说明的是,x射线粉末衍射特征峰的衍射强度随着晶体制备技术、样品安装方法和测量仪器的不同可以有微量变化,也应该在本发明的保护范围之内。此外,仪器的差异和其它因素可能影响衍射2θ值,所以上述有特征峰的衍射角2θ值可以在现有值
±
0.2
°
内变化。
[0020]
作为本发明的第二个方面,本发明还提供了一种倍他米松磷酸酯二水合物的制备方法,包括如下步骤:将倍他米松磷酸酯溶于与水互溶的有机溶剂和水的混合溶剂中,加热待溶液澄清后,降温析晶,得到倍他米松磷酸酯二水合物。
[0021]
进一步的,所述与水互溶的有机溶剂选自低级醇类溶剂、丙酮、四氢呋喃或乙腈中的一种或几种。
[0022]
进一步的,所述低级醇类溶剂选自甲醇、乙醇或异丙醇中的一种或几种。
[0023]
进一步的,所述混合溶剂中与水互溶的有机溶剂与水的体积比为1:(1-6)。
[0024]
在本发明中,与水互溶的有机溶剂与水的典型但非限制性的体积比例可以为1:1、1:2、1:3、1:4、1:5或1:6。
[0025]
进一步的,所述倍他米松磷酸酯与混合溶剂的质量体积比为1:(20-50)g/ml。
[0026]
在本发明中,倍他米松磷酸酯与混合溶剂的典型但非限制性的质量体积比例如可以为1:20g/ml、1:22.5g/ml、1:25g/ml、1:27.5g/ml、1:30g/ml、1:32.5g/ml、1:35g/ml、1:37.5g/ml、1:40g/ml、1:42.5g/ml、1:45g/ml、1:47.5g/ml或1:50g/ml。
[0027]
进一步的,以(5-10)℃/h的速率降温至-5-10℃,保温0.5-2h,得到倍他米松磷酸酯二水合物。
[0028]
在本发明中,析晶过程中典型但非限制性的降温速率例如可以为5℃/h、6℃/h、7℃/h、8℃/h、9℃/h、10℃/h;析晶过程中典型但非限制性的温度例如可以为-5℃、-4℃、-3℃、-2℃、-1℃、0℃、1℃、2℃、3℃、4℃、5℃、6℃、7℃、8℃、9℃、10℃;析晶过程中典型但非限制性的保温时间为0.5h、0.6h、0.7h、0.8h、0.9h、1h、1.1h、1.2h、1.3h、1.4h、1.5h、1.6h、1.7h、1.8h、1.9h或2h。
[0029]
作为本发明的第三个方面,本发明还提供了倍他米松磷酸酯二水合物单晶的制备方法,包括如下步骤:
[0030]
将倍他米松磷酸酯溶于与水互溶的有机溶剂和水的混合溶剂中,待溶液澄清后,过滤至容器中,静置挥发,得到倍他米松磷酸酯二水合物单晶;
[0031]
或,将倍他米松磷酸酯溶于与水互溶的有机溶剂和水的混合溶剂中,待溶液澄清后,过滤至容器中,置于弱极性溶剂环境中气相扩散,得到倍他米松磷酸酯二水合物单晶。
[0032]
进一步的,所述与水互溶的有机溶剂选自低级醇类溶剂、丙酮、四氢呋喃或乙腈中的一种或几种。
[0033]
进一步的,所述c6-8烷烃类溶剂选自正己烷或环己烷。
[0034]
进一步的,所述低剂醚类溶剂选自乙醚、甲基叔丁基醚或异丙醚。
[0035]
进一步的,所述混合溶剂中与水互溶的有机溶剂与水的体积比为(1-8):1。
[0036]
在本发明中,与水互溶的有机溶剂与水的典型但非限制性的体积比例如可以为1:1、2:1、3:1、4:1、5:1、6:1、7:1或8:1。
[0037]
进一步的,所述倍他米松磷酸酯与混合溶剂的质量体积比为1:(5-30)g/ml。。
[0038]
在本发明中,倍他米松磷酸酯与混合溶剂的典型但非限制性的质量体积比例如可以为1:5g/ml、1:6g/ml、1:7g/ml、1:8g/ml、1:9g/ml、1:10g/ml、1:11g/ml、1:12g/ml、1:13g/ml、1:14g/ml、1:15g/ml、1:16g/ml、1:17g/ml、1:18g/ml、1:19g/ml、1:20g/ml、1:21g/ml、1:22g/ml、1:23g/ml、1:24g/ml、1:25g/ml、1:26g/ml、1:27g/ml、1:28g/ml、1:29g/ml或1:30g/ml。
[0039]
作为本发明的第四个方面,本发明还提供了倍他米松磷酸酯二水合物或上述制备方法制备得到的倍他米松磷酸酯二水合物在制备倍他米松磷酸盐中的应用。
[0040]
进一步的,所述倍他米松磷酸盐选自倍他米松磷酸钠。
[0041]
与现有技术相比,本发明具有以下有益效果:
[0042]
本发明提供的全新的倍他米松磷酸酯二水合物,与倍他米松磷酸酯无水物相比,结晶度好,纯度高,解决了后续工序中产品提纯困难的问题,可作为倍他米松磷酸钠制剂的高质量原料,有利于倍他米松磷酸钠制剂质量的提升。
[0043]
本发明提供的倍他米松磷酸酯二水合物的制备方法,工艺过程简单且易于操作,所用溶剂都是常规的工业化试剂,优选出了特定的溶剂和比例,十分有利于倍他米松磷酸酯二水合物的工业化。此外,与现有技术中的方法比较,无需使用强酸、强碱,操作简单,更利于工业化。
附图说明
[0044]
为了更清楚地说明本发明具体实施方式或现有技术中的技术方案,下面将对具体实施方式或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图是本发明的一些实施方式,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
[0045]
图1为实施例1-1得到的倍他米松磷酸酯二水合物单晶的立体结构椭球图;
[0046]
图2为实施例1-1得到的倍他米松磷酯二水合物单晶晶胞分子堆积图;
[0047]
图3为实施例1-1得到的倍他米松磷酯二水合物单晶通过单晶数据模拟得到pxrd;
[0048]
图4为实施例3-1得到的倍他米松磷酯二水合物的pxrd图谱;
[0049]
图5为实施例3-1得到的倍他米松磷酯二水合物的dsc图谱;
[0050]
图6为对比实施例1得到的倍他米松磷无水合物的pxrd图谱;
[0051]
图7为对比实施例1得到的倍他米松磷无水合物的dsc图谱。
具体实施方式
[0052]
下面将结合实施例对本发明的实施方案进行详细描述,但是本领域技术人员将会理解,下列实施例仅用于说明本发明,而不应视为限制本发明的范围。实施例中未注明具体条件者,按照常规条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市售购买获得的常规产品。
[0053]
实施例1倍他米松磷酸酯单晶的制备
[0054]
实施例1-1
[0055]
将1.0g倍他米松磷酸酯粗品加入到甲醇(13.3ml)和纯化水(6.7ml)的混合溶剂中,加热到55℃溶解,将溶液用0.45um滤膜过滤至西林瓶中,保鲜膜密封后,扎1小孔,室温静置挥发,长出适合大小晶体。
[0056]
将得到的晶体通过单晶x射线衍射(sxrd)分析得到分子的立体结构如图1所示;晶胞分子堆积图如图2所示。该单晶属于正交晶系,空间群为p212121,晶胞参数:,晶胞参数:α=90
°
,β=90
°
,γ=90
°
,晶胞体积晶胞内分子数z=4,单位晶格的独立区域内含有1个分子。
[0057]
通过单晶数据模拟得到pxrd,测得特征峰位置为2θ=11.5
°±
0.2
°
、13.8
°±
0.2
°
、14.2
°±
0.2
°
、14.8
°±
0.2
°
、15.1
°±
0.2
°
、16.2
°±
0.2
°
、17.8
°±
0.2
°
、19.1
°±
0.2
°
、21.7
°±
0.2
°
、27.7
°±
0.2
°
,如图3所示。
[0058]
实施例1-2
[0059]
将1.0g倍他米松磷酸酯粗品加入到丙酮(2.5ml)和纯化水(2.5ml)的混合溶剂中,加热到50℃溶解,将溶液用0.45um滤膜过滤至西林瓶中,保鲜膜密封后,扎1小孔,室温静置挥发,长出适合大小晶体。
[0060]
实施例1-3
[0061]
将1.0g倍他米松磷酸酯粗品加入到乙腈(26.7ml)和纯化水(3.3ml)的混合溶剂中,加热到60℃溶解,将溶液用0.45um滤膜过滤至西林瓶中,保鲜膜密封后,扎1小孔,室温静置挥发,长出适合大小晶体。
[0062]
将实施例1-2至实施例1-3得到的晶体进行单晶x射线衍射分析,与实施例1-1得到
的晶体具有相同的晶胞结构,且与实施例1-1得到的晶体的特征峰的衍射角2θ值与实施例1-1得到的晶体的衍射角2θ值在
±
0.2
°
内变化,证明与实施例1-1得到的晶体均为二水合物,且具有相同的晶型。
[0063]
实施例2倍他米松磷酸酯单晶的制备
[0064]
实施例2-1
[0065]
将1.0g倍他米松磷酸酯粗品加入到甲醇(13.3ml)和纯化水(6.7ml)的混合溶剂中,55℃溶解澄清,溶液过0.45um滤膜至西林瓶中,用保鲜膜密封并扎1小孔,置于正己烷环境中气相扩散结晶,长出大小合适的晶体。
[0066]
实施例2-2
[0067]
将1.0g倍他米松磷酸酯粗品加入到四氢呋喃(15ml)和纯化水(15ml)的混合溶剂中,50℃溶解澄清,溶液过0.45um滤膜至西林瓶中,用保鲜膜密封并扎1小孔,置于环己烷环境中气相扩散结晶,长出大小合适的单晶。
[0068]
实施例2-3
[0069]
将1.0g倍他米松磷酸酯粗品加入到异丙醇(4.4ml)和纯化水(0.6ml)的混合溶剂中,52℃溶解澄清,溶液过0.45um滤膜至西林瓶中,用保鲜膜密封并扎1小孔,置于乙醚环境中气相扩散结晶,长出大小合适的晶体。
[0070]
将实施例2-1至实施例2-3得到的晶体进行单晶x射线衍射分析,与实施例1-1得到的晶体具有相同的晶胞结构,且与实施例1-1得到的晶体的特征峰的衍射角2θ值与实施例1-1得到的晶体的衍射角2θ值在
±
0.2
°
内变化,证明与实施例1-1得到的晶体均为二水合物,且具有相同的晶型。
[0071]
实施例3倍他米松磷酸酯的制备
[0072]
实施例3-1
[0073]
将10g倍他米松磷酸酯粗品加入到甲醇(67ml)和纯化水(133ml)的混合溶剂中,加热至50℃溶解澄清后,滤除不溶物,以10℃/h的速率降温至2℃,保温1h,晶浆抽滤,滤饼水洗,38℃鼓风干燥得白色结晶性粉末,收率89.1%,hplc纯度99.92%。
[0074]
将得到的白色结晶性粉末进行x射线粉末衍射测定,测得特征峰的衍射角2θ值与实施例1-1得到的晶体的衍射角2θ值在
±
0.2
°
内变化,证明与实施例1-1得到的晶体均为二水合物,且具有相同的晶型,如图4所示。
[0075]
将得到的晶体进行dsc测定,脱水吸热峰峰值约为94℃,如图5所示。
[0076]
实施例3-2
[0077]
将10g倍他米松磷酸酯粗品加入到丙酮(250ml)和纯化水(250ml)的混合溶剂中,加热至52℃溶解澄清后,滤除不溶物,以5℃/h的速率降温至-5℃,保温2h,晶浆抽滤,滤饼水洗,35℃鼓风干燥得白色结晶性粉末,收率85.3%,hplc纯度99.92%。
[0078]
实施例3-3
[0079]
将10g倍他米松磷酸酯粗品加入到乙醇(43ml)和纯化水(257ml)的混合溶剂中,加热至55℃溶解澄清后,滤除不溶物,以8℃/h的速率降温至10℃,保温0.5h,晶浆抽滤,滤饼水洗,40℃鼓风干燥得白色结晶性粉末,收率85.5%,hplc纯度99.88%。
[0080]
实施例3-4
[0081]
将10g倍他米松磷酸酯粗品加入到四氢呋喃(62.5ml)和纯化水(187.5ml)的混合
溶剂中,加热至50℃溶解澄清后,滤除不溶物,以10℃/h的速率降温至8℃,保温2h,晶浆抽滤,滤饼水洗,40℃鼓风干燥得白色结晶性粉末,收率87.5%,hplc纯度99.90%。
[0082]
实施例3-5
[0083]
将10g倍他米松磷酸酯粗品加入到异丙醇(114.3ml)和纯化水(285.7ml)的混合溶剂中,加热至50℃溶解澄清后,滤除不溶物,以7℃/h的速率降温至8℃,保温1.5h,晶浆抽滤,滤饼水洗,40℃鼓风干燥得白色结晶性粉末,收率88.1%,hplc纯度99.90%。
[0084]
实施例3-6
[0085]
将10g倍他米松磷酸酯粗品加入到乙腈(58.3ml)和纯化水(291.7ml)的混合溶剂中,加热至50℃溶解澄清后,滤除不溶物,以6℃/h的速率降温至0℃,保温1h,晶浆抽滤,滤饼水洗,40℃鼓风干燥得白色结晶性粉末,收率85.6%,hplc纯度99.89%。
[0086]
实施例3-7
[0087]
将10g倍他米松磷酸酯粗品加入到甲醇(50ml)、乙醇(50ml)和纯化水(350ml)的混合溶剂中,加热至55℃溶解澄清后,滤除不溶物,以9℃/h的速率降温至10℃,保温2h,晶浆抽滤,滤饼水洗,40℃鼓风干燥得白色结晶性粉末,收率87.5%,hplc纯度99.91%。
[0088]
实施例3-8
[0089]
将10g倍他米松磷酸酯粗品加入到丙酮(50ml)、乙腈(31.8ml)和纯化水(368.2ml)的混合溶剂中,加热至55℃溶解澄清后,滤除不溶物,以5℃/h的速率降温至10℃,保温2h,晶浆抽滤,滤饼水洗,40℃鼓风干燥得白色结晶性粉末,收率85.5%,hplc纯度99.90%。
[0090]
将实施例3-2至实施例3-8得到的晶体进行x射线粉末衍射测定,测得特征峰的衍射角2θ值与实施例1-1得到的晶体的衍射角2θ值在
±
0.2
°
内变化,证明与实施例1-1得到的晶体均为二水合物,且具有相同的晶型。
[0091]
对比实施例1
[0092]
将10g倍他米松磷酸酯粗品加入到甲醇(200ml)中,加热至50℃溶解澄清后,滤除不溶物,以10℃/h的速率降温至2℃,保温1h,晶浆抽滤,滤饼水洗,38℃鼓风干燥得白色固体,收率86.1%,hplc纯度99.78%。
[0093]
将得到的固体进行x射线衍射测定,测得特征峰的衍射角2θ值与实施例1-1得到的晶体的衍射角2θ值的特征峰不同,确定为结晶度较差的无水物,如图6所示。
[0094]
将得到的固体进行dsc测定,脱水吸热峰峰值约为180℃,如图7所示。
[0095]
对比实施例2(专利cn 106432390 a)
[0096]
将10g倍他米松磷酸酯粗品加入纯化水中,缓慢加入氢氧化钠溶液使倍他米松磷酸酯全溶;加入硫酸铝,用盐酸调ph值至7.5-7.0,搅拌30min,复测ph值不变后,升温至70℃,然后降温静置,静置不少于8h;过滤,将滤液倒入酸化罐内,在20℃慢慢加入盐酸,调ph值至0.5-1.0,继续搅拌2h,复测ph值不变后静置,静置不少于8h,过滤,干燥得白色固体,收率85.8%,hplc纯度99.75%。
[0097]
将得到的固体进行x射线衍射测定,测得特征峰的衍射角2θ值与实施例1-1得到的晶体的衍射角2θ值的特征峰不同,确定为结晶度较差的无水物。
[0098]
对比实施例3(专利cn104744543a)
[0099]
将10g倍他米松磷酸钠粗品溶于2000ml水中,加入6000ml乙酸乙酯,得到两相体系,再用盐酸调节ph值至2.0-3.0,放置使析晶,过滤,干燥得白色固体,收率88.9%,hplc含
量99.82%。
[0100]
将得到的固体进行x射线衍射测定,测得特征峰的衍射角2θ值与实施例1-1得到的晶体的衍射角2θ值的特征峰不同,确定为结晶度较差的无水物。
[0101]
对比实施例4(cn 110964075 a)
[0102]
将10g倍他米松磷酸酯粗品加入200ml乙醇/三氯甲烷(1:10)中,升温至48℃,50℃保温搅拌,反应结束后缓慢降至室温,过滤,干燥得白色固体,收率88.1%,hplc含量99.80%。
[0103]
将得到的固体进行x射线衍射测定,测得特征峰的衍射角2θ值与实施例1-1得到的晶体的衍射角2θ值的特征峰不同,确定为结晶度较差的无水物。
[0104]
试验例
[0105]
采用同一批号的倍他米松磷酸酯粗品分别采用实施例3-1至3-8和对比实施例1至4的方法来进行精制。采用hplc法测定倍他米松磷酸酯中杂质的含量,用溶液颜色检查法(中国药典2015版附录ix a第一法)检查各样品溶液的颜色,用澄清度检查法(中国药典2015版附录ix b)检查各样品溶液的澄清度。检测结果见表1。
[0106]
hplc检测条件:色谱柱:c18(4.6
×
250mm,5μm);流动相:磷酸二氢钾已胺溶液(取磷酸二氢钾1.36g与已胺0.60g混匀,放置10min后,加水185ml使溶解)-乙睛=74:26;检测波长:254nm;柱温:30℃;流速:1.0ml/min;进样量:20μl。
[0107]
表1倍他米松磷酸酯溶液澄清度、颜色及杂质含量
[0108][0109]
从试验结果可以看出:采用本发明方法制备得到的倍他米松磷酸酯二水合物与采用对比实施例1至4得到的倍他米松磷酸酯无水物相比,杂质较更少、溶液澄清度和颜色更佳,更适合作为高质量的原料。
[0110]
最后应说明的是:以上各实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述各实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依
然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分或者全部技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1