一种近红外发光的三苯胺衍生物荧光分子及其制备方法与应用
1.本发明涉及一种近红外发光的三苯胺衍生物荧光分子及其制备方法与应用,属于有机荧光小分子技术领域。
背景技术:
2.近红外(650
‑
900nm)波段的光波具有stokes位移大、荧光信号稳定、生物组织穿透能力强的优点,能有效地减少生物体自荧光和自吸收的影响,降低对生物体的损伤,提高检测的准确性和灵敏度。有机荧光分子通常表现出荧光性能好、设计性强、合成简便且易于应用的优势。因此,有机近红外荧光分子在生物检测与医学诊断领域具有巨大的应用潜力。
3.三苯胺荧光团具有易于设计、合成简便、荧光信号稳定等特点,是制备有机荧光分子的良好发色团。近年来,随着有机荧光小分子研究与发展,一些以三苯胺为发色团的近红外有机荧光分子引起了诸多研究者的关注。然而,基于三苯胺的有机荧光分子通常发射波长短、stokes位移小、量子产率低等问题。
4.中国专利文件cn108892654a提供了一种含4
‑
二氰基甲基苯并吡喃单元的近红外荧光成像剂,其荧光发射波长为680~800nm,在该荧光化合物的三苯胺上引入可修饰基团,增加的可修饰位点可用于连接不同的生物活性物质,进而改善其水溶性与生物相容性,扩大其在生物医学领域的应用范围,上述近红外荧光成像剂具有下式所示结构,但是上述荧光分子为d
‑
π
‑
a结构,与d
‑
π
‑
a
‑
π
‑
d结构相比,其量子产率一般不高,并且不利于后续的功能化。
[0005][0006]
因此,充分发挥三苯胺发色团的优势,设计并合成具有优异荧光性能的近红外荧光分子并对其荧光性能进行表征是实现医学精准诊断的基础和前提;设计并合成新型的具有优异荧光性能的三苯胺衍生物具有重大的理论和实际意义。
技术实现要素:
[0007]
针对现有技术的不足,尤其是已报道的有机近红外荧光分子大多表现出stokes位移小、发射波长短、量子产率低、光稳定性差、合成过程复杂等问题,本发明提供了一种近红外发光的三苯胺衍生物荧光分子及其制备方法与应用。本发明以分子内电子转移(ict)过程为设计基础合成出一系列的近红外发光的三苯胺衍生物有机荧光分子,本发明制备的有机荧光功能分子具有近红外荧光发射、斯托克斯位移大、光化学稳定性高等特点同时其合
成步骤简单,反应产率高、量子产率高等优势。
[0008]
本发明的技术方案如下:
[0009]
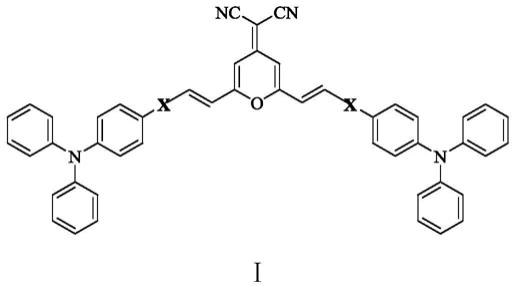
一种近红外发光的三苯胺衍生物荧光分子,其结构式如下式ⅰ所示:
[0010][0011]
式ⅰ中,x为:
[0012]
根据本发明优选的,当x为时,为具有苯基结构的近红外发光的三苯胺衍生物荧光分子:2
‑
(2,6
‑
二((e)
‑2‑
(4
‑
(4
‑
(二苯基氨基)苯基)苯基
‑1‑
乙烯基)
‑
4h
‑
吡喃
‑4‑
亚烷基)丙二腈,其结构式如式
ⅰ‑
1所示:
[0013][0014]
根据本发明优选的,当x为时,为具有萘基结构的近红外发光的三苯胺衍生物荧光分子:2
‑
(2,6
‑
二((e)
‑2‑
(4
‑
(4
‑
(二苯基氨基)苯基)萘基
‑1‑
乙烯基)
‑
4h
‑
吡喃
‑4‑
亚烷基)丙二腈,其结构式如式
ⅰ‑
2所示:
[0015][0016]
根据本发明优选的,当x为时,为具有呋喃基结构的近红外发光的三苯胺衍生物荧光分子:2
‑
(2,6
‑
二((e)
‑2‑
(4
‑
(4
‑
(二苯基氨基)苯基)呋喃基
‑1‑
乙烯基)
‑
4h
‑
吡喃
‑4‑
亚烷基)丙二腈,其结构式如式
ⅰ‑
3所示:
[0017][0018]
根据本发明优选的,当x为时,为具有噻吩基结构的近红外发光的三苯胺衍生物荧光分子:2
‑
(2,6
‑
二((e)
‑2‑
(4
‑
(4
‑
(二苯基氨基)苯基)呋喃基
‑1‑
乙烯基)
‑
4h
‑
吡喃
‑4‑
亚烷基)丙二腈,其结构式如式
ⅰ‑
4所示:
[0019][0020]
根据本发明,上述近红外发光的三苯胺衍生物荧光分子的制备方法,包括步骤如下:
[0021]
(1)将2,6
‑
二甲基
‑4‑
吡喃酮与丙二腈反应,得到2,6
‑
二甲基
‑4‑
吡喃亚基丙二腈;
[0022]
(2)将4
‑
(二苯基氨基)苯硼酸与醛类化合物进行反应,得到式ⅱ化合物;所述的醛
类化合物为4
‑
溴苯甲醛、4
‑
溴
‑1‑
萘甲醛、5
‑
溴呋喃
‑2‑
甲醛或5
‑
溴噻吩
‑2‑
甲醛;
[0023]
(3)将步骤(1)中得到的2,6
‑
二甲基
‑4‑
吡喃亚基丙二腈与步骤(2)中得到的式ⅱ化合物进行反应,得到近红外发光的三苯胺衍生物荧光分子。
[0024]
根据本发明优选的,步骤(1)中,2,6
‑
二甲基
‑4‑
吡喃酮与丙二腈的反应步骤如下:
[0025]
将2,6
‑
二甲基
‑4‑
吡喃酮和丙二腈溶于醋酸酐后,139
‑
140℃下回流反应11
‑
15h;反应结束后减压蒸馏除去溶剂,得到粗产物;将粗产物通过硅胶柱色谱提纯,得到淡黄色固体产物2,6
‑
二甲基
‑4‑
吡喃亚基丙二腈;
[0026]
优选的,所述的2,6
‑
二甲基
‑4‑
吡喃酮与丙二腈的摩尔比为1:1
‑
1.5;所述2,6
‑
二甲基
‑4‑
吡喃酮的摩尔数与醋酸酐的体积之比为1mmol:1
‑
5ml。
[0027]
优选的,所述的硅胶柱色谱提纯的洗脱剂为二氯甲烷与甲醇的混合溶剂,二氯甲烷与甲醇的体积比为9
‑
10:1。
[0028]
根据本发明,所述的2,6
‑
二甲基
‑4‑
吡喃亚基丙二腈具有下式ⅲ所示结构:
[0029][0030]
根据本发明优选的,步骤(2)中4
‑
(二苯基氨基)苯硼酸与醛类化合物的反应步骤如下:
[0031]
将4
‑
(二苯基氨基)苯硼酸、醛类化合物、k3po
4 7h2o和pd(oac)2加入溶剂中,在25
‑
35℃下搅拌反应30
‑
120min;反应完成后经后处理得到粗产物,将所得粗产物通过硅胶柱色谱提纯,得到式ⅱ化合物。
[0032]
优选的,所述的4
‑
(二苯基氨基)苯硼酸与醛类化合物的摩尔比为1
‑
1.2:1;所述的醛类化合物、k3po
4 7h2o和pd(oac)2的摩尔比为1:1.5
‑
2.5:0.01
‑
0.1。
[0033]
优选的,所述的溶剂为水和异丙醇的混合溶剂,其中水和异丙醇的体积比为1:2
‑
2.5。
[0034]
优选的,所述的醛类化合物的摩尔数与溶剂的体积之比为1mmol:10
‑
40ml。
[0035]
优选的,所述的后处理步骤为:向反应所得混合物中加入去离子水,然后用ch2cl2萃取三次,合并有机相,将有机相使用无水na2so4干燥12h,过滤,之后除去溶剂,得到粗产物;所述的去离子水的加入体积与醛类化合物的摩尔数之比为15
‑
25ml:1mmol。
[0036]
优选的,所述的硅胶柱色谱提纯的洗脱剂为乙酸乙酯和石油醚的混合溶剂,混合溶剂中乙酸乙酯和石油醚的体积比为1:30
‑
50。
[0037]
根据本发明,步骤(2)中所述的醛类化合物为4
‑
溴苯甲醛时,所得式ⅱ化合物为4'
‑
(二苯氨基)
‑
[1,1'
‑
联苯基]
‑4‑
甲醛,其结构式如式
ⅱ‑
1所示:
[0038][0039]
根据本发明,步骤(2)中所述的醛类化合物为4
‑
溴
‑1‑
萘甲醛时,所得式ⅱ化合物为4
‑
(4
‑
(二苯氨基)苯基)
‑1‑
萘甲醛,其结构式如
ⅱ‑
2所示:
[0040][0041]
根据本发明,步骤(2)中所述的醛类化合物为5
‑
溴呋喃
‑2‑
甲醛时,所得式ⅱ化合物为5
‑
(4
‑
(二苯氨基)苯基)呋喃
‑2‑
甲醛,其结构式如式
ⅱ‑
3所示:
[0042][0043]
根据本发明,步骤(2)中所述的醛类化合物为5
‑
溴噻吩
‑2‑
甲醛时,所得式ⅱ化合物为5
‑
(4
‑
(二苯氨基)苯基)噻吩
‑2‑
甲醛,其结构式如式
ⅱ‑
4所示:
[0044][0045]
根据本发明优选的,步骤(3)中,2,6
‑
二甲基
‑4‑
吡喃亚基丙二腈与式ⅱ化合物的反应步骤如下:
[0046]
将2,6
‑
二甲基
‑4‑
吡喃亚基丙二腈与式ⅱ化合物溶于乙腈中,加入哌啶,n2条件下,78
‑
82℃下回流反应24h;反应结束后,经后处理得到粗产物,所得粗产物通过硅胶柱色谱提纯,得到近红外发光的三苯胺衍生物荧光分子。
[0047]
优选的,所述的2,6
‑
二甲基
‑4‑
吡喃亚基丙二腈与式ⅱ化合物的摩尔比为1:2
‑
2.5;所述的2,6
‑
二甲基
‑4‑
吡喃亚基丙二腈的摩尔数与乙腈的体积之比为1mmol:50
‑
100ml;所述的2,6
‑
二甲基
‑4‑
吡喃亚基丙二腈的摩尔数与哌啶的体积之比为1mmol:0.1
‑
0.8ml。
[0048]
优选的,所述的后处理步骤为:将反应所得混合物冷却至室温,之后于
‑
4~0℃下静置12h,过滤,得到粗产品。
[0049]
优选的,所述的硅胶柱色谱提纯步骤为:首先使用乙酸乙酯和石油醚的混合溶剂为洗脱剂,混合溶剂中乙酸乙酯和石油醚的体积比为1:10
‑
40;待产物流出后使用二氯甲烷为洗脱剂。
[0050]
根据本发明,上述近红外发光的三苯胺衍生物荧光分子的应用,作为荧光成像试剂使用。
[0051]
本发明的技术特点及有益效果如下:
[0052]
1、本发明制备的近红外发光的三苯胺衍生物荧光分子具有近红外荧光发射波长较长、斯托克斯位移大、光化学稳定性高等特点。
[0053]
2、本发明的近红外发光的三苯胺衍生物荧光分子的制备及分离提纯过程相对简单、产率以及量子产率较高。
[0054]
3、本发明的近红外发光的三苯胺衍生物荧光分子具有近红外荧光性能,可以对细胞进行荧光标记,是一种有用的荧光成像试剂。
附图说明
[0055]
图1为实施例1制备的近红外发光的三苯胺衍生物荧光分子
ⅰ‑
1的1h nmr图谱。
[0056]
图2为实施例2制备的近红外发光的三苯胺衍生物荧光分子
ⅰ‑
2的1h nmr图谱。
[0057]
图3为实施例3制备的近红外发光的三苯胺衍生物荧光分子
ⅰ‑
3的1h nmr图谱。
[0058]
图4为实施例4制备的近红外发光的三苯胺衍生物荧光分子
ⅰ‑
4的1h nmr图谱。
[0059]
图5为实施例1
‑
4制备的近红外发光的三苯胺衍生物荧光分子的荧光光谱图。
具体实施方式
[0060]
下面通过具体实施并结合附图对本发明进一步阐述,但本发明的保护范围并不受限于此。在不背离本发明实质的情况下,对本发明方法、步骤或条件所作的修改和替换,均属于本发明的范围。
[0061]
实施例中所用原料和设备均为本领域技术人员熟知,且均为市场上能够购买到或容易获得或制得。
[0062]
实施例1
[0063]
一种近红外发光的三苯胺衍生物荧光分子的制备方法,包括步骤如下:
[0064]
(1)2,6
‑
二甲基
‑4‑
吡喃亚基丙二腈的合成
[0065]
将2.6
‑
二甲基
‑4‑
吡喃酮(248.28mg,2mmol)和丙二腈(132.12mg,2mmol)溶解在6ml的醋酸酐中,搅拌加热至140℃,140℃下持续回流反应12h,反应结束后,减压蒸馏除去溶剂,得到粗产物,使用硅胶柱色谱提纯(洗脱剂为二氯甲烷与甲醇的混合溶剂,混合溶剂中二氯甲烷与甲醇的体积比为9
‑
10:1,硅胶为200
‑
300目),得到淡黄色固体粉末2,6
‑
二甲基
‑4‑
吡喃亚基丙二腈,产率74%。
[0066]
合成路线如下:
[0067][0068]
(2)4'
‑
(二苯氨基)
‑
[1,1'
‑
联苯基]
‑4‑
甲醛的合成
[0069]4‑
(二苯基氨基)苯硼酸(1.1mmol,318.1mg),4
‑
溴苯甲醛(1.0mmol,185.2mg),k3po
4 7h2o(2.0mmol,676.7mg)和pd(oac)2(0.01mmol,2.2mg)加入到三口烧瓶中,加入水/异丙醇(v/v,1/2.1)混合溶剂16.2ml,25℃下搅拌反应60min,反应体系由灰白色变为灰绿色;反应结束后,向反应所得混合物中加入20ml去离子水,然后用ch2cl2萃取三次(30ml
×
3),合并有机相,将有机相使用无水na2so4干燥12h,过滤,之后减压蒸馏除去溶剂,得到粗产物,将所得粗产物使用硅胶柱色谱提纯(洗脱剂:乙酸乙酯和石油醚的混合溶剂,乙酸乙酯和石油醚的体积比为1:40,硅胶为200
‑
300目),得到黄绿色固体粉末4'
‑
(二苯氨基)
‑
[1,1'
‑
联苯基]
‑4‑
甲醛,产率86%。
[0070]
合成路线如下:
[0071][0072]
(3)2
‑
(2,6
‑
二((e)
‑2‑
(4
‑
(4
‑
(二苯基氨基)苯基)苯基
‑1‑
乙烯基)
‑
4h
‑
吡喃
‑4‑
亚烷基)丙二腈(
ⅰ‑
1)的合成
[0073]
4'
‑
(二苯氨基)
‑
[1,1'
‑
联苯基]
‑4‑
甲醛(0.42mmol,146.7mg)和2,6
‑
二甲基
‑4‑
吡喃亚基丙二腈(0.2mmol,34.4mg)溶解在15ml乙腈中,加入哌啶0.05ml,n2条件下,在80℃下回流反应24h;反应结束后,冷却至室温,在
‑
4℃下静置12h,减压抽滤,得粗产品。将所得粗
产品使用硅胶柱色谱进行提纯(首先洗脱剂为乙酸乙酯和石油醚的混合溶剂,乙酸乙酯和石油醚的体积比为1:20,待红色产物流出时使用二氯甲烷为洗脱剂),得到目标产物近红外发光的三苯胺衍生物荧光分子
ⅰ‑
1,深红色固体粉末,产率为67%。
[0074]
合成路线如下:
[0075][0076]
近红外发光的三苯胺衍生物荧光分子
ⅰ‑
1的数据表征如下:
[0077]1h nmr(400mhz,cd2cl2,ppm):δ=6.64(s,2h),6.78(d,j=16.0hz,2h),6.99(t,j=8.0hz,4h),7.05(d,j=6.0hz,12h),7.21(t,j=8.0hz,8h),7.47(d,j=8.0hz,4h),7.55(d,j=16.0hz,2h),7.60(s,8h)。
[0078]
hr
‑
ms:calcd for c
60
h
42
n4o,[m+h]
+
=835.3392,found 835.3383。
[0079]
本实施例制备的近红外发光的三苯胺衍生物荧光分子的1h nmr图谱如图1所示。
[0080]
实施例2
[0081]
一种近红外发光的三苯胺衍生物荧光分子的制备方法,包括步骤如下:
[0082]
(1)2,6
‑
二甲基
‑4‑
吡喃亚基丙二腈的合成
[0083]
同实施例1步骤(1)。
[0084]
(2)4
‑
(4
‑
(二苯氨基)苯基)
‑1‑
萘甲醛的合成
[0085]4‑
(二苯基氨基)苯硼酸(1.1mmol,318.1mg),4
‑
溴
‑1‑
萘甲醛(1.0mmol,235.1mg),k3po4·
7h2o(2.0mmol,676.7mg)和pd(oac)2(0.01mmol,2.2mg)加入到三口烧瓶中,加入水/异丙醇(v/v,1/2.1)混合溶剂20.6ml,在25℃下搅拌反应60min,反应体系由灰白色变为灰绿色;反应结束后,向反应所得混合物中加入20ml去离子水,然后用ch2cl2萃取三次(40ml
×
3),合并有机相,将有机相使用无水na2so4干燥12h,过滤,之后减压蒸馏除去溶剂,得到粗产物,将所得粗产物使用柱色谱提纯(洗脱剂:乙酸乙酯和石油醚的混合溶剂,乙酸乙酯和石油醚的体积比为1:40,硅胶为200
‑
300目),得到黄绿色固体粉末4
‑
(4
‑
(二苯氨基)苯基)
‑1‑
萘甲醛,产率71%。
[0086]
合成路线如下:
[0087][0088]
(3)2
‑
(2,6
‑
二((e)
‑2‑
(4
‑
(4
‑
(二苯基氨基)苯基)萘基
‑1‑
乙烯基)
‑
4h
‑
吡喃
‑4‑
亚烷基)丙二腈(
ⅰ‑
2)的合成
[0089]4‑
(4
‑
(二苯氨基)苯基)
‑1‑
萘甲醛(0.42mmol,167mg)和2,6
‑
二甲基
‑4‑
吡喃亚基
丙二腈(0.2mmol,34.4mg)溶解在20ml乙腈中,加入哌啶0.05ml,n2条件下,在80℃下回流反应24h;反应结束后,冷却至室温,置于
‑
4℃下静置12h,减压抽滤,得粗产品;将所得粗产品使用硅胶柱色谱进行提纯(首先洗脱剂为乙酸乙酯和石油醚的混合溶剂,乙酸乙酯和石油醚的体积比为1:15,待红色产物流出后,改用ch2cl2作为洗脱剂),得到目标产物近红外发光的三苯胺衍生物荧光分子
ⅰ‑
2,所得产物为红色固体粉末。
[0090]
合成路线如下:
[0091][0092]
近红外发光的三苯胺衍生物荧光分子
ⅰ‑
2的数据表征如下:
[0093]1h nmr(400mhz,cd2cl2,ppm):δ=6.76(s,2h),6.97(d,j=12.0hz 2h),7.00(t,j=8.0hz,4h),7.11(d,j=8.0hz,10h),7.24(t,j=8.0hz,8h),7.33(d,j=8.0hz,4h),7.46
‑
7.51(m,4h),7.56
‑
7.63(m,4h),7.99(d,j=8.0hz,2h),8.07(d,j=8.0hz,2h),8.33(d,j=8.0hz,2h),8.55(d,j=12.0hz,2h)。
[0094]
hr
‑
ms:calcd for c
68
h
46
n4o,[m+na]
+
=958.3564,found 958.6498.
[0095]
本实施例制备的近红外发光的三苯胺衍生物荧光分子的1h nmr图谱如图2所示。
[0096]
实施例3
[0097]
一种近红外发光的三苯胺衍生物荧光分子的制备方法,包括步骤如下:
[0098]
(1)2,6
‑
二甲基
‑4‑
吡喃亚基丙二腈的合成
[0099]
同实施例1步骤(1)。
[0100]
(2)5
‑
(4
‑
(二苯氨基)苯基)呋喃
‑2‑
甲醛的合成
[0101]4‑
(二苯基氨基)苯硼酸(1.0mmol,289.1mg),5
‑
溴代呋喃
‑2‑
甲醛(1.0mmol,175.0mg),k3po4·
7h2o(2.0mmol,676.7mg)和pd(oac)2(0.05mmol,11.2mg)置于50ml三口烧瓶中,加入水/异丙醇混合溶剂(v/v,1.0/2.2)25.6ml,在35℃条件下搅拌反应1.5h,混合物由灰白色变为灰绿色;反应结束后,向反应所得混合物中加入20ml去离子水,然后使用ch2cl2萃取三次(40ml
×
3),合并有机相,将有机相使用无水na2so4干燥12h,过滤,之后减压蒸馏除去溶剂,得到粗产物,将所得粗产物使用硅胶柱色谱提纯(洗脱剂:乙酸乙酯和石油醚的混合溶剂,乙酸乙酯和石油醚的体积比为1:40,硅胶为200
‑
300目),得到5
‑
(4
‑
(二苯氨基)苯基)呋喃
‑2‑
甲醛。
[0102]
合成路线如下:
[0103][0104]
(3)2
‑
(2,6
‑
二((e)
‑2‑
(4
‑
(4
‑
(二苯基氨基)苯基)呋喃基
‑1‑
乙烯基)
‑
4h
‑
吡喃
‑4‑
亚烷基)丙二腈(
ⅰ‑
3)的合成
[0105]5‑
(4
‑
(二苯氨基)苯基)呋喃
‑2‑
甲醛(1.1mmol,373.3mg)和2,6
‑
二甲基
‑4‑
吡喃亚基丙二腈(0.5mmol,86.1mg)溶解在50ml的乙腈中,加入哌啶0.3ml,n2条件下,在80℃下回流反应24h;反应结束后,冷却至室温,在
‑
4℃下静置12h,减压抽滤,得粗产品。将所得粗产品使用硅胶柱色谱进行提纯(洗脱剂为乙酸乙酯与石油醚的混合溶剂,乙酸乙酯与石油醚的体积比为1:20,待红色产物流出后使用二氯甲烷为洗脱剂),得到目标产物近红外发光的三苯胺衍生物荧光分子
ⅰ‑
3,产率为46%。
[0106]
合成路线如下:
[0107][0108]
近红外发光的三苯胺衍生物荧光分子
ⅰ‑
3的数据表征如下:
[0109]1h nmr(400mhz,cd3cl),δ(ppm):6.566(s,2h),6.597(d,2h,j=4.8hz),6.621(d,2h,j=7.6hz),6.705(d,2h,j=3.2hz),6.994
‑
7.081(m,16h),7.166(d,2h,j=16hz),7.224(t,8h,j=8.4hz),7.547(d,4h,j=8.4hz).
[0110]
hr
‑
ms m/z calcd for c
56
h
38
n4o3,[m]
+
=815.2977,found:815.2907.
[0111]
本实施例制备的近红外发光的三苯胺衍生物荧光分子的1h nmr图谱如图3所示。
[0112]
实施例4
[0113]
一种近红外发光的三苯胺衍生物荧光分子的制备方法,包括步骤如下:
[0114]
(1)2,6
‑
二甲基
‑4‑
吡喃亚基丙二腈的合成
[0115]
同实施例1步骤(1)。
[0116]
(2)5
‑
(4
‑
(二苯氨基)苯基)噻吩
‑2‑
甲醛的合成
[0117]4‑
(二苯基氨基)苯硼酸(1.0mmol,289.1mg),5
‑
溴代呋喃
‑2‑
甲醛(1.0mmol,191.0mg),k3po4·
7h2o(2.0mmol,676.7mg)和pd(oac)2(0.05mmol,11.2mg)置于50ml三口烧瓶中,加入水/异丙醇混合溶剂(v/v,1.0/2.2)25.6ml,在35℃条件下搅拌反应1.5h,混合物由灰白色变为灰绿色;反应结束后,向反应所得混合物中加入20ml去离子水,然后用ch2cl2萃取三次(30ml
×
3),合并有机相,所得有机相用无水na2so4干燥12h,过滤,之后减压蒸馏除去溶剂,得到粗产物,将所得粗产物使用硅胶柱色谱提纯(洗脱剂:乙酸乙酯和石油醚的混合溶剂,乙酸乙酯和石油醚的体积比为1:50,硅胶为200
‑
300目),得到目标产物5
‑
(4
‑
(二苯氨基)苯基)噻吩
‑2‑
甲醛。
[0118]
合成路线如下:
[0119][0120]
(3)2
‑
(2,6
‑
二((e)
‑2‑
(4
‑
(4
‑
(二苯基氨基)苯基)噻吩基
‑1‑
乙烯基)
‑
4h
‑
吡喃
‑4‑
亚烷基)丙二腈(
ⅰ‑
4)的合成
[0121]5‑
(4
‑
(二苯氨基)苯基)呋喃
‑2‑
甲醛(1.1mmol,390.5mg)和2,6
‑
二甲基
‑4‑
吡喃亚基丙二腈(0.5mmol,86.1mg)溶解在50ml的ch3cn中,加入哌啶0.3ml,n2条件下,在80℃下回流反应24h;反应结束后,冷却至室温,在
‑
4℃下静置12h,减压抽滤,得粗产品;将所得粗产品使用硅胶柱色谱进行提纯(首先洗脱剂为乙酸乙酯和石油醚的混合溶剂,乙酸乙酯和石油醚的体积比为1:20,待红色产物流出后使用二氯甲烷为洗脱剂),得到目标产物近红外发光的三苯胺衍生物荧光分子
ⅰ‑
4,产率为50%。
[0122]
合成路线如下:
[0123][0124]
近红外发光的三苯胺衍生物荧光分子
ⅰ‑
4的数据表征如下:
[0125]1h nmr(500mhz,cd3cl),6.398(d,2h,j=26.3hz),6.553(s,2h),6.991
‑
7.037(m,6h),7.084
‑
7.145(m,6h),7.145
‑
7.254(m,16h),7.405(d,4h,j=14.5hz),7.471(d,2h,j=26.3hz).
[0126]
ms m/z calcd for c
56
h
38
n4os2,[m+h]
+
=847.2521,found:847.2528.
[0127]
本实施例制备的近红外发光的三苯胺衍生物荧光分子的1h nmr图谱如图4所示。
[0128]
试验例1
[0129]
采用f
‑
7000型荧光光谱仪对实施例1
‑
4制备的近红外发光的三苯胺衍生物荧光分子进行测定,其荧光光谱图如图5所示。
[0130]
测试结果:
[0131]
近红外荧光分子
ⅰ‑
1和
ⅰ‑
2的激发波长分别为475和480nm,发射波长分别为680和682nm,在乙醇
‑
水(v/v,1:1)溶液中,stokes位移高达200nm。近红外荧光分子
ⅰ‑
3和
ⅰ‑
4在四氢呋喃中的激发波长为525nm,发射波长接近660nm,斯托克斯位移高达130nm;因此,本发明的近红外荧光分子具有优异的荧光性能。
[0132]
本领域的技术人员容易理解,此处所描述的具体实施例仅仅用于解释本发明,并不用以限定本发明。凡本发明的精神和原则之内所作的任何修改、等同替换或改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1