用于有机电致发光元件的有机铱配合物
1.本发明涉及一种提供适合用作有机电致发光元件的磷光材料的有机铱配合物的技术。具体涉及一种用作红色磷光材料、发光量子产率(φ
pl
)高且色纯度优异的有机铱配合物。
背景技术:
2.有机电致发光元件(oled)作为下一代显示器或照明的技术开发备受期待。其特征为:消耗电力少,可薄型化,并具有反应速度优异,以及在暗处和明处均可清晰地显示图像等优点。图7为用于说明oled结构的一个例子的图。在图7的示例中,oled以阴极/电子传输层/发光层/空穴传输层/阳极/玻璃基板的多层结构为基本构成。近年来,也有人提出在具有这种基本结构的元件中适当地追加用于提高空穴(电子)注入层、缓冲层、层间绝缘膜等的发光特性的功能层。
3.在具有上述那样的结构的oled发光层中,可以使用各种发光材料。在此,oled的发光材料大致分为荧光材料和磷光材料,但是近年来,关于磷光材料的新型材料的开发有所进展。这是因为,要求发光材料具有高发光效率以能够应对节能化,并且与荧光材料相比,磷光材料基于其发光原理可以满足这些要求。并且,作为oled用的磷光材料,至今已研究了各种有机化合物,但是近年来,由以铂、铱等贵金属为中心金属的有机金属配合物构成的磷光材料的研究例成为主流。
4.在此,当研究oled在显示器等的应用时,作为关于磷光材料所应研究的重要事项,可列举出其发光色的纯度。在oled中,通过调节来自发光材料的红(r)绿(g)蓝(b)光三原色发光的发光强度,可以显示所有的颜色。为了适当地显示该颜色,要求高纯度且高效率地发出目标的发光色。在这方面,根据本技术的发明人,显示发光波长最长的红色发光的磷光材料倾向于具有低的发光量子产率(φ
pl
),因此相对于其他发光色而成为关注度高的材料。
5.需要说明的是,发光量子产率(pl量子效率(φ
pl
))是指与发光材料的量子效率相关联的因素之一。在将发光材料的量子效率大致分为“外部量子效率”和“内部量子效率”的情况下,pl量子效率是决定内部量子效率的因素。发光材料需要提高内部量子效率,因此目的在于实现高的pl量子效率。
6.如上所述,作为磷光材料,有很多关于铂、铱等贵金属的有机金属配合物的报告例。并且,迄今为止,已报道了一些显示红色发光的红色磷光性有机铱。本技术人也公开了一些关于红色磷光性有机铱的现有技术。作为这些红色磷光性有机铱,提出了将具有杂环且具备c
‑
n结构的配位体(c
‑
n配位体)和二酮基类辅助配位体(β
‑
二酮类配位体)配位于作为中心金属的铱上而得到的有机金属配合物。
7.具体而言,专利文献1公开了由下述化学式1所示的铱配合物,其中使用1
‑
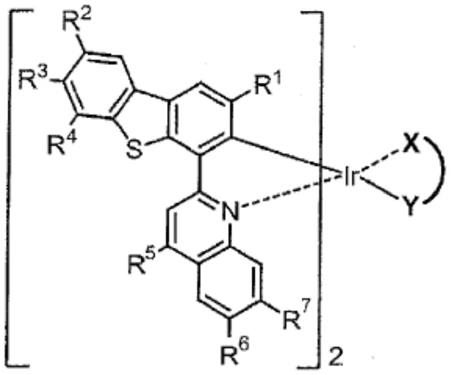
(二苯并[b,d]呋喃4
‑
基)异喹啉作为c
‑
n配位体,并使引入有丁氧基的二苯甲酰基甲烷作为二酮基类辅助配位体进行配位而得到的。此外,专利文献2公开了由下述化学式2所示的铱配合物,其中使用2
‑
(二苯并[b,d]噻吩
‑4‑
基)喹啉作为c
‑
n配位体,并使引入有叔丁基的二苯甲酰
基甲烷作为二酮基类辅助配位体进行配位而得到的。
[0008]
[化学式1]
[0009][0010]
[化学式2]
[0011][0012]
现有技术文献
[0013]
专利文献
[0014]
专利文献1:日本特开2008
‑
222635号公报
[0015]
专利文献2:日本特开2016
‑
15468号公报
技术实现要素:
[0016]
发明所要解决的课题
[0017]
上述现有的有机铱配合物均为呈现可以划分为红色的发光色的磷光材料。然而,这些有机铱配合物在发光效率(发光量子产率(φ
pl
))和色纯度这两方面不尽如人意。即,专利文献1的有机铱配合物在聚甲基丙烯酸甲酯薄膜(pmma薄膜)中的发光波长(λ
pl
)为635nm,可以呈现大致为纯红色的发光。但是,φ
pl
低至0.17,从发光效率的观点来看是不够的。另一方面,虽然专利文献2的有机铱配合物在pmma薄膜中的φ
pl
比较高,为0.61,但是发光波长(λ
pl
)为612nm,呈现比纯红色的波长更短的发光。
[0018]
如上述专利文献1、2所记载的有机铱配合物所示,对于红色发光性磷光材料,难以在保持色纯度优异的纯红色的同时还提高发光量子产率(φ
pl
)。目前的现状是,还没有两种特性的平衡良好的红色发光性磷光材料。
[0019]
本发明是鉴于以上背景而完成的,目的在于提供一种适合用作oled用的红色发光性磷光材料、发光量子产率(φ
pl
)高且色纯度优异的有机铱配合物。此外,如后所述,该有机铱配合物在其合成工艺中也具有特征。本发明还公开了用于有效地制备该有机铱配合物的方法。
[0020]
用于解决课题的方案
[0021]
为了解决上述课题,本发明人研究了将2
‑
(二苯并[b,d]噻吩
‑4‑
基)喹啉的衍生物作为c
‑
n配位体而得的有机铱配合物的效果。具体而言,对将作为取代基的甲基引入2
‑
(二苯并[b,d]噻吩
‑4‑
基)喹啉的特定位置而得的有机铱配合物进行了研究。结果发现,相对于现有的有机铱,可以得到高效率的纯红色磷光性有机铱配合物,从而想到了本发明。
[0022]
解决上述课题的本发明是如下式所示的有机铱配合物,其中,在将c
‑
n配位体和辅助配位体配位于铱而形成的用于有机电致发光元件的有机铱配合物中,将引入有至少一个甲基的2
‑
(二苯并[b,d]噻吩
‑4‑
基)喹啉配位体作为所述c
‑
n配位体进行配位。
[0023]
[化学式3]
[0024][0025]
(在上式中,r1、r2、r3、r4、r5、r6、r7各自为甲基或氢原子中的任一者。但是,r1、r2、r3、r4中的至少任意一者为甲基。x
‑
y为辅助配位体。)
[0026]
以下,对本发明进行详细地说明。本发明涉及的红色磷光性有机铱配合物的发明点在于在作为其c
‑
n配位体的2
‑
(二苯并[b,d]噻吩
‑4‑
基)喹啉中引入甲基。特别是,其特征在于,在该c
‑
n配位体的二苯并噻吩部位的特定位置上引入一个以上的甲基。
[0027]
在由具有c
‑
n配位体的有机铱配合物构成的磷光材料中,可列举出分子的热失活作为pl量子效率φ
pl
降低的原因。作为该热失活的原因之一,可列举出c
‑
n配位体上的芳香族的c
‑
h键的伸缩振动。在本发明中,通过在c
‑
n配位体的特定位置(c
‑
h键)上引入甲基(
‑
ch3),可以使显示高能量的伸缩振动的c
‑
h键被低能量的c
‑
c键取代。由此可以抑制振动失活,并可以谋求φ
pl
的提高。此外,本发明人进行了以下研究:通过在c
‑
n配位体上引入甲基,以使配合物分子的最高占据轨道(homo)与最低空轨道(lumo)的能量差发生变化,从而使发光波长λ
pl
向长波长侧位移而显示出纯红色的磷光。
[0028]
这样,本发明通过在作为c
‑
n配位体的2
‑
(二苯并[b,d]噻吩
‑4‑
基)喹啉的特定部位即2’位(r1)、6’位(r2)、7’位(r3)、8’位(r4)引入甲基,可以谋求φ
pl
的提高和λ
pl
的长波长化,从而形成合适的红色磷光性有机铱配合物。
[0029]
此外,在根据本发明的有机铱配合物中,关于将甲基引入2
‑
(二苯并[b,d]噻吩
‑4‑
基)喹啉中,除了将甲基引入二苯并噻吩部位以外,将甲基引入喹啉部位(r5、r6、r7)也是有用的。这是因为,在喹啉部位也产生由上述芳香族的c
‑
h键的伸缩振动所引起的失活,因此通过将c
‑
h键置换为c
‑
c键,可以期待φ
pl
的提高。
[0030]
如上所述,在根据本发明的有机铱配合物中,将甲基引入2
‑
(二苯并[b,d]噻吩
‑4‑
基)喹啉的二苯并噻吩部位是必须的。若列举本发明的优选方式,则可列举出:仅在2’位引入甲基的方式(r1=甲基、r2、r3、r4=氢)、在2’、7’位引入甲基的方式(r1、r3=甲基、r2、r4=氢)、在6’、8’位引入甲基的方式(r2、r4=甲基、r1、r3=氢)、在2’、6’、8’位引入甲基的方式(r1、r2、r4=甲基、r3=氢)。在这些方式当中,特别优选的方式是将甲基引入2’位为必须的方式(r1=甲基、r2~r4=氢或甲基中的任一者)。
[0031]
另外,在根据本发明的有机铱配合物中,与c
‑
n配位体一起配位至铱的辅助配位体(x
‑
y配位体)优选列举出以下配位体。辅助配位体如下构成:虽然其不会直接有助于有机铱配合物的发光特性的提高,但是会使有机铱配合物的发光特性产生一些变动。下述配位体具有低分子量且具有作为辅助配位体的所述作用。
[0032]
[化学式4]
[0033][0034]
(在上式中,带有*的原子是与铱原子配位的配位原子。)
[0035]
在上述辅助配位体中,更优选的配位体是作为β
‑
二酮配位体的乙酰丙酮(2,4
‑
戊二酮)、或二叔戊酰甲烷(2,2,6,6
‑
四甲基
‑
3,5
‑
庚二酮)。并且,最优选的配位体为二叔戊酰甲烷。如上所述,辅助配位体不会直接有助于有机铱配合物的发光特性的提高。但是,在本发明的目的中,优选谋求辅助配位体的最优化。从该观点出发,二叔戊酰甲烷作为辅助配位体是最合适的。二叔戊酰甲烷是具有比乙酰丙酮更大的立体位阻的配位体。在oled这样的
固体介质中的发光中,优选可以降低由分子间相互作用所引起的无辐射失活,因此,将二叔戊酰甲烷这样的立体位阻大的配位体作为辅助配位体是合适的。
[0036]
[化学式5]
[0037][0038]
(在上式中,带有*的原子是与铱原子配位的配位原子。)
[0039]
相对于现有的有机铱配合物,以上所说明的、根据本发明的红色磷光性有机铱配合物的色纯度优异并且发光效率也高。因此,作为oled的发光层是有用的。通过在高分子薄膜中掺杂该有机铱配合物,可以形成发光层。
[0040]
接着,对根据本发明的有机铱配合物的制造方法进行说明。对于本发明这样的、将c
‑
n配位体和β
‑
二酮化合物等辅助配位体配位于铱的有机铱配合物的合成方法,其制造方法的大致框架是已知的。即,首先,可以使铱盐与构成c
‑
n配位体的含氮化合物进行加热反应以得到前驱体,然后使该前驱体与辅助配位体(β
‑
二酮化合物等)进行加热反应来合成有机铱配合物。此外,除了前述步骤以外,也可以通过首先使铱盐与辅助配位体发生反应,然后再使含氮化合物(c
‑
n配位体)进行反应,从而合成有机铱配合物。
[0041]
上述合成工艺也适用于本发明的有机铱配合物。但是,本发明的特征是,在作为c
‑
n配位体的2
‑
(二苯并[b,d]噻吩
‑4‑
基)喹啉的特定位置(2’位、6’位、7’位、8’位)引入甲基。
[0042]
在这方面,未取代的2
‑
(二苯并[b,d]噻吩
‑4‑
基)喹啉的合成是比较容易的并且是已知的。例如,使用二苯并[b,d]噻吩
‑4‑
硼酸作为初始原料,通过使其与喹啉的卤代化合物进行交叉耦合,以合成2
‑
(二苯并[b,d]噻吩
‑4‑
基)喹啉。由于作为初始原料的二苯并[b,d]噻吩
‑4‑
硼酸可以作为市售商品而获得,因此可以容易地合成作为c
‑
n配位体的含氮化合物。
[0043]
与此相对,对于本发明涉及的有机铱配合物即在特定位置引入有甲基的2
‑
(二苯并[b,d]噻吩
‑4‑
基)喹啉,迄今为止没有其合成例子的报告。
[0044]
从本发明人想到作为本发明的主题事项的、将甲基引入2
‑
(二苯并[b,d]噻吩
‑4‑
基)喹啉配位体的阶段开始,就对该合成方法进行了深入的研究。并且,作为其具体的方法,对于c
‑
n配位体的合成,发现了以下这样的工艺。
[0045]
在本发明的合成工艺中,使用二溴苯胺或引入有甲基的二溴苯胺衍生物作为初始原料。并且,包括:通过使二溴苯胺(衍生物)、与苯基硼酸或具有甲基的苯基硼酸衍生物进行铃木
‑
宮浦交叉耦合反应,以得到由二苯胺衍生物构成的中间体1的步骤;以及将该中间体1的氨基进行碘化以合成中间体2的步骤。此外,还包括:用间氯过氧苯甲酸等过氧酸氧化所述中间体2,以合成作为环状二苯并碘鎓化合物的中间体3的步骤。该中间体3的合成是本
发明的合成方法中的特征步骤。
[0046]
然后,通过使中间体3与硫代乙酸盐发生反应以合成作为溴二苯并噻吩衍生物的中间体4,然后通过将中间体4锂化并与硼酸酯反应、以及使其水解,以合成作为引入了1个以上的甲基的二苯并噻吩
‑
硼酸衍生物的中间体5。通过这种方式所生成的、作为中间体5的多甲基取代的二苯并噻吩衍生物是新物质,与上述中间体3的合成一起,可以说都是本发明的配合物合成方法的特征。
[0047]
在合成了作为中间体5的二苯并噻吩
‑
硼酸衍生物之后,与未取代的2
‑
(二苯并[b,d]噻吩
‑4‑
基)喹啉配位体的合成方法是同样的。即,通过使中间体5与2
‑
氯喹啉进行铃木
‑
宮浦交叉耦合反应,可以合成作为本发明的c
‑
n配位体的、引入有甲基的2
‑
(二苯并[b,d]噻吩
‑4‑
基)喹啉配位体。
[0048]
需要说明的是,当合成在喹啉部位引入了甲基的c
‑
n配位体时,通过使中间体5与2
‑
氯喹啉或2
‑
氯喹啉衍生物(2
‑
氯
‑4‑
甲基喹啉等)反应,可以合成所期望的c
‑
n配位体。对于2
‑
氯喹啉衍生物,可以使用市售品或通过已知方法合成的化合物。
[0049]
如上所述,在本发明中,经由中间体1~中间体5的合成,从而合成了作为c
‑
n配位体的、引入有取代基的2
‑
(二苯并[b,d]噻吩
‑4‑
基)喹啉配位体。采用这样的反应路线是为了合成在目标位置引入了甲基的c
‑
n配位体。本发明的合成工艺无需担心异构化和位置选择性的问题就可以得到目标化合物。由于oled用的发光材料要求具有高纯度,因此本发明方法的一大优点是不用担心因异构化等而使纯度降低。
[0050]
然后,在合成了引入有甲基的c
‑
n配位体之后,如上述那样,通过将铱盐与c
‑
n配位体进行加热反应以合成前驱体,进一步通过使前驱体与辅助配位体反应,可以制备本发明的有机铱配合物。也可以先使铱盐与辅助配位体反应,然后再使c
‑
n配位体进行反应。
[0051]
需要说明的是,用于合成上述前驱体的加热反应优选在80℃~130℃进行12~24小时。此外,与辅助配位体的加热反应优选在60℃~130℃下进行0.5~12小时。这些反应优选在溶剂存在下进行。
[0052]
以上所说明的本发明的有机铱配合物作为oled发光层的发光掺杂剂是有用的。发光层构成如下:除了本发明的有机铱配合物以外,还适当地混合了电子传输材料、空穴传输主材料等成分。发光层的形成是通过旋转涂布法、真空沉积法等方法而适当地形成的。
[0053]
发明效果
[0054]
本发明的有机铱配合物的pl量子产率及耐热性高于传统的配合物,适合用作oled的发光材料。
附图说明
[0055]
[图1]为示出本实施方式所合成的有机铱配合物ir
‑
1~ir
‑
4在二氯甲烷溶液中的发光光谱的图。
[0056]
[图2]为示出本实施方式所合成的有机铱配合物ir
‑
1~ir
‑
4在甲苯溶液中的发光光谱的图。
[0057]
[图3]为示出本实施方式所合成的有机铱配合物ir
‑
1~ir
‑
4在pmma薄膜中的发光光谱的图。
[0058]
[图4]为用于说明本实施方式中制作的oled的构成的图。
[0059]
[图5]为示出本实施方式中制作的oled(devicel~device 4)的电致发光光谱的图。
[0060]
[图6]为示出本实施方式中制作的oled(devicel~device 4)的j
‑
v
‑
l曲线的图。
[0061]
[图7]为用于说明oled的结构的一个例子的图。
具体实施方式
[0062]
以下,对本发明的实施方式进行说明。在本实施方式中,合成了以下4种有机铱配合物并对其发光特性进行了评价,该4种有机铱配合物是通过将2
‑
(二苯并[b,d]噻吩
‑4‑
基)喹啉及其衍生物作为c
‑
n配位体进行配位,并且将二叔戊酰甲烷(2,2,6,6
‑
四甲基
‑
3,5
‑
庚二酮)作为辅助配位体进行配位而得到的。在下述4种有机铱配合物中,以未取代的2
‑
(二苯并[b,d]噻吩
‑4‑
基)喹啉作为c
‑
n配位体的ir
‑
1是作为比较例的现有的有机ir配合物。另一方面,在2
‑
(二苯并[b,d]噻吩
‑4‑
基)喹啉的二苯并噻吩部位引入了甲基的ir
‑
2、ir
‑
3、ir
‑
4是本实施方式的有机铱配合物。
[0063]
[化学式6]
[0064][0065]
(i)有机铱配合物(ir
‑
1~ir
‑
4)的合成
[0066]
在上述有机铱配合物的合成中,(i)在进行c
‑
n配位体(c
‑
n配位体(1)~c
‑
n配位体(4))的合成之后,(ii)使c
‑
n配位体与铱盐(氯化铱)发生反应以合成前驱体(前驱体(1)~前驱体(4))。然后,使前驱体与作为辅助配位体的二叔戊酰甲烷发生反应以合成有机铱配合物(ir
‑
1~ir
‑
4)。
[0067]
本实施方式中的有机铱配合物(ir
‑
1~ir
‑
4)的合成方案如下所示。在下述方案中,作为现有例的有机铱配合物(ir
‑
1)的合成是以市售的二苯并噻吩
‑
硼酸为初始原料来合成c
‑
n配位体(1),然后经由前驱体(1)的合成来合成有机铱配合物(ir
‑
1)。
[0068]
另一方面,对于本实施方式的有机铱配合物(ir
‑
2~ir
‑
4),首先,使用二溴苯胺衍
生物作为初始原料依次合成中间体1a~1c、中间体2a~2c、中间体3a~3c、中间体4a~4c、中间体5a~5c,从而合成c
‑
n配位体(2)~(4)。然后,以与ir
‑
1相同的方式合成前驱体(2)~(4),从而合成了有机铱配合物(ir
‑
2~ir
‑
4)。
[0069]
[化学式7]
[0070][0071]
需要说明的是,在本实施方式中,初始原料及合成所使用的试剂和溶剂均使用未进行精制的市售的试剂等级的。对于干燥thf,购入市售的脱水thf直接使用。此外,使用nacalai tesque,inc制的球状硅胶(中性)或关东化学公司制的球状硅胶(中性)作为柱层
析法所使用的充填剂。
[0072]
此外,使用质子核磁共振(1h nmr)光谱及质量分析(质谱(ms))来鉴别所合成的化合物。1h nmr光谱的测定中使用了jeol公司制的jnm
‑
ecx400分光光度计(400mhz)或jeol公司制的jnm
‑
ecs400分光光度计(400mhz)。ms谱是以α
‑
氰基
‑4‑
羟基肉桂酸(chca)为基质,通过飞行时间(tof)型质量分析,对以基质辅助激光解吸离子化法(maldi法)进行离子化后的试样进行测定(maldi
‑
tof
‑
ms谱)。使用shimadzu
‑
kratos axima
‑
cfr plus tof mass质量分析装置进行测量。对于元素分析,以乙酰苯胺为标准物质,通过j
‑
science lab co.,ltd.制造的jm
‑
10元素分析装置进行测定。
[0073]
有机铱配合物ir
‑
1的合成
[0074]
(i)c
‑
n配位体(1)(2
‑
(二苯并[b,d]噻吩
‑4‑
基)喹啉的合成
[0075]
将碳酸钾(8.90g、64.4mmol)溶解在水(225ml)中而得的水溶液加入到1000ml四口圆底烧瓶中,进一步将二苯并[b,d]噻吩
‑4‑
基硼酸(4.72g、20.7mmol)、2
‑
氯喹啉(3.64g、22.3mmol)、四(三苯基膦)钯(0)(0.94g、0.813mmol)、1,2
‑
二甲氧基乙烷(225ml)、乙醇(90ml)加入到圆底烧瓶中,在氮气气氛下于100℃加热搅拌18小时。冷却后,加入氯仿(200ml)并在分液漏斗中进行震荡,除去水层。然后,用水(300ml)和饱和食盐水(300ml)清洗有机层,以无水硫酸镁使有机层干燥后,使用旋转蒸发器蒸馏除去溶剂。通过硅胶柱层析法(展开溶剂:氯仿/己烷、v/v、4/1)对残渣进行纯化。通过以上操作,合成了5.44g(17.5mmol)的2
‑
(二苯并[b,d]噻吩
‑4‑
基)喹啉(c
‑
n配位体(1))。该化合物为白色固体,产率为84%。
[0076]
(ii)前驱体(1)的合成
[0077]
将上述所合成的c
‑
n配位体(1)(5.23g,16.7mmol)、氯化铱三水合物(2.95g,8.36mmol)、2
‑
乙氧基乙醇(330ml)及水(65ml)加入到1000ml四口圆底烧瓶中,在氮气气氛下于100℃加热搅拌20小时。冷却后,通过抽吸过滤回收反应体系中所生成的深红色沉淀。通过以上操作,合成了c
‑
n配位体(1)与铱的前驱体(1)(产率为76%(5.40g,3.18mmol))。
[0078]
(iii)有机铱配合物(ir
‑
1)的合成
[0079]
将上述所合成的前驱体(1)(2.01g,1.18mmol)、二叔戊酰甲烷(0.45ml,2.29mmol)、碳酸钠(2.04g,19.2mmol)及2
‑
乙氧基乙醇(300ml)加入到500ml三口圆底烧瓶中,在氮气气氛下于85℃加热搅拌3小时。冷却后,加入二氯甲烷(200ml)和水(200ml)并在分液漏斗中进行震荡,除去水层。然后,用饱和食盐水(200ml)清洗有机层,以无水硫酸钠使有机层干燥后,使用旋转蒸发器蒸馏除去溶剂。通过硅胶柱层析法(展开溶剂:二氯甲烷/己烷,v/v,4/1)对残渣进行纯化后,加入己烷,并在75℃下搅拌以进行清洗,通过抽吸过滤进行回收,从而得到了作为目标化合物的有机铱配合物(ir
‑
1)。该有机铱配合物为红色固体且产率为68%(1.59g,1.60mmol)。此外,在以上步骤中所合成的化合物的通过nmr等所获得的特性如下。
[0080]1h nmr(400mhz,cdcl3)δ0.59(s,18h),4.84(s,1h),6.83(d,j=7.8hz,2h),7.30
‑
7.52(m,10h),7.83
‑
7.85(dd,j=8.2,1.4hz,2h),7.89
‑
7.93(m,2h),7.97
‑
7.99(m,2h),8.29(d,j=8.7hz,2h),8.38(d,j=8.7hz,2h),8.70(d,j=8.7hz,2h).
13
c nmr(100mhz,cdcl3)δ27.76,40.62,88.78,119.02,120.21,121.44,122.05,124.52,125.35,125.99,126.08,126.90,127.32,130.76,131.25,133.91,136.20,136.45,136.53,137.99,141.48,
149.09,156.56,169.50,194.28.maldi tof
‑
ms:m/z[m+h]+calcd.for c
53
h
44
irn2o2s2:997.24,found:997.27.anal.calcd.for c
53
h
43
irn2o2s2:c,63.90;h,4.35;n,2.81.found:c,64.09;h,4.48;n,2.81.
[0081]
(a)有机铱配合物ir
‑
2的合成
[0082]
(a
‑
i)c
‑
n配位体(2)(2
‑
(甲基二苯并[b,d]噻吩
‑4‑
基)喹啉)的合成
[0083]
c
‑
n配位体(2)的合成是以2,6
‑
二溴
‑4‑
甲基苯胺为初始原料,经由中间体1a、2a、3a、4a、5a而合成的。
[0084]
<中间体1a(3
‑
溴
‑5‑
甲基
‑
[1,1
’‑
联苯]
‑2‑
胺)的合成>
[0085]
将碳酸钾(33.2g,0.240mol)溶解在水(120ml)中而得的水溶液加入到2000ml四口圆底烧瓶中,进一步将2,6
‑
二溴
‑4‑
甲基苯胺(20.1g,76.0mmol)、苯基硼酸(6.35g,52.1mmol)、四(三苯基膦)钯(0)(4.33g,3.74mmol)、甲苯(200ml)、乙醇(120ml)加入到圆底烧瓶中,在氮气气氛下,于100℃加热搅拌13小时。冷却后,加入氯仿(200ml)并在分液漏斗中进行震荡,除去水层。进一步用水(200ml)和饱和食盐水(200ml)清洗有机层,以无水硫酸镁使有机层干燥后,使用旋转蒸发器蒸馏除去溶剂。通过硅胶柱层析法(展开溶剂:氯仿/己烷,v/v,1/3)对残渣进行纯化,从而以82%的产率得到了作为目标化合物的中间体1a(3
‑
溴
‑5‑
甲基
‑
[1,1
’‑
联苯]
‑2‑
胺)的白色固体(11.2g,42.7mmol)。
[0086]
<中间体2a(3
‑
溴
‑2‑
碘
‑5‑
甲基
‑
1,1
’‑
联苯)的合成>
[0087]
将中间体1a(3
‑
溴
‑5‑
甲基
‑
[1,1
’‑
联苯]
‑2‑
胺)(6.02g,23.0mmol)、4m盐酸(65ml)以及四氢呋喃(65ml)加入到1000ml的四口圆底烧瓶中,将该烧瓶浸入装有制冷剂甲醇/丙酮及液氮的杜瓦瓶中,以将反应体系保持在0℃。使用滴液漏斗,向其中缓慢滴加将亚硝酸钠(2.01g,29.1mmol)溶解在水(24ml)中而得的水溶液。
[0088]
将反应体系在0℃下搅拌20分钟之后,加入将碘化钾(9.53g,57.4mmol)溶解在水(36ml)中而得的水溶液。在0℃下搅拌10分钟之后,除去杜瓦瓶,在缓缓恢复至室温的同时进一步搅拌1小时。此时,体系中为茶色。向其中添加饱和硫代硫酸钠水溶液直至体系中的颜色变为淡黄色,搅拌15分钟。之后,加入乙酸乙酯(200ml)并在分液漏斗中进行震荡,除去水层。进一步用水(300ml)及饱和食盐水(300ml)清洗有机层,以无水硫酸钠干燥后,使用旋转蒸发器蒸馏除去溶剂。
[0089]
然后,将残渣溶解在氯仿中,在其中加入50g硅胶后,使用旋转蒸发器蒸馏除去溶剂,从而使残渣吸附在硅胶上。然后,将含有展开溶剂的250g硅胶装入色谱柱管(φ:60mm)中,将刚才吸附有残渣的硅胶装在顶部,通过迅速地展开溶剂(展开溶剂:己烷)来进行纯化。通过以上操作,以38%的产率得到了作为目标化合物的中间体2a(3
‑
溴
‑2‑
碘
‑5‑
甲基
‑
1,1
’‑
联苯)的白色固体(3.28g,8.81mmol)。
[0090]
<中间体3a(4
‑
溴
‑2‑
甲基二苯并[b,d]碘
‑5‑
鎓三氟甲烷磺酸盐)的合成>
[0091]
将中间体2a(3
‑
溴
‑2‑
碘
‑5‑
甲基
‑
1,1
’‑
联苯)(3.07g,8.25mmol)、三氟甲烷磺酸(3.0ml,33.9mmol)、间氯过氧苯甲酸(含水约30%)(3.03g,12.3mmol)、脱水二氯甲烷(36ml)加入到100ml的三口圆底烧瓶中,在氮气气氛下,在室温下搅拌1小时。之后,使用旋转蒸发器蒸馏除去大约80%的溶剂。向其中添加乙醚(45ml),再在室温下进行搅拌,然后通过抽吸过滤以回收沉淀物。通过以上操作,以96%的产率得到了作为环状二苯并碘鎓化合物的中间体3a(4
‑
溴
‑2‑
甲基二苯并[b,d]碘
‑5‑
鎓三氟甲烷磺酸盐)的白色固体(4.13g,
7.93mmol)。
[0092]
<中间体4a(4
‑
溴
‑2‑
甲基二苯并[b,d]噻吩)的合成>
[0093]
将中间体3a(4
‑
溴
‑2‑
甲基二苯并[b,d]碘
‑5‑
鎓三氟甲烷磺酸盐)(4.25g,8.15mmol)、硫代乙酸钾(1.86g,16.3mmol)、氯化铜(ii)(无水)(43.5mg,0.324mmol)、用氢化钙干燥后通过蒸馏而得的脱水二甲基亚砜(30ml)加入到100ml的三口圆底烧瓶中,在氮气气氛下,于110℃加热搅拌20小时。冷却后,加入氯仿(30ml)和水(30ml)并在分液漏斗中进行震荡,除去水层。进一步用水(30ml)和饱和食盐水(30ml)清洗有机层,以无水硫酸钠干燥后,使用旋转蒸发器蒸馏除去溶剂。将残渣溶解在氯仿中,在其中加入20g硅胶后,使用旋转蒸发器蒸馏除去溶剂,以使残渣吸附在硅胶上。然后,将含有展开溶剂的100g硅胶装入色谱柱管(φ:46mm)中,将刚才吸附了残渣的硅胶装在顶部,通过迅速地展开溶剂(展开溶剂:己烷)来进行纯化。通过以上操作,以77%的产率得到了作为溴代二苯并噻吩衍生物的中间体4a(4
‑
溴
‑2‑
甲基二苯并[b,d]噻吩)的淡黄色固体(1.74g,6.27mmol)。
[0094]
<中间体5a((2
‑
甲基苯并[b,d]噻吩
‑4‑
基)硼酸)的合成>
[0095]
将中间体4a(4
‑
溴
‑2‑
甲基二苯并[b,d]噻吩)(4.24g,15.3mmol)、脱水四氢呋喃(45.0ml)加入到置于氮气气氛下的100ml三口圆底烧瓶中,并浸入到装有液氮及作为制冷剂的甲醇/丙酮的杜瓦瓶中,以将反应体系冷却至
‑
78℃。使用滴液漏斗向其中缓慢滴入1.6m的正丁基锂(12.0ml,19.2mmol),在
‑
78℃下搅拌1.5小时。之后,使用滴液漏斗滴加硼酸三异丙酯(11.0ml,71.3mmol),然后缓慢地恢复至室温并搅拌18小时。之后,加入75ml 4m盐酸,并进一步在室温下搅拌1小时。反应后,加入二氯甲烷(50ml)并在分液漏斗中进行震荡,除去水层,用无水硫酸钠进行干燥后,使用旋转蒸发器蒸馏除去溶剂。将残渣溶于10ml的二氯甲烷之后,加入50ml的己烷,则通过再沉淀析出白色固体。通过抽吸过滤回收这些沉淀。在tlc上仅确认到r
f
=0(展开溶剂:氯仿)的斑点。不用进行进一步的纯化,即用于下一步反应。通过以上操作,以67%的产率合成了中间体5a((2
‑
甲基苯并[b,d]噻吩
‑4‑
基)硼酸)(2.49g,10.3mmol)。
[0096]
·
c
‑
n配位体(2)的合成
[0097]
将碳酸钾(13.5g,97.7mmol)溶解在水(280ml)中而得的水溶液加入到2l的三口圆底烧瓶中,进一步将中间体5a((2
‑
甲基苯并[b,d]噻吩
‑4‑
基)硼酸)(6.76g,27.9mmol)、2
‑
氯喹啉(4.88g,29.8mmol)、四(三苯基膦)钯(0)(1.70g,1.47mmol)、1.2
‑
二甲氧基乙烷(280ml)、乙醇(120ml)加入到圆底烧瓶中,在氮气气氛下,于100℃加热搅拌18小时。冷却后,加入氯仿(200ml)并在分液漏斗中进行震荡,除去水层。进一步用水(200ml)及饱和食盐水(200ml)清洗有机层,以无水硫酸镁干燥后,使用旋转蒸发器蒸馏除去溶剂。通过硅胶柱层析法(展开溶剂:氯仿/己烷,v/v,2/3)对残渣进行纯化,从而以97%的产率得到了作为c
‑
n配位体(2)的2
‑
(甲基二苯并[b,d]噻吩
‑4‑
基)喹啉的淡黄色固体(8.83g,27.1mmol)。
[0098]
(a
‑
ii)前驱体(2)的合成
[0099]
将上述所合成的c
‑
n配位体(2)(0.732g,2.25mmol)、氯化铱三水合物(0.397g,1.12mmol)、2
‑
乙氧基乙醇(40ml)及水(8ml)添加到100ml的双口圆底烧瓶中,在氮气气氛下,于100℃加热搅拌20小时。冷却后,通过抽吸过滤回收反应体系中所生成的橙色沉淀。通过以上操作,合成了c
‑
n配位体(2)与铱的前驱体(2)(产率为56%(0.554g,0.316mmol))。
[0100]
(a
‑
iii)有机铱配合物(ir
‑
2)的合成
[0101]
将上述所合成的前驱体(2)(152mg,0.0867mmol)、二叔戊酰甲烷(0.1ml,0.488mmol)、碳酸钠(96.2mg,0.911mmol)以及2
‑
乙氧基乙醇(20ml)加入到50ml的双口圆底烧瓶中,在氮气气氛下,于85℃加热搅拌3小时。冷却后,加入二氯甲烷(20ml)和水(30ml)并在分液漏斗中进行震荡,除去水层。进一步,以饱和食盐水(30ml)清洗有机层,用无水硫酸钠使有机层干燥之后,使用旋转蒸发器蒸馏除去溶剂。通过硅胶柱层析法(展开溶剂:二氯甲烷/己烷,v/v,1/1)对残渣进行纯化后,加入己烷,在75℃下搅拌以进行清洗,通过抽吸过滤进行回收,从而得到了作为目标化合物的有机铱配合物(ir
‑
2)。该有机铱配合物为红色固体且产率为5.2%(9.1mg,0.00864mmol)。此外,由以上步骤所合成的化合物的通过nmr等获得的特性如下。
[0102]1h nmr(400mhz,cdcl3)δ0.55(s,18h),1.51(s,6h),4.59(s,1h),7.20
‑
7.24(m,2h),7.36
‑
7.41(m,2h),7.42
‑
7.46(m,4h),7.56(s,2h),7.68(d,j=9.2hz,2h),7.74(d,j=6.8hz,2h),7.93
‑
7.96(m,4h),8.08
‑
8.10(m,4h),8.29(d,j=9.2hz,2h),8.73(d,j=9.2hz,2h).
13
cnmr(100mhz,cdcl3)δ23.77,28.14,29.79,88.39,119.79,120.45,120.94,122.21,124.57,125.42,125.45,125.70,126.34,127.34,130.49,132.10,133.33,136.15,137.04,143.89,149.50,154.43,170.36,193.55.esi
‑
ms:m/z[m+h]+calcd.for c
55
h
48
irn2o2s2:1025.28,found:1025.27.anal.calcd.for c
55
h
47
irn2o2s2:c,64.49;h,4.63;n,2.73.found:c,64.24;h,4.67;n,2.70.
[0103]
(b)有机铱配合物ir
‑
3的合成
[0104]
(b
‑
i)c
‑
n配位体(3)(2
‑
(6,8
‑
二甲基苯并[b,d]噻吩
‑4‑
基)喹啉)的合成
[0105]
c
‑
n配位体(3)的合成是以2,6
‑
二溴苯胺为初始原料,经由中间体1b、2b、3b、4b、5b而合成的。
[0106]
<中间体1b(3
‑
溴
‑3’
,5
’‑
二甲基
‑
[1,1
’‑
联苯]
‑2‑
胺)的合成>
[0107]
将碳酸钾(13.0g,93.7mmol)溶解在水(60ml)中而得的水溶液加入到1000ml四口圆底烧瓶中,进一步将2,6
‑
二溴苯胺(10.1g,40.1mmol)、3,5
‑
二甲基苯基硼酸(4.05g,27.0mmol)、四(三苯基膦)钯(0)(1.61g,1.39mmol)、甲苯(200ml)、乙醇(60ml)加入到圆底烧瓶中,在氮气气氛下,于100℃加热搅拌20小时。冷却后,加入氯仿(150ml)并在分液漏斗中进行震荡,除去水层。进一步用水(200ml)及饱和食盐水(200ml)清洗有机层,以无水硫酸镁使有机层干燥后,使用旋转蒸发器蒸馏除去溶剂。通过硅胶柱层析法(展开溶剂:氯仿/己烷,v/v,2/3)对残渣进行纯化,从而以42%的产率得到了作为目标化合物的中间体1b(3
‑
溴
‑3’
,5
’‑
二甲基
‑
[1,1
’‑
联苯]
‑2‑
胺)的白色固体(3.13g,11.3mmol)。
[0108]
<中间体2b(3
‑
溴
‑2‑
碘
‑3’
,5
’‑
甲基
‑
1,1
’‑
联苯)的合成>
[0109]
将中间体1b(3
‑
溴
‑3’
,5
’‑
二甲基
‑
[1,1
’‑
联苯]
‑2‑
胺)(3.08g,11.2mmol)、4m的盐酸(32ml)以及四氢呋喃(32ml)加入到500ml的三口圆底烧瓶中,浸入到装有液氮及作为制冷剂的甲醇/丙酮的杜瓦瓶中,以将反应体系冷却至0℃。使用滴液漏斗向其中缓慢滴加将亚硝酸钠(1.05g,15.2mmol)溶解在水(12ml)中而得的水溶液。
[0110]
将反应体系在0℃下搅拌20分钟后,加入将碘化钾(4.74g,28.6mmol)溶解在水(18ml)中而得的水溶液。在0℃下搅拌10分钟后,除去杜瓦瓶,缓慢地恢复至室温,并进一步搅拌1小时。此时,体系中为茶色。向其中添加饱和硫代硫酸钠水溶液,直至体系中的颜色变为淡黄色,搅拌15分钟。之后,加入乙酸乙酯(100ml)并在分液漏斗中进行震荡,除去水层。
进一步用水(100ml)及饱和食盐水(100ml)清洗有机层,以无水硫酸钠使有机层干燥后,使用旋转蒸发器蒸馏除去溶剂。
[0111]
然后,将残渣溶解在氯仿中,向其中加入25g硅胶后,使用旋转蒸发器蒸馏除去溶剂,以吸附在硅胶上。然后,将含有展开溶剂的125g硅胶装入色谱柱管(φ:46mm)中,将刚刚吸附了残渣的硅胶装在顶部,通过迅速展开溶剂(展开溶剂:己烷)来进行纯化。以49%的产率得到了作为目标化合物的中间体2b(3
‑
溴
‑2‑
碘
‑3’
,5
’‑
甲基
‑
1,1
’‑
联苯)(2.11g,5.46mmol)。该化合物为高粘度的透明液体。
[0112]
<中间体3b(6
‑
溴
‑
2,4
‑
二甲基二苯并[b,d]碘
‑5‑
鎓三氟甲烷磺酸盐)的合成>
[0113]
将中间体2b(3
‑
溴
‑2‑
碘
‑3’
,5
’‑
甲基
‑
1,1
’‑
联苯)(2.08g,5.37mmol)、三氟甲烷磺酸(1.4ml,15.9mmol)、间氯过氧苯甲酸(含水约30%)(1.78g,7.22mmol)、脱水二氯甲烷(20ml)加入到100ml三口圆底烧瓶中,在氮气气氛下,于室温搅拌1小时。之后,使用旋转蒸发器蒸馏除去大致80%的溶剂。向其中加入二乙醚(20ml),于室温搅拌30分钟后,通过抽吸过滤回收所产生的沉淀物。通过以上操作,以90%的产率得到了作为环状二苯并碘鎓化合物的中间体3b(6
‑
溴
‑
2,4
‑
二甲基二苯并[b,d]碘
‑5‑
鎓三氟甲烷磺酸盐)的白色固体(2.57g,4.83mmol)。
[0114]
<中间体4b(6
‑
溴
‑
2,4
‑
甲基二苯并[b,d]噻吩)的合成>
[0115]
将中间体3b(6
‑
溴
‑
2,4
‑
二甲基二苯并[b,d]碘
‑5‑
鎓三氟甲烷磺酸盐)(2.50g,4.69mmol)、硫代乙酸钾(1.08g,9.46mmol)、氯化铜(ii)(无水)(25.3mg,0.188mmol)、由氢化钙干燥后并通过蒸馏而得到的脱水二甲基亚砜(15ml)加入到50ml双口圆底烧瓶中,在氮气气氛下,于110℃加热搅拌20小时。冷却后,加入氯仿(20ml)和水(20ml)并在分液漏斗中进行震荡,除去水层。进一步用水(20ml)和饱和食盐水(30ml)清洗有机层,以无水硫酸钠使有机层干燥后,使用旋转蒸发器蒸馏除去溶剂。将残渣溶解在氯仿中,向其中加入20g硅胶后,使用旋转蒸发器蒸馏除去溶剂,从而使残渣吸附在硅胶上。然后,在色谱柱管(φ:32mm)中装入含有展开溶剂的100g硅胶,将刚才吸附有残渣的硅胶装入顶部,通过迅速展开溶剂(展开溶剂:己烷)来进行纯化。将所得到的白色固体在乙醇中进行重结晶。通过以上操作,以43%的产率得到了作为溴代二苯并噻吩衍生物的中间体4b(6
‑
溴
‑
2,4
‑
甲基二苯并[b,d]噻吩)的白色固体(0.585g,2.01mmol)。
[0116]
<中间体5b((6,8
‑
二甲基苯并[b,d]噻吩
‑4‑
基)硼酸)的合成>
[0117]
将中间体4b(6
‑
溴
‑
2,4
‑
甲基二苯并[b,d]噻吩)(0.579g,1.99mmol)、脱水四氢呋喃(8ml)加入到置于氮气气氛下的100ml三口圆底烧瓶中,将该烧瓶浸入装有液氮及作为制冷剂的甲醇/丙酮的杜瓦瓶中,并将反应体系冷却至
‑
78℃。使用滴液漏斗向其中缓慢滴加1.6m的正丁基锂(1.6ml,2.56mmol),在
‑
78℃下搅拌1.5小时。然后,使用滴液漏斗滴加硼酸三异丙酯(3.2ml,14.0mmol)后,缓慢地恢复至室温,并搅拌18小时。之后,加入8ml 4m的盐酸,进一步在室温下搅拌1小时。反应后,加入二氯甲烷(30ml)并在分液漏斗中进行震荡,除去水层,以无水硫酸钠进行干燥后,使用旋转蒸发器蒸馏除去溶剂。将残渣溶于5ml的二氯甲烷中之后,加入30ml的己烷,则通过再沉淀析出白色固体。通过抽吸过滤回收这些沉淀。在tlc上仅确认到r
f
=0(展开溶剂:氯仿)的斑点。不用进行进一步的纯化,即用于下一步反应。通过以上操作,以60%的产率合成了中间体5b((6,8
‑
二甲基苯并[b,d]噻吩
‑4‑
基)硼酸)(0.306g,1.19mmol)。
[0118]
·
c
‑
n配位体(3)的合成
[0119]
将碳酸钾(0.516g,3.69mmol)溶解在水(12ml)中而得的水溶液加入到100ml双口圆底烧瓶中,进一步将中间体5b(6,8
‑
二甲基苯并[b,d]噻吩
‑4‑
基)硼酸)(0.298g,1.16mmol)、2
‑
氯喹啉(0.192g,1.17mmol)、四(三苯基膦)钯(0)(62.3mg,0.0537mmol)、1,2
‑
二甲氧基乙烷(12ml)、乙醇(5ml)加入到圆底烧瓶中,在氮气气氛下,在100℃下加热搅拌18小时。冷却后,加入氯仿(20ml)并在分液漏斗中进行震荡,除去水层。进一步用水(20ml)和饱和食盐水(20ml)清洗有机层,以无水硫酸镁使有机层干燥后,使用旋转蒸发器蒸馏除去溶剂。通过硅胶柱层析法(展开溶剂:氯仿/己烷,v/v,2/3)对残渣进行纯化,从而以73%的产率得到了作为c
‑
n配位体(3)的2
‑
(6,8
‑
二甲基苯并[b,d]噻吩
‑4‑
基)喹啉的白色固体。
[0120]
(b
‑
ii)前驱体(3)的合成
[0121]
将上述所合成的c
‑
n配位体(3)(0.270g,0.795mmol)、氯化铱三水合物(0.140g,0.396mmol)、2
‑
乙氧基乙醇(15ml)及水(3ml)加入到50ml双口圆底烧瓶中,在氮气气氛下,在100℃下加热搅拌20小时。冷却后,通过抽吸过滤回收反应体系中所生成的深红色沉淀。通过以上操作,合成了c
‑
n配位体(3)与铱的前驱体(3)(产率为48%(172mg,0.0954mmol))。
[0122]
(b
‑
iii)有机铱配合物(ir
‑
3)的合成
[0123]
将上述所合成的前驱体(3)(156mg,0.0862mmol)、二叔戊酰甲烷(0.1ml,0.488mmol)、碳酸钠(96.2mg,0.911mmol)及2
‑
乙氧基乙醇(20ml)加入到50ml双口圆底烧瓶中,在氮气气氛下,在100℃下加热搅拌15分钟。冷却后,加入二氯甲烷(30ml)和水(30ml)并在分液漏斗中进行震荡,除去水层。进一步用饱和食盐水(30ml)清洗有机层,以无水硫酸钠使有机层干燥后,使用旋转蒸发器蒸馏除去溶剂。以硅胶柱层析法(展开溶剂:二氯甲烷/己烷,v/v,1/1)对残渣进行纯化。然后,加入环己烷,通过在85℃下搅拌以进行溶剂清洗,冷却后,通过抽吸过滤回收沉淀的固体,从而得到了作为目标化合物的有机铱配合物(ir
‑
3)。该有机铱配合物为红色固体且产率为40%(72.5mg,0.0690mmol)。此外,以上步骤所合成的化合物的通过nmr等获得的特性如下。
[0124]1h nmr(400mhz,cd2cl2)δ0.52(s,18h),2.36(s,6h),2.58(s,6h),4.82(s,1h),6.71(d,j=8.2hz,2h),7.01(s,2h),7.27
‑
7.33(m,4h),7.43(t,j=7.1hz,2h),7.57(s,2h),7.82(d,j=7.8hz 2h),8.25(d,j=7.8hz 2h),8.38(d,j=7.8hz 2h),8.72(d,j=7.8hz,2h).
13
c nmr(100mhz,cdcl3)δ20.43,21.62,27.78,40.62,88.69,118.01,119.14,121.64,125.89,126.07,127.00,127.29,127.40,130.66,131.14,131.83,133.56,133.90,134.58,136.24,136.72,137.89,141.46,149.11,156.33,169.61,194.23.maldi tof
‑
ms:m/z[m]+calcd.for c
57
h
51
irn2o2s2:1052.30,found:1052.32.anal.calcd.for c
57
h
51
irn2o2s2:c,65.05;h,4.88;n,2.66.found:c,64.93;h,4.83;n,2.52.
[0125]
(c)有机铱配合物ir
‑
4的合成
[0126]
(c
‑
i)c
‑
n配位体(4)(2
‑
(6,6,8
‑
三甲基苯并[b,d]噻吩
‑4‑
基)喹啉)的合成
[0127]
c
‑
n配位体(4)的合成是以2,6
‑
二溴
‑4‑
甲基苯胺为初始原料,并经由中间体1c、2c、3c、4c、5c而合成的。
[0128]
<中间体1c(3
‑
溴
‑3’
,5,5
’‑
三甲基
‑
[1,1
’‑
联苯]
‑2‑
胺)的合成>
[0129]
将碳酸钾(3.96g,0.286mol)溶解于水(90ml)中而得的水溶液加入到1000ml四口圆底烧瓶中,进一步将2,6
‑
二溴
‑4‑
甲基苯胺(15.06g,56.9mmol)、3,5
‑
二甲基苯基硼酸
(5.66g,37.8mmol)、四(三苯基膦)钯(0)(2.14g,1.85mmol)、甲苯(450ml)、乙醇(90ml)加入到圆底烧瓶中,在氮气气氛下,在100℃下加热搅拌20小时。冷却后,加入氯仿(200ml)并在分液漏斗中进行震荡,除去水层。进一步以水(200ml)和饱和食盐水(200ml)清洗有机层,以无水硫酸镁使有机层干燥后,使用旋转蒸发器蒸馏除去溶剂。通过硅胶柱层析法(展开溶剂:氯仿/己烷,v/v,2/3)对残渣进行纯化。通过将所得到的淡黄色固体在乙醇中进行重结晶,从而以38%的产率得到了作为目标化合物的中间体1c(3
‑
溴
‑3’
,5,5
’‑
三甲基
‑
[1,1
’‑
联苯]
‑2‑
胺)的白色固体(4.16g,14.4mmol)。
[0130]
<中间体2c(3
‑
溴
‑2‑
碘
‑3’
,5,5
’‑
三甲基
‑
[1,1
’‑
联苯)的合成>
[0131]
将中间体1c(3
‑
溴
‑3’
,5,5
’‑
三甲基
‑
[1,1
’‑
联苯]
‑2‑
胺)(4.10g,14.1mmol)、4m的盐酸(40ml)及四氢呋喃(40ml)添加到500ml三口圆底烧瓶中,浸入到装有制冷剂甲醇/丙酮及液氮的杜瓦瓶中,以将反应体系冷却至0℃。用滴液漏斗向其中缓慢滴加将亚硝酸钠(1.21g,17.5mmol)溶于水(15ml)中而得的水溶液。
[0132]
将反应体系在0℃下搅拌20分钟后,加入将碘化钾(5.95g,35.9mmol)溶于水(25ml)而得的水溶液。在0℃下搅拌10分钟后,除去杜瓦瓶,缓慢恢复至室温同时进一步搅拌1小时。此时,体系中为茶色。向其中加入饱和硫代硫酸钠水溶液,直至体系中的颜色变为淡黄色,搅拌15分钟。之后,加入乙酸乙酯(150ml)并在分液漏斗中进行震荡,除去水层。进一步以水(200ml)和饱和食盐水(200ml)清洗有机层,以无水硫酸钠使有机层干燥后,使用旋转蒸发器蒸馏除去溶剂。
[0133]
然后,将残渣溶解在氯仿中,向其中加入50g硅胶后,使用旋转蒸发器蒸馏除去溶剂,从而使残渣吸附在硅胶上。之后,将含有展开溶剂的250g硅胶装入色谱柱管(φ:60mm)中,将刚才吸附了残渣的硅胶装在顶部。迅速展开溶剂,以进行纯化。展开溶剂使用己烷。通过以上步骤,以49%的产率得到了作为目标化合物的中间体2c(3
‑
溴
‑2‑
碘
‑3’
,5,5
’‑
三甲基
‑
1,1
’‑
联苯)(2.77g,6.90mmol)。该化合物为高粘性的透明液体。
[0134]
<中间体3c(4
‑
溴
‑
2,6,8
‑
三甲基二苯并[b,d]碘
‑5‑
鎓三氟甲烷磺酸盐)的合成>
[0135]
将中间体2c(3
‑
溴
‑2‑
碘
‑3’
,5,5
’‑
三甲基
‑
1,1
’‑
联苯)(2.65g,6.61mmol)、三氟甲烷磺酸(1.7ml,19.3mmol)、间氯过氧苯甲酸(含水约30%)(2.14g,8.66mmol)、脱水二氯甲烷(25ml)加入到100ml三口圆底烧瓶中,在氮气气氛下,于室温搅拌1小时。之后,使用旋转蒸发器蒸馏除去大约80%的溶剂。向其中加入二乙醚(25ml),在室温下搅拌30分钟,然后通过抽吸过滤回收所产生的沉淀。通过以上操作,以93%的产率得到了作为环状二苯并碘鎓化合物的中间体3c(4
‑
溴
‑
2,6,8
‑
三甲基二苯并[b,d]碘
‑5‑
鎓三氟甲烷磺酸盐)的白色固体(3.38g,6.15mmol)。
[0136]
<中间体4c(4
‑
溴
‑
2,6,8
‑
三甲基二苯并[b,d]噻吩)的合成>
[0137]
将中间体3c(4
‑
溴
‑
2,6,8
‑
三甲基二苯并[b,d]碘
‑5‑
鎓三氟甲烷磺酸盐)(3.35g,6.10mmol)、硫代乙酸钾(1.40g,12.2mmol)、氯化铜(ii)(无水)(32.4mg,0.241mmol)、由氢化钙干燥后并通过蒸馏所得到的脱水二甲基亚砜(25ml)添加到50ml双口圆底烧瓶中,在氮气气氛下,在110℃下加热搅拌20小时。冷却后,加入氯仿(30ml)和水(30ml)并在分液漏斗中进行震荡,除去水层。进一步以水(40ml)和饱和食盐水(30ml)清洗有机层,以无水硫酸钠使有机层干燥后,使用旋转蒸发器蒸馏除去溶剂。将残渣溶解在氯仿中,向其中加入20g硅胶后,使用旋转蒸发器蒸馏除去溶剂,以使残渣吸附在硅胶上。然后,将含有展开溶剂的
100g硅胶装入色谱柱管(φ:32mm)中,并将刚才吸附了残渣的硅胶装在顶部,通过迅速地展开溶剂(展开溶剂:己烷)以进行纯化。将所得到的白色固体在乙醇中进行重结晶。通过以上操作,以47%的产率得到了作为溴代二苯并噻吩衍生物的中间体4c(4
‑
溴
‑
2,6,8
‑
三甲基二苯并[b,d]噻吩)的白色固体(0.876g,2.87mmol)。
[0138]
<中间体5c((2,6,8
‑
三甲基二苯并[b,d]噻吩
‑4‑
基)硼酸)的合成>
[0139]
将中间体4c(4
‑
溴
‑
2,6,8
‑
三甲基二苯并[b,d]噻吩)(0.865g,2.83mmol)、脱水四氢呋喃(12ml)加入到置于氮气气氛下的100ml三口圆底烧瓶中,浸入到装有液氮及作为制冷剂的甲醇/丙酮的杜瓦瓶中,以将反应体系冷却至
‑
78℃。使用滴液漏斗向其中缓慢滴加1.6m的正丁基锂(2.5ml,3.88mmol),并在
‑
78℃下搅拌1.5小时。之后,使用滴液漏斗滴加硼酸三异丙酯(4.8ml,20.9mmol)后,除去杜瓦瓶,在缓慢恢复至室温的同时搅拌18小时。之后,加入12ml4m的盐酸,进一步在室温下搅拌1小时。反应后,加入二氯甲烷(30ml)并在分液漏斗中进行震荡,除去水层,然后以无水硫酸钠进行干燥,并使用旋转蒸发器蒸馏除去溶剂。将残渣溶于10ml的二氯甲烷后,加入50ml的己烷,通过抽吸过滤以回收沉淀的白色固体。以62%的产率合成了中间体5c((2,6,8
‑
三甲基二苯并[b,d]噻吩
‑4‑
基)硼酸)(0.474g,1.75mmol)。
[0140]
·
c
‑
n配位体(4)的合成
[0141]
将碳酸钾(0.782g,5.66mmol)溶解在水(20ml)中而得的水溶液加入到100ml三口圆底烧瓶中,进一步将中间体5c((2,6,8
‑
三甲基二苯并[b,d]噻吩
‑4‑
基)硼酸)(0.470g,1.74mmol)、2
‑
氯喹啉(0.290g,1.77mmol)、四(三苯基膦)钯(0)(92.1mg,0.0797mmol)、1,2
‑
二甲氧基乙烷(20ml)、乙醇(8ml)加入到圆底烧瓶中,在氮气气氛下于100℃加热搅拌18小时。冷却后,加入氯仿(50ml)并在分液漏斗中进行震荡,除去水层。进一步用水(50ml)和饱和食盐水(50ml)清洗有机层,用无水硫酸镁使有机层干燥后,使用旋转蒸发器蒸馏除去溶剂。通过硅胶柱层析法(展开溶剂:氯仿/己烷,v/v,4/1)对残渣进行纯化,从而以73%的产率得到了作为c
‑
n配位体(3)的2
‑
(2,6,8
‑
三甲基苯并[b,d]噻吩
‑4‑
基)喹啉的白色固体(0.466g,1.27mmol)。
[0142]
(c
‑
ii)前驱体(4)的合成
[0143]
将上述所合成的c
‑
n配位体(4)(0.410g,1.15mmol)、氯化铱三水合物(0.204g,0.581mmol)、2
‑
乙氧基乙醇(30ml)及水(6ml)加入到100ml双口圆底烧瓶中,在氮气气氛下于110℃加热搅拌20小时。冷却后,通过抽吸过滤回收在反应体系中所产生的红色沉淀。通过以上操作,合成了c
‑
n配位体(4)与铱的前驱体(4)(产率为47%(0.252g,0.135mmol))。
[0144]
(c
‑
iii)有机铱配合物(ir
‑
4)的合成
[0145]
将上述所合成的前驱体(4)(212mg,0.114mmol)、二叔戊酰甲烷(0.1ml,0.488mmol)、碳酸钠(110mg,1.04mmol)及2
‑
乙氧基乙醇(30ml)加入到50ml双口圆底烧瓶中,在氮气气氛下于85℃加热搅拌3小时。冷却后,加入二氯甲烷(30ml)和水(30ml)并在分液漏斗中进行震荡,除去水层。进一步用饱和食盐水(30ml)清洗有机层,以无水硫酸钠使有机层干燥后,使用旋转蒸发器蒸馏除去溶剂。通过硅胶柱层析法(展开溶剂:二氯甲烷/己烷,v/v,1/1)对残渣进行纯化后,加入己烷,在75℃下搅拌以进行溶剂清洗,通过抽吸过滤得到了作为目标化合物的有机铱配合物(ir
‑
4)。该有机铱配合物为红色固体且产率为3.8%(9.4mg,0.0866mmol)。此外,由以上步骤所合成的化合物的通过nmr等所获得的特性
如下。
[0146]1h nmr(400mhz,cdcl3)δ0.54(s,18h),1.49(s,3h),2.49(s,3h),2.68(s,3h),4.58(s,1h),7.08(s,2h),7.16
‑
7.21(m,2h),7.35
‑
7.37(m,2h),7.51(s,2h),7.66(d,j=8.7hz,2h),7.72
‑
7.74(m,4h),8.28(d,j=8.7hz,2h),8.79(d,j=9.2hz,2h).
13
c nmr(100mhz,cdcl3)δ20.54,21.59,23.75,28.15,40.61,88.36,118.20,119.90,121.17,124.85,125.58,126.39,127.30,127.48,130.39,131.38,132.69,134.14,134.60,136.04,136.84,142.31,143.80,148.81,159.68,168.49,187.84,193.55.esi
‑
ms:m/z[m+h]+calcd.for c
59
h
56
irn2o2s2:1081.34,found:1081.37.anal.calcd.for c
59
h
55
irn2o2s2:c,65.59;h,5.13;n,2.59.found:c,65.37;h,4.77;n,2.59.
[0147]
(ii)各有机铱配合物的特性评价
[0148]
对以上述方式所合成的有机铱配合物(ir
‑
1~ir
‑
4)进行光学特性(发光光谱、pl量子产率)的测定和评价。此外,使用各铱配合物制作oled,并对el特性进行评价。
[0149]
[有机铱配合物的发光特性的测定/评价]
[0150]
对于各有机铱配合物,测定其发光(pl)光谱及pl量子产率φ
pl
。使用“堀場製作所社”制的fluorolog
‑
3分光光度计来进行pl光谱的测定。使用“浜松
ホトニクス
社”制的c9920
‑
12量子产率测定装置来进行pl量子产率的测定。
[0151]
对于发光特性的评价,进行了以下评价:在作为介质的有机溶剂(二氯甲烷(ch2cl2)、甲苯)中的特性评价、以及在高分子薄膜(聚甲基丙烯酸甲酯(pmma))中的特性评价。有机溶剂均为光谱分析用二氯甲烷和光谱分析用甲苯,通过将样品放入光路长为1cm的样品池中进行测定。
[0152]
此外,pmma薄膜是通过如下方法制得的:在氩气氛下,将1gpmma和0.04mmol有机铱配合物溶于脱水甲苯中,该溶液经由注射器式过滤器而滴下,并通过旋涂来制膜(2000rpm,2秒;4000rpm,60秒)。然后,在115℃下烧制1小时,并作为pmma薄膜进行测定。
[0153]
<在有机溶剂中的发光特性>
[0154]
图1、图2示出本实施方式中所合成的有机铱配合物ir
‑
1~ir
‑
4在有机溶剂(二氯甲烷溶液、甲苯溶液)中的发光光谱。此外,将在有机溶剂中的发光特性的测定结果汇总并示于表1、表2。
[0155]
[表1]
[0156][0157]
[表2]
[0158][0159]
本实施方式所合成的有机铱配合物ir
‑
1~ir
‑
4的发光波长(λ
pl
)在二氯甲烷溶液中为619nm~629nm(表1)、在甲苯溶液中为616nm~627nm(表2),显示红橙色或红色的发光。另外,在任意一种有机溶剂中,相对于未取代的有机铱配合物(ir
‑
1),本实施方式涉及的引入有甲基的有机铱配合物(ir
‑
2~ir
‑
4)均显示出发光向长波长侧移动(红移)。此外,若参照发光的半峰全宽(fwhm),则与ir
‑
1相比,ir
‑
2~ir
‑
4倾向于更小。因此可以确认,本实施方式涉及的引入有甲基的有机铱配合物(ir
‑
2~ir
‑
4)通过引入甲基而显示出色纯度得以提高的红色磷光。
[0160]
若参照pl量子效率(φ
pl
)的测定结果,则任意一种有机铱配合物均显示出超过0.7的高值。另外,可以确认,在任意一种有机溶剂中,相对于未取代的有机铱配合物(ir
‑
1),本实施方式所涉及的引入有甲基的有机铱配合物(ir
‑
2~ir
‑
4)的φ
pl
均得以提高。
[0161]
<pmma薄膜中的发光特性>
[0162]
图3示出包含本实施方式所合成的有机铱配合物ir
‑
1~ir
‑
4的pmma薄膜的发光光谱。此外,将在有机溶剂中的发光特性的测定结果汇总并显示于表3。
[0163]
[表3]
[0164][0165]
本实施方式所合成的有机铱配合物ir
‑
1~ir
‑
4在pmma薄膜中的发光特性与在上述有机溶剂中的发光特性的倾向类似。它们的λ
pl
显示出616nm~627nm的红橙色或红色的发光。另外,相对于作为现有例的未取代的有机铱配合物(ir
‑
1),本实施方式所涉及的引入有甲基的有机铱配合物(ir
‑
2~ir
‑
4)的发光发生了红移。并且,如参照φ
pl
,则任意一种有机铱配合物均显示出超过0.75的值。特别是,相对于ir
‑
1,ir
‑
2、ir
‑
4的φ
pl
得以提高,为超过0.85的高值。
[0166]
此外,从ir
‑
3的结果来看,fwhm比ir
‑
1的宽,φ
pl
也是比ir
‑
1低的值。由此可以认为,作为甲基的引入位置,应优先引入至2’位。虽然ir
‑
3的发光特性稍差,但是由于其相对于未取代的配合物具有红移,因此是有意义的配合物。
[0167]
[有机铱配合物的电致发光特性的评价]
[0168]
为了研究本实施方式所合成的有机铱配合物(ir
‑
1~ir
‑
4)的电致发光特性,制作有机电致发光元件(oled)并对其性能进行评价。
[0169]
图4示出本实施方式所制作的oled的结构。在oled的制作中,通过旋涂,在形成有阳极(ito)膜的玻璃基板(20
×
25mm)上,涂布作为空穴注入层的、溶于异丙醇/水=1/1(v/v)中的pedot:pss(聚(3,4
‑
亚乙基二氧噻吩)
‑
聚(苯乙烯磺酸)),并进行烧制。
[0170]
接着,将作为发光掺杂剂的有机铱配合物(ir
‑
1~ir
‑
4)、作为电子传输材料的pbd(2
‑
(4
‑
叔丁基苯基)
‑5‑
(4
‑
联苯基)
‑
1,3,4
‑
噁二唑)、以及作为具有空穴传输性的主体材料的pvcz(聚(n
‑
乙烯基咔唑))以0.4:6:10的质量比进行混合并溶解在甲苯中而得到溶液,通过旋涂来涂布该溶液,并进行烧制,从而形成发光层。
[0171]
然后,通过真空沉积在发光层上依次层叠氟化铯、铝,从而形成阴极。将使用由以上步骤所制作的ir
‑
1~ir
‑
4的oled分别称为device 1~device 4。
[0172]
<oled特性的评价>
[0173]
使用载玻片和uv固化环氧树脂,对由上述步骤所得到的oled(device 1~device 4)进行密封,从而制作有机el特性评价用的样品。并且,对电致发光特性进行评价。在本实施方式中,通过亮度配光特性测定装置(“浜松
ホトニクス
社”制、c
‑
9920
‑
11)来测定各device的el光谱、最大亮度l
max
(cd/m2)、最大外部量子效率η
ext
,max(%)、及cie表色系统(x,y)等oled特性。
[0174]
表4示出device 1~device 4的发光起始电压v
turn
‑
on
(v)、最大亮度l
max
(cd/m2)、最大外部量子效率η
ext,max
(%)、最大电流效率η
j,max
(cd/a)、最大功率η
p,max
(lm/w)、el光谱中的峰值波长λ
el
(nm)、以及cie(x,y)的结果。发光起始电压v
turn
‑
on
表示亮度达到1cd/m2的电压。此外,对于l
max
等,测量时的施加电压(@v)也同时显示在[]内。需要说明的是,cie(x,y)是最大亮度时的发光cie色度坐标。
[0175]
此外,图5、图6示出在各oled器件的最大亮度l
max
时所测定的电致发光(el)光谱和j
‑
v
‑
l曲线。
[0176]
[表4]
[0177][0178]
当对el光谱和电致发光波长(λ
el
)进行研究时,device 1~device 4显示出λ
el
为
615nm~633nm的红色电致发光。该结果与在上述有机溶剂中和pmma薄膜中所显示的光谱是一样的。另外,与device 1(ir
‑
1)相比,掺杂引入有甲基的有机铱配合物(ir
‑
2~ir
‑
4)而得的decice 2~device 4获得了更长波长的电致发光。此外,el光谱的fwhm也变窄。因此确认了,本实施方式的有机铱配合物(ir
‑
2~ir
‑
4)作为红色磷光材料且作为oled发光层的发光掺杂剂是有效的。
[0179]
工业实用性
[0180]
本发明的有机铱配合物是pl量子效率(φ
pl
)高且色纯度(红色)优异的红色磷光材料。根据本发明,在使用有机电致发光元件的显示器等中,作为用于红色发光的掺杂剂是有用的。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1