树脂组合物、固化物、衍射光学元件、多层型衍射光学元件的制作方法
1.本发明涉及一种树脂组合物。本发明尤其涉及一种包含ito粒子的树脂组合物。本发明还涉及一种使用上述树脂组合物获得的固化物、衍射光学元件及多层型衍射光学元件。
背景技术:
2.利用衍射光学元件,可获得波长越长则焦点距离越短,并且显现与以往的折射型透镜相反的色差的透镜。与为了校正色差而需要多个透镜的折射型透镜不同,能够通过改变透镜的衍射结构的周期来校正色差,因此能够利用衍射光学元件来设计更紧凑且高性能的透镜单元。
3.在具有由相互不同的2种材料分别形成的衍射光学元件在彼此的晶格面接触的结构的多层型衍射光学元件中,由相对高折射率且高阿贝数的材料形成其中一个衍射光学元件,由相对低折射率且低阿贝数的材料形成另一个衍射光学元件,由此抑制透镜内的光斑的产生等,能够充分地利用色差减少作用。此时,若具有在更长的波长下2个衍射光学元件的折射率差更大的光学特性,则能够在宽的波长范围内获得色差减少作用。
4.近年来,如上所述,为了在宽的波长范围内获得色差减少作用,提出了在用作用于多层型衍射光学元件中的低阿贝数衍射光学元件的制作的材料的固化性树脂组合物中添加ito(氧化铟锡)粒子。例如,在专利文献1中公开了一种固化性树脂组合物,该固化性树脂组合物的特征在于,作为用于制作衍射光学元件的固化性树脂组合物,在含有光聚合引发剂、分散剂及两个以上的丙烯酰基、甲基丙烯酸基或乙烯基或者这些不饱和乙烯基的混合体的树脂中分散有ito微粒。
5.以往技术文献
6.专利文献
7.专利文献1:日本特开2006
‑
220689号公报
技术实现要素:
8.发明要解决的技术课题
9.在专利文献1中所记载的技术中,通过添加ito粒子,由固化性树脂组合物获得的固化物的近红外波长区域的折射率下降并且改善了色差减少作用。但是,已知在利用近红外波长区域的光的光学系统中由添加ito粒子导致的近红外波长区域的透射率下降成为问题。
10.本发明的课题在于提供一种适合于制造衍射光学元件的树脂组合物。更详细而言,本发明的课题在于提供一种树脂组合物,其包含ito粒子,该树脂组合物中,在保持与专利文献1相同程度的折射率的波长依赖性的状态下,近红外波长区域的透射率高。
11.用于解决技术课题的手段
12.为了解决上述课题,本发明人等进行深入研究,通过提高短波长侧的折射率并调
节折射率的波长依赖性,可减少ito粒子的添加量而提高近红外波长区域的透射率,基于该构思进一步重复进行研究而完成了本发明。
13.即,用于解决上述课题的具体方式如下。
14.[1]一种树脂组合物,其包含氧化铟锡粒子,上述树脂组合物包含近紫外光吸收性有机化合物,
[0015]
上述近紫外光吸收性有机化合物中,在从波长800nm进行吸光度测定时最初显示极大值的最初波长在340nm~400nm,并且,
[0016]
在将340nm~400nm处的最大吸光度设为abs(λmax),将波长410nm处的吸光度设为abs(410nm),将波长430nm处的吸光度设为abs(430nm)时,同时满足以下2个式。
[0017]
(abs(λmax)
‑
abs(410nm))/abs(λmax)≥0.97
[0018]
1.00≥(abs(λmax)
‑
abs(410nm))/(abs(λmax)
‑
abs(430nm))≥0.97
[0019]
[2]根据[1]所述的树脂组合物,其中,
[0020]
上述近紫外光吸收性有机化合物中的下述p1为0.005以上。
[0021]
p1=(abs(λmax)
‑
abs(410nm))/(410
‑
λmax)
[0022]
[3]根据[1]或[2]所述的树脂组合物,其中,
[0023]
上述近紫外光吸收性有机化合物为由下述通式1表示的化合物;
[0024]
[化学式1]
[0025]
pol1‑
sp1‑
l1‑
ar
‑
l2‑
sp2‑
pol2ꢀꢀꢀꢀ
(通式1)
[0026]
式中,ar为由下述通式2
‑
1~2
‑
4表示的任一个芳香环基;
[0027]
[化学式2]
[0028][0029]
式中,q1表示
‑
s
‑
、
‑
o
‑
或nr
11
‑
,r
11
表示氢原子或碳原子数1~6的烷基,
[0030]
y1表示可以具有取代基的碳原子数1~6的烷基、可以具有取代基的碳原子数6~12的芳香族烃基或可以具有取代基的碳原子数3~12的芳香族杂环基,
[0031]
z1、z2及z3分别独立地表示氢原子、可以具有取代基的碳原子数1~20的脂肪族烃基、可以具有取代基的碳原子数1~20的烷氧基、可以具有取代基的碳原子数3~20的脂环式烃基、可以具有取代基的1价的碳原子数6~20的芳香族烃基、卤原子、氰基、硝基、
‑
nr
12
r
13
或sr
12
,z1与z2可以彼此键合而形成可以具有取代基的芳香族烃环或可以具有取代基的芳香族杂环,r
12
及r
13
分别独立地表示氢原子或碳原子数1~6的烷基,
[0032]
a1及a2各自独立地表示选自包含
‑
o
‑
、
‑
nr
21
‑
、
‑
s
‑
及co
‑
的组中的基团,r
21
表示氢原子或取代基,
[0033]
x表示o、s、氢原子或取代基所键合的c、或者氢原子或取代基所键合的n,
[0034]
ax表示具有选自包含芳香族烃环及芳香族杂环的组中的至少一种芳香环的碳原子数1~30的有机基团,ay表示氢原子、可以具有取代基的碳原子数1~6的烷基或具有选自包含芳香族烃环及芳香族杂环的组中的至少一种芳香环的碳原子数1~30的有机基团,ax及ay所具有的芳香环可以具有取代基,ax与ay可以彼此键合而形成可以具有取代基的环,
[0035]
q2表示氢原子或可以具有取代基的碳原子数1~6的烷基,
[0036]
l1、l2分别独立地表示单键或选自包含
‑
o
‑
、
‑
s
‑
、
‑
c(=o)
‑
、
‑
oc(=o)
‑
、
‑
c(=o)o
‑
、
‑
oc(=o)o
‑
、
‑
nr
101
c(=o)
‑
、
‑
c(=o)nr
102
‑
、
‑
oc(=o)nr
103
‑
、
‑
nr
104
c(=o)o
‑
、
‑
sc(=o)
‑
或
‑
c(=o)s
‑
的组中的连结基团,r
101
、r
102
、r
103
、r
104
分别独立地表示
‑
sp3‑
pol3或卤原子,
[0037]
sp1、sp2分别独立地表示单键或选自包含可以具有取代基的碳原子数1至30的直链亚烷基及在可以具有取代基的碳原子数2至30的直链亚烷基中一个或者不相邻的两个以上的
‑
ch2‑
被
‑
o
‑
、
‑
s
‑
、
‑
c(=o)
‑
、
‑
oc(=o)
‑
、
‑
c(=o)o
‑
、
‑
oc(=o)o
‑
、
‑
nr
201
c(=o)
‑
、
‑
c(=o)nr
202
‑
、
‑
oc(=o)nr
203
‑
、
‑
nr
204
c(=o)o
‑
、
‑
sc(=o)
‑
或
‑
c(=o)s
‑
替换的基团的组中的连结基团,
[0038]
r
201
、r
202
、r
203
、r
204
分别独立地表示
‑
sp4‑
pol4或卤原子,
[0039]
sp3、sp4分别独立地表示单键或2价连结基团,
[0040]
pol1、pol2、pol3、pol4分别独立地表示氢原子或聚合性基团,
[0041]
由通式1表示的化合物具有至少一个聚合性基团。
[0042]
[4]根据[3]所述的树脂组合物,其中,
[0043]
ar为由通式2
‑
2表示的芳香环基。
[0044]
[5]根据[3]或[4]所述的树脂组合物,其中,
[0045]
l1及l2均为
‑
o
‑
、
‑
oc(=o)
‑
、
‑
oc(=o)o
‑
或o
‑
c(=o)nh
‑
。
[0046]
[6]根据[5]所述的树脂组合物,其中,
[0047]
l1及l2均为
‑
o
‑
。
[0048]
[7]根据[3]至[6]中任一项所述的树脂组合物,其中,
[0049]
上述聚合性基团均为(甲基)丙烯酰氧基。
[0050]
[8]根据[3]至[7]中任一项所述的树脂组合物,其中,
[0051]
pol1或pol2中的任一者为(甲基)丙烯酰氧基。
[0052]
[9]一种固化物,其为[1]至[8]中任一项所述的树脂组合物的固化物,其中,
[0053]
波长486.13nm处的折射率为1.42以上且1.60以下,阿贝数为15以上且25以下。
[0054]
[10]一种衍射光学元件,其包含[9]所述的固化物,
[0055]
包含由上述固化物形成的具有衍射光栅形状的面。
[0056]
[11]一种多层型衍射光学元件,其包含第1衍射光学元件和第2衍射光学元件,
[0057]
第1衍射光学元件为[10]所述的衍射光学元件,
[0058]
第1衍射光学元件的具有衍射光栅形状的上述面与第2衍射光学元件的具有衍射光栅形状的面对置。
[0059]
[12]根据[11]所述的多层型衍射光学元件,其中,
[0060]
第2衍射光学元件的波长486.13nm处的折射率为1.55以上且1.70以下,并且比第1衍射光学元件的波长486nm处的折射率大,第2衍射光学元件的阿贝数为35以上且60以下。
[0061]
[13]根据[11]或[12]所述的多层型衍射光学元件,其中,
[0062]
第1衍射光学元件的具有衍射光栅形状的上述面与第2衍射光学元件的具有衍射光栅形状的上述面接触。
[0063]
[14]根据[11]至[13]中任一项所述的多层型衍射光学元件,其包含透明基板,
[0064]
依次配置有第1衍射光学元件、第2衍射光学元件及上述透明基板。
[0065]
发明效果
[0066]
根据本发明,提供一种包含ito粒子的新型树脂组合物。本发明的树脂组合物的固化物能够用作用于多层型衍射光学元件中的低阿贝数衍射光学元件的制作的材料而赋予优异的衍射效率,并且近红外波长区域中的光的透射率高。
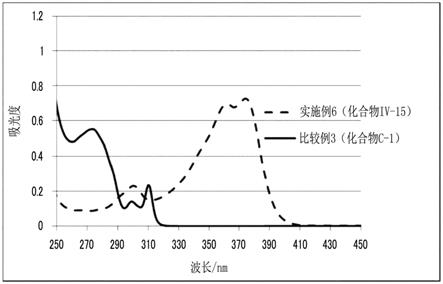
附图说明
[0067]
图1是表示实施例6(化合物iv
‑
15)及比较例3(化合物c
‑
1)的吸收光谱的图。
具体实施方式
[0068]
以下,对本发明详细地进行说明。以下所记载的构成要件的说明有时基于具有代表性的实施方式或具体例完成,但本发明并不限定于这种实施方式。另外,在本说明书中使用“~”表示的数值范围是指包含记载于“~”前后的数值作为下限值及上限值的范围。
[0069]
另外,本说明书中,“(甲基)丙烯酸酯”表示丙烯酸酯及甲基丙烯酸酯中的任一个或两个,“(甲基)丙烯酰基”表示丙烯酰基及甲基丙烯酰基中的任一个或两个。本发明中的单体与低聚物及聚合物有区分,是指重均分子量为1,000以下的化合物。
[0070]
在本说明书中,在称为脂肪族烃基时,表示从直链或者支链的烷烃、直链或者支链的烯烃或直链或者支链的炔烃去除一个任意的氢原子而得到的基团。在本说明书中,脂肪族烃基优选为从直链或者支链的烷烃去除一个任意的氢原子而得到的烷基。
[0071]
在本说明书中,在称为烷基时,表示直链或者支链的烷基。作为烷基,可举出甲基、乙基、丙基、异丙基、丁基、异丁基、仲丁基、叔丁基、戊基、1
‑
甲基丁基、3
‑
甲基丁基、己基、1
‑
甲基戊基、4
‑
甲基戊基、庚基、1
‑
甲基己基、5
‑
甲基己基、辛基、1
‑
甲基庚基、壬基、1
‑
甲基辛基、癸基、十一烷基、十二烷基、十三烷基、十四烷基、十五烷基、十六烷基、十七烷基、十八烷基、十九烷基、二十烷基等。关于含有烷基的基团(烷氧基、烷氧基羰基、酰基等)中的烷基也相同。
[0072]
并且,在本说明书中,作为直链亚烷基的例子,可举出上述烷基中从直链烷基分别去除一个与末端的碳键合的氢原子而得到的基团。
[0073]
在本说明书中,作为脂环式烃环的例子,可举出环丙烷、环丁烷、环戊烷、环己烷、环庚烷、环辛烷、环壬烷、环癸烷。
[0074]
在本说明书中,作为不饱和烃环的例子,可举出茚、茚满、芴。
[0075]
在本说明书中,在称为脂环式烃基时,表示从环烷烃去除一个任意的氢原子而得到的环烷基。作为脂环式烃基的例子,可举出环丙基、环丁基、环戊基、环己基、环庚基、环辛基、环壬基、环癸基等,优选为碳原子数3~12的环烷基。
[0076]
在本说明书中,亚环烷基表示从环烷烃去除两个任意的氢原子而得到的2价基团。作为亚环烷基的例子,可举出亚环己基。
[0077]
在本说明书中,在称为芳香环时,是指芳香族烃环及芳香族杂环中的任一个或这
两个。
[0078]
在本说明书中,作为芳香族烃环的例子,可举出苯、联苯、亚联苯、萘、蒽、菲。
[0079]
在本说明书中,在称为芳香族烃基时,表示从芳香族烃环去除一个任意的氢原子而得到的1价的基团。作为例子,可举出苯基、联苯基、1
‑
萘基、2
‑
萘基、1
‑
蒽基、2
‑
蒽基、3
‑
蒽基、4
‑
蒽基、9
‑
蒽基、1
‑
菲基、2
‑
菲基、3
‑
菲基、4
‑
菲基、9
‑
菲基等。在本说明书中,在称为2价的芳香族烃基时,表示从芳香族烃环去除两个任意的氢原子而得到的2价基团,作为例子,可举出从上述(1价的)芳香族烃基去除一个任意的氢原子而得到的2价基团。
[0080]
在本说明书中,作为芳香族杂环的例子,可举出呋喃、噻吩、吡咯、咪唑、异噻唑、异噁唑、吡啶、吡嗪、喹啉、苯并呋喃、苯并噻唑、苯并噁唑等。
[0081]
在本说明书中,在称为芳香族杂环基时,表示从芳香族杂环去除一个任意的氢原子而得到的1价的基团。作为1价的芳香族杂环基的例子,可举出呋喃基、噻吩基(优选为2
‑
噻吩基)、吡咯基、咪唑基、异噻唑基、异噁唑基、吡啶基、吡嗪基、喹啉基、苯并呋喃基(优选为2
‑
苯并呋喃基)、苯并噻唑基(优选为2
‑
苯并噻唑基)、苯并噁唑基(优选为2
‑
苯并噁唑基)等。在本说明书中,在称为2价的芳香族杂环基时,表示从芳香族杂环去除两个任意的氢原子而得到的2价基团,作为例子,可举出从上述(1价的)芳香族杂环基去除一个任意的氢原子而得到的2价基团。
[0082]
<<树脂组合物>>
[0083]
本发明的树脂组合物包含氧化铟锡粒子及近紫外光吸收性有机化合物。本发明的树脂组合物除了这些成分以外,还可以包含其他成分。对以下各成分进行说明。
[0084]
<近紫外光吸收性有机化合物>
[0085]
本发明的树脂组合物包含在近紫外波长区域中显示光吸收的近紫外光吸收性有机化合物。近紫外光吸收性有机化合物的上述光吸收并不涉及到可见光区域,近紫外光吸收性有机化合物在波长430~800nm处实质上不显示光吸收。通过在包含氧化铟锡粒子的树脂组合物中添加这种近紫外光吸收性有机化合物,即使氧化铟锡粒子添加量少,在用作多层型衍射光学元件中的低折射率且低阿贝数的材料时,也能够在宽的波长范围内获得色差减少作用。并且,能够减少氧化铟锡粒子添加量,因此能够抑制近红外波长区域中的透射率下降。
[0086]
具体而言,近紫外光吸收性有机化合物中,在从波长800nm进行吸光度测定时最初显示极大值的波长在340nm~400nm。即,在340nm~800nm的波长区域的吸收光谱中仅在340nm~400nm的范围内,具备具有极大值的吸光度的峰值。在340nm~400nm的范围内的极大值可以是一个,也可以是两个以上。在从波长800nm进行吸光度测定时,最初显示极大值的波长优选在340nm~385nm,更优选在350nm~380nm。并且,340nm~400nm的范围的极大值中显示最大吸光度的极大值优选在340nm~385nm,更优选在350nm~380nm。在此,吸收光谱只要是用近紫外光吸收性有机化合物的溶液测定即可,通过将只有溶剂的比色池放置于试样光路、对照光路并将吸光度调节至零之后,将试样光路侧比色池替换为近紫外光吸收性有机化合物的溶液并进行测定而获得。
[0087]
并且,将上述在340nm~400nm的范围内的极大值中最大的极大值(在340nm~400nm的范围内的极大值中吸光度最大的波长下的吸光度)设为abs(λmax),将上述吸收光谱的波长410nm处的吸光度设为abs(410nm),将上述吸收光谱的波长430nm处的吸光度设为
abs(430nm)时,近紫外光吸收性有机化合物只要满足以下式即可。
[0088]
(abs(λmax)
‑
abs(410nm))/abs(λmax)≥0.97
[0089]
1.00≥(abs(λmax)
‑
abs(410nm))/(abs(λmax)
‑
abs(430nm))≥0.97
[0090]
优选为只要满足以下式即可。
[0091]
(abs(λmax)
‑
abs(410nm))/abs(λmax)≥0.98
[0092]
1.00≥(abs(λmax)
‑
abs(410nm))/(abs(λmax)
‑
abs(430nm))≥0.98
[0093]
即,abs(410nm)及abs(430nm)均为小于abs(λmax)的值,并且为实质上接近0的值。
[0094]
上述吸收光谱的测定条件并无特别限定。作为一例,能够使用近紫外光吸收性有机化合物的20mg/l溶液,并利用shimadzu corporation制造的uv
‑
2550进行测定。另外,优选在该条件下满足以下内容。
[0095]
(abs(λmax)
‑
abs(410nm))/(410
‑
λmax)≥0.005
[0096]
关于测定上述吸收光谱时的溶剂,只要是能够溶解近紫外光吸收性有机化合物的溶剂,则并无特别限定,例如,能够使用四氢呋喃。
[0097]
本发明的树脂组合物中所包含的近紫外光吸收性有机化合物优选为聚合性化合物。即,近紫外光吸收性有机化合物优选为具有聚合性基团的化合物。作为聚合性基团,例如,可举出由以下式pol
‑
1~式pol
‑
6表示的聚合性基团。
[0098]
[化学式3]
[0099][0100]
*表示键合位置。
[0101]
这些中,优选为(甲基)丙烯酰氧基(pol
‑
1、pol
‑
2)。近紫外光吸收性有机化合物只要具有1个以上的聚合性基团即可,优选具有1~4个,更优选具有1~2个。
[0102]
本发明的树脂组合物中所包含的近紫外光吸收性有机化合物也优选为包含芳香环作为部分结构的化合物。
[0103]
[由通式1表示的化合物]
[0104]
本发明的树脂组合物中所包含的近紫外光吸收性有机化合物优选为由下述通式1表示的化合物。由通式1表示的化合物在其结构中包含苯并二硫醇或苯并噻唑等苯和杂环的稠环或具有腙等作为取代基的苯环。本发明人等发现了:由通式1表示的化合物具有上述光谱特性,并且由含有由通式1表示的化合物的树脂组合物形成的固化物的阿贝数(νd)低。本发明人等还发现了:由含有由通式1表示的化合物的树脂组合物形成的固化物的耐热冲击性高。另外,在本说明书中,耐热冲击性表示固化物的热变化时的缓解应力的能力。
[0105]
[化学式4]
[0106]
pol1‑
sp1‑
l1‑
ar
‑
l2‑
sp2‑
pol2ꢀꢀ
(通式1)
[0107]
式中,ar为由下述通式2
‑
1~2
‑
4表示的任一个芳香环基。
[0108]
[化学式5]
[0109][0110]
通式2
‑
1~2
‑
4中,q1表示
‑
s
‑
、
‑
o
‑
或nr
11
‑
,r
11
表示氢原子或碳原子数1~6的烷基,
[0111]
y1表示可以具有取代基的碳原子数1~6的烷基、可以具有取代基的碳原子数6~12的芳香族烃基或可以具有取代基的碳原子数3~12的芳香族杂环基,
[0112]
z1、z2及z3分别独立地表示氢原子、可以具有取代基的碳原子数1~20的脂肪族烃基、可以具有取代基的碳原子数1~20的烷氧基、可以具有取代基的碳原子数3~20的脂环式烃基、可以具有取代基的1价的碳原子数6~20的芳香族烃基、卤原子、氰基、硝基、
‑
nr
12
r
13
或sr
12
,z1与z2可以彼此键合而形成可以具有取代基的芳香族烃环或可以具有取代基的芳香族杂环,r
12
及r
13
分别独立地表示氢原子或碳原子数1~6的烷基,
[0113]
a1及a2各自独立地表示选自包含
‑
o
‑
、
‑
nr
21
‑
(r
21
表示氢原子或取代基。)、
‑
s
‑
及
‑
c(=o)
‑
的组中的基团,x表示o、s、键合有氢原子或者取代基的c或键合有氢原子或者取代基的n,
[0114]
ax表示具有选自包含芳香族烃环及芳香族杂环的组中的至少一种芳香环的碳原子数1~30的有机基团,ay表示氢原子、可以具有取代基的碳原子数1~6的烷基或具有选自包含芳香族烃环及芳香族杂环的组中的至少一种芳香环的碳原子数1~30的有机基团,ax及ay所具有的芳香环可以具有取代基,ax与ay可以彼此键合而形成可以具有取代基的环,
[0115]
q2表示氢原子或可以具有取代基的碳原子数1~6的烷基。
[0116]
另外,*表示与l1或l2的键合位置。
[0117]
关于通式2
‑
1~2
‑
4中的各取代基的定义及优选的范围,能够分别针对y1、z1、z2参考日本特开2012
‑
21068号公报中所记载的关于化合物(a)的与y1、q1、q2有关的记载,能够分别针对a1、a2及x参考日本特开2008
‑
107767号公报中所记载的关于由通式(i)表示的化合物的与a1、a2及x有关的记载,能够分别针对通式2
‑
3的ax、ay、q2参考wo2013/018526中所记载的关于由通式(i)表示的化合物的与a
x
、a
y
、q1有关的记载,能够分别针对通式2
‑
4的ax、ay、q2参考wo2013/018526中所记载的关于由通式(ii)表示的化合物的与a
a
、a
b
、q
11
有关的记载。关于z3能够参考日本特开2012
‑
21068号公报中所记载的关于化合物(a)的与q1有关的记载。
[0118]
通式2
‑
2中的x优选为键合有两个取代基的c,a1、a2优选均为
‑
s
‑
。通式2
‑
3中作为ax、ay彼此键合而形成可以具有取代基的环时的环,优选为脂环式烃环、芳香族烃环或芳香族杂环,更优选为芳香族杂环。通式2
‑
4中作为ax、ay彼此键合而形成可以具有取代基的环时的环,优选为不饱和烃环。
[0119]
通式1中的ar优选为由通式2
‑
2表示的芳香环基。
[0120]
作为由通式2
‑
2表示的芳香环基,尤其优选为由下述通式2
‑2‑
1表示的芳香环基。
[0121]
[化学式6]
[0122][0123]
式中,rz表示取代基。作为rz所表示的取代基的例子,可举出后述的作为sp1的取代基例示的取代基等。两个rz可以相同,也可以不同。并且,两个rz也可以键合而形成环。此时所形成的环优选为5元环或6元环,更优选为包含氮或氧作为构成环的元素的5元环或6元环。尤其优选为由以下任一个式表示的环。
[0124]
[化学式7]
[0125][0126]
上述式中,*分别表示通式2
‑2‑
1中两个rz所键合的碳原子的位置。并且,由上述式表示的环可以在氮或碳中具有取代基。作为此时的取代基,优选为碳原子数1~6的烷基,更优选为直链的碳原子数1~4的烷基。
[0127]
作为由通式2
‑2‑
1表示的芳香环基,优选为至少任一个rz为氰基的芳香环基或两个rz键合而形成环的芳香环基,更优选为两个rz均为氰基的芳香环基。
[0128]
通式1中,l1、l2分别独立地表示单键或选自包含
‑
o
‑
、
‑
s
‑
、
‑
c(=o)
‑
、
‑
oc(=o)
‑
、
‑
c(=o)o
‑
、
‑
oc(=o)o
‑
、
‑
nr
101
c(=o)
‑
、
‑
c(=o)nr
102
‑
、
‑
oc(=o)nr
103
‑
、
‑
nr
104
c(=o)o
‑
、
‑
sc(=o)
‑
或c(=o)s
‑
的组中的连结基团。另外,在上述连结基团的记载中,左侧与ar键合,右侧与sp1或sp2键合。r
101
、r
102
、r
103
、r
104
分别独立地表示
‑
sp3‑
pol3或卤原子。优选l1、l2分别独立地为
‑
o
‑
、
‑
oc(=o)
‑
、
‑
c(=o)o
‑
、
‑
oc(=o)o
‑
、
‑
nr
101
c(=o)
‑
、
‑
c(=o)nr
102
‑
、
‑
oc(=o)nr
103
‑
、
‑
nr
104
c(=o)o
‑
,更优选为
‑
o
‑
、
‑
oc(=o)
‑
、
‑
oc(=o)o
‑
、
‑
oc(=o)nr
103
‑
,进一步优选为
‑
o
‑
。优选r
101
、r
102
、r
103
、r
104
分别独立地为氢原子、碳原子数1~4的烷基或卤原子。
[0129]
l1及l2可以相同,也可以不同,但是优选为相同。
[0130]
r
101
、r
102
、r
103
、r
104
中的sp3分别独立地表示单键或2价连结基团。作为2价连结基团,可举出以下连结基团及选自包含以下连结基团的两个以上的组合的组中的连结基团:
[0131]
可以具有取代基的直链亚烷基;可以具有取代基的亚环烷基(例如,反式
‑
1,4
‑
亚环己基);可以具有取代基的2价的芳香族烃基(例如,1,4
‑
亚苯基);可以具有取代基的2价的芳香族杂环基;
‑
o
‑
;
‑
s
‑
;
‑
c(=o)
‑
;
‑
oc(=o)
‑
;
‑
c(=o)o
‑
;
‑
oc(=o)o
‑
;
‑
nr
201
c(=o)
‑
;
‑
c(=o)nr
202
‑
;
‑
oc(=o)nr
203
‑
;
‑
nr
204
c(=o)o
‑
;
‑
sc(=o)
‑
;
‑
c(=o)s
‑
。
[0132]
作为2价连结基团的sp3的例子,可举出可以具有取代基的直链亚烷基、可以具有取代基的亚环烷基、可以具有取代基的2价的芳香族烃基、可以具有取代基的2价的芳香族杂环基以及选自包含可以具有取代基的直链亚烷基、可以具有取代基的亚环烷基、可以具有取代基的2价的芳香族烃基及可以具有取代基的2价的芳香族杂环基的组中的两个以上的连结基团经由单键或选自包含
‑
o
‑
、
‑
c(=o)
‑
、
‑
oc(=o)
‑
、
‑
c(=o)o
‑
、
‑
oc(=o)o
‑
、
‑
nr
201
c(=o)
‑
及c(=o)nr
202
‑
的组中的连结基团键合而成的连结基团等。
[0133]
作为由sp3表示的2价连结基团,优选为单键或可以具有取代基的碳原子数1至10
的直链亚烷基,更优选为可以具有取代基的碳原子数1至5的直链亚烷基,进一步优选为可以具有取代基的碳原子数1至3的直链亚烷基,尤其优选为未取代的直链亚烷基。
[0134]
r
101
、r
102
、r
103
、r
104
中的pol3与下述pol1含义相同。
[0135]
作为
‑
sp3‑
pol3,优选为氢原子或可以具有取代基的碳原子数1~4的烷基,更优选为氢原子或碳原子数1~4的烷基。
[0136]
通式1中,sp1、sp2分别独立地表示单键或选自包含可以具有取代基的碳原子数1至30的直链亚烷基及在可以具有取代基的碳原子数2至30的直链亚烷基中一个或者不相邻的两个以上的
‑
ch2‑
被
‑
o
‑
、
‑
s
‑
、
‑
c(=o)
‑
、
‑
oc(=o)
‑
、
‑
c(=o)o
‑
、
‑
oc(=o)o
‑
、
‑
nr
201
c(=o)
‑
、
‑
c(=o)nr
202
‑
、
‑
oc(=o)nr
203
‑
、
‑
nr
204
c(=o)o
‑
、
‑
sc(=o)
‑
或
‑
c(=o)s
‑
替换的基团的组中的连结基团。在此,r
201
、r
202
、r
203
、r
204
分别独立地表示
‑
sp4‑
pol4或卤原子。sp4及pol4分别与上述sp3及pol3含义相同,优选的范围也相同。优选r
201
、r
202
、r
203
、r
204
分别独立地为氢原子、碳原子数1~4的烷基或卤原子。
[0137]
sp1及sp2可以相同,也可以不同,但是优选为相同。
[0138]
在碳原子数2至30的直链亚烷基中
‑
ch2‑
被选自包含上述
‑
o
‑
、
‑
s
‑
、
‑
c(=o)
‑
、
‑
oc(=o)
‑
、
‑
c(=o)o
‑
、
‑
oc(=o)o
‑
、
‑
nr
201
c(=o)
‑
、
‑
c(=o)nr
202
‑
、
‑
oc(=o)nr
203
‑
、
‑
nr
204
c(=o)o
‑
、
‑
sc(=o)
‑
及
‑
c(=o)s
‑
的组中的其他2价基团替换的基团的sp1及sp2中,优选被取代的其他2价基团不直接与l1或l2键合。即,优选被上述其他2价基团取代的部位不是sp1的l1侧末端及sp2的l2侧末端。
[0139]
作为由sp1、sp2表示的2价连结基团,分别独立地更优选为可以具有取代基的碳原子数1至20的直链亚烷基及选自包含可以具有取代基的碳原子数2至20的直链亚烷基中一个或不相邻的两个以上的
‑
ch2‑
被
‑
o
‑
、
‑
c(=o)
‑
、
‑
oc(=o)
‑
、
‑
c(=o)o
‑
、
‑
oc(=o)o
‑
、
‑
nr
201
c(=o)
‑
、
‑
c(=o)nr
202
‑
、
‑
oc(=o)nr
203
‑
、
‑
nr
204
c(=o)o
‑
取代而得的基团的组中的连结基团,进一步优选为可以具有取代基的碳原子数1至10的直链亚烷基及选自包含可以具有取代基的碳原子数2至10的直链亚烷基中一个或不相邻的两个以上的
‑
ch2‑
被
‑
o
‑
、
‑
c(=o)
‑
、
‑
oc(=o)
‑
、
‑
c(=o)o
‑
取代而得的基团的组中的连结基团,尤其优选为不具有取代基或具有甲基作为取代基的碳原子数1至10的直链亚烷基及选自包含不具有取代基或具有甲基作为取代基的碳原子数2至10的直链亚烷基中一个或不相邻的两个以上的
‑
ch2‑
被
‑
o
‑
、
‑
c(=o)
‑
、
‑
oc(=o)
‑
、
‑
c(=o)o
‑
取代而得的基团的组中的连结基团。
[0140]
另外,在上述连结基团的记载中,左侧与l1、l2或n(在sp3的情况下)键合,右侧与pol1、pol2或pol3键合。
[0141]
关于sp1、sp2、sp3及通式2
‑
1~2
‑
4中的取代基,在称为“可以具有取代基”时的取代基并无特别限定,例如可举出烷基、环烷基、烷氧基、酰基、酰氧基、烷氧基羰基、酰胺基、氨基、卤原子、硝基及氰基以及选自包含组合两个以上的上述取代基构成的基团的组中的取代基。取代基可以为由
‑
sp5‑
pol5表示的基团。sp5及pol5分别与sp1及pol1含义相同,优选的范围也相同。取代基的数量并无特别限定,可以具有1~4个取代基。在具有两个以上的取代基时,两个以上的取代基可以彼此相同或不同。
[0142]
通式1中,pol1、pol2分别独立地表示氢原子或聚合性基团。作为聚合性基团,例如可举出由以下式pol
‑
1~式pol
‑
6表示的聚合性基团。
[0143]
[化学式8]
[0144][0145]
*表示键合位置。
[0146]
这些中,优选为(甲基)丙烯酰氧基(pol
‑
1、pol
‑
2)。
[0147]
优选聚合性基团pol1或pol2中的任一个是(甲基)丙烯酰氧基,更优选两者是(甲基)丙烯酰氧基。
[0148]
pol1及pol2可以相同,也可以不同,但是优选为相同。
[0149]
由通式1表示的化合物具有至少一个聚合性基团。由通式1表示的化合物优选具有至少两个聚合性基团。
[0150]
作为pol1‑
sp1‑
l1‑
或pol2‑
sp2‑
l2‑
的具体结构的例子,可举出以下结构。
[0151]
另外,pol1‑
sp1‑
l1‑
及pol2‑
sp2‑
l2‑
可以相同,也可以不同,但是优选为相同。
[0152]
[化学式9]
[0153][0154]
(r为氢原子或甲基。并且,*表示与ar的键合位置。)
[0155]
另外,在本说明书中,以下结构表示甲基分别与乙烯基的任一个碳键合的两个部分结构混合存在的情况。
[0156]
[化学式10]
[0157][0158]
如此,在由通式1表示的化合物中,在具有取代基在直链亚烷基上取代的结构的情况下,可能存在该取代基的取代位置不同的结构异构体。由通式1表示的化合物可以为这种结构异构体的混合物。
[0159]
由通式1表示的化合物优选为非液晶性化合物。
[0160]
以下,列举优选用于本发明的树脂组合物的由通式1表示的化合物的具体例,但并不限定于以下化合物。另外,以下结构式中的me表示甲基,et表示乙基,npr表示正丙基,ipr表示异丙基,nbu表示正丁基,tbu表示叔丁基。
[0161]
[化学式11]
[0162][0163]
[化学式12]
[0164][0165]
[化学式13]
[0166]
[0167]
[化学式14]
[0168][0169]
[化学式15]
[0170][0171]
[化学式16]
[0172][0173]
[化学式17]
[0174][0175]
由通式1表示的化合物有时具有一个或两个以上的不对称碳,对于这种不对称碳的立体化学,能够分别独立地采用(r)体或(s)体中的任一个。并且,由通式1表示的化合物可以为光学异构体或非对映异构体等立体异构体的混合物。即,由通式1表示的化合物可以为任意一种立体异构体,也可以为立体异构体的任意的混合物,还可以为外消旋体。
[0176]
[近紫外光吸收性有机化合物的含量等]
[0177]
关于树脂组合物中的近紫外光吸收性有机化合物的含量,只要根据近紫外光吸收性有机化合物的上述abs(λmax)值,并且根据是否为近紫外光吸收性有机化合物的聚合性化合物来调节即可。典型地,相对于树脂组合物的总质量(在包含溶剂的情况下,除溶剂以外的固体成分质量),优选为1~60质量%,更优选为5~50质量%,进一步优选为10质量%~40质量%,尤其优选为15~35质量%。通过将近紫外光吸收性有机化合物的含量设在上述范围内,能够充分地获得在近紫外光区域中提高折射率的效果。
[0178]
在树脂组合物中可以含有两种以上的近紫外光吸收性有机化合物。在含有两种以上的近紫外光吸收性有机化合物的情况下,优选总含量在上述范围内。
[0179]
<氧化铟锡粒子>
[0180]
本发明的树脂组合物包含氧化铟锡(ito)粒子。通过在树脂组合物中添加ito粒子,能够在可见光区域中获得波长越长则折射率越低的固化物。
[0181]
ito粒子的粒径优选为5nm~50nm。通过设为50nm以下,能够防止由瑞利散射引起的透射率劣化。并且,能够以5nm以上没有技术难度地制造。关于ito粒子的粒径,能够通过将利用透射型电子显微镜(transmission electron microscopy:tem)测定的粒径进行平均来求出。即,关于利用tem拍摄的电子显微照片中的一个粒子,测定短径和长径,将其平均值作为一个粒子的粒径而求出。在本说明书中,将50个以上的粒子的粒径进行平均,作为平均一次粒径而求出。
[0182]
关于ito粒子,优选在分散于溶剂中的状态下,与上述2官能以上的(甲基)丙烯酸酯化合物及后述酸性聚合物进行混合,成为本发明的树脂组合物。混合之后,溶剂可以通过蒸馏除去等而被除去,也可以不被除去,但是优选被除去。
[0183]
关于ito粒子,通过制成经表面修饰的ito粒子而能够提高溶剂中的分散性。关于ito粒子的表面修饰,优选利用例如碳原子数6~20的一元羧酸进行。关于基于一元羧酸的ito粒子的表面修饰,优选通过源自一元羧酸的羧基经由ito粒子表面的氧原子共价键合而形成酯键来进行。作为碳原子数6~20的一元羧酸,例如,可举出油酸(碳原子数18)、硬脂酸(碳原子数18)、十六烷酸(碳原子数16)、肉豆蔻酸(碳原子数14)、癸烷酸(碳原子数10),优选为油酸(碳原子数18)。
[0184]
在树脂组合物中,通过上述表面修饰与ito粒子键合的部位(例如,源自碳原子数6~20的一元羧酸的基团)可以直接与ito粒子键合,也可以一部分替换为源自酸性聚合物的基团,还可以全部替换为源自酸性聚合物的基团。在本发明的树脂组合物中,优选在ito粒子表面键合有源自碳原子数6~20的一元羧酸的基团及源自酸性聚合物的基团这两者。
[0185]
作为上述溶剂,优选为sp值的极性项的成分(δp)是0~6mpa
(1/2)
的溶剂。
[0186]
sp值的极性项的成分(δp)为通过汉森溶解度参数计算的值。汉森溶解度参数由分子之间的分散能(δd)、分子之间的极性能(δp)及分子之间的氢键能(δh)构成。在本说明书中,汉森溶解度参数设为使用hspip(version 4.1.07)软件计算出的值。
[0187]
具体而言,溶剂优选为甲苯(1.4)、二甲苯(1.0)或己烷(0),更优选为甲苯。另外,括号内为δp的值。
[0188]
ito粒子的制造方法并无特别限定,但是例如能够按照acs nano 2016,10,6942
‑
6951中所记载的步骤来制造。通过acs nano 2016,10,6942
‑
6951的步骤,可获得经表面修饰的ito粒子的分散液。
[0189]
具体而言,能够将碳原子数6~20的一元羧酸、铟盐(例如乙酸铟)及锡盐(例如乙酸锡)进行混合而得的溶液滴加到在高温下进行了加热的醇(油醇等长链醇)中,保持高温而形成粒子。然后,加入高分子的溶解度低的不良溶剂(乙醇等低级醇)而使粒子沉淀之后,通过去除上清液,并再分散于上述甲苯等溶剂中而能够获得经表面修饰的ito粒子的分散液。
[0190]
在本发明的树脂组合物中,相对于树脂组合物的总质量(在包含溶剂的情况下,除溶剂以外的固体成分质量),ito粒子优选为15质量%以上且40质量%以下,更优选为20质量%以上且38质量%以下,进一步优选为25质量%以上且36质量%以下。
[0191]
[氧化铟锡粒子分散剂]
[0192]
本发明的树脂组合物优选包含用于将ito粒子分散于组合物中的分散剂。作为分散剂,能够使用阳离子系、非离子系或两种表面活性剂。具体而言,能够使用具有羧酸、磷
酸、胺、多羧酸、多磷酸、氢硬脂酸、酰胺磺酸、丙烯酸、聚丙烯酸或它们的盐的分散剂。
[0193]
作为具体例,在disperbyk系列(byk japan kk制造)中可举出disperbyk
‑
106、108、110、111、118、140、142、145、161、162、163、164、167、168、180、2013、2055、2155,在phosmer系列(uni
‑
chemical co.,ltd.制造)中可举出phosmer m、phosmer pe、phosmer mh、phosmer pp等。但是,本发明的分散剂并不限定于这些。
[0194]
在本发明的树脂组合物中,相对于树脂组合物中的ito粒子的总质量,分散剂优选为5质量%~40质量%以下,更优选为6质量%以上且30质量%以下,进一步优选为7质量%以上且20质量%以下。
[0195]
<其他成分>
[0196]
树脂组合物除了近紫外光吸收性有机化合物、ito粒子及分散剂以外,还可以包含其他成分。作为其他成分,具体而言,例如,可举出选自(甲基)丙烯酸酯单体、聚合物、光自由基聚合引发剂及热自由基聚合引发剂中的至少1种等。
[0197]
[(甲基)丙烯酸酯单体]
[0198]
树脂组合物可以包含(甲基)丙烯酸酯单体。尤其,在近紫外光吸收性有机化合物不是聚合性化合物的情况下,优选树脂组合物通过包含(甲基)丙烯酸酯单体而成为固化性组合物。(甲基)丙烯酸酯单体可以为分子中具有两个以上的(甲基)丙烯酰基的多官能(甲基)丙烯酸酯单体,也可以为分子中具有一个(甲基)丙烯酰基的单官能(甲基)丙烯酸酯单体。
[0199]
作为(甲基)丙烯酸酯单体的具体例,例如,能够举出以下单体1(苯氧乙基丙烯酸酯)、单体2(丙烯酸苄酯)、单体3(三环癸烷二甲醇二丙烯酸酯)、单体4(丙烯酸二环戊酯)及日本特开2012
‑
107191号公报的0037~0046段中所记载的(甲基)丙烯酸酯单体等。(甲基)丙烯酸酯单体的分子量优选为100~500。
[0200]
[化学式18]
[0201][0202]
对于(甲基)丙烯酸酯单体的获取方法,并无特别限制,可以通过商业手段获取,也可以通过合成来制造。在通过商业手段获取的情况下,例如,能够优选使用viscort#192pea(单体1)(osaka organic chemical ind.ltd.制造)、viscort#160bza(单体2)(osaka organic chemical ind.ltd.制造)、a
‑
dcp(单体3)(shin
‑
nakamura chemical co.,ltd.制造)、fa
‑
513as(单体4)(hitachi chemical co.,ltd.制造)。
[0203]
并且,在需要提高固化物表面的硬度、耐磨性的情况下,树脂组合物优选包含在分子中具有3个以上的(甲基)丙烯酰基的多官能(甲基)丙烯酸酯单体。通过包含在分子中具
有3个以上的(甲基)丙烯酰基的多官能(甲基)丙烯酸酯单体,能够有效地提高固化物的交联密度,因此能够在保持高的部分色散比的状态下提高表面硬度、耐磨性。在分子中具有3个以上的(甲基)丙烯酰基的多官能(甲基)丙烯酸酯单体的(甲基)丙烯酰基的数量的上限并无特别限定,但是优选为8个,更优选为6个。在通过商业手段获取的情况下,例如,能够优选使用a
‑
tmpt(单体5)、a
‑
tmmt(单体6)、ad
‑
tmp(单体7)、a
‑
dph(单体8)(shin
‑
nakamura chemical co.,ltd.制造)。
[0204]
[化学式19]
[0205][0206]
在树脂组合物含有(甲基)丙烯酸酯单体的情况下,相对于树脂组合物的总质量,(甲基)丙烯酸酯单体的含量优选为1~60质量%,更优选为2~50质量%,进一步优选为3~40质量%。在近紫外光吸收性有机化合物不是聚合性化合物的情况下,优选为20~70质量%,更优选为30~60质量%,进一步优选为40~50质量%。通过调节树脂组合物中的(甲基)丙烯酸酯单体量能够调节固化物在热变化时的缓和应力的功能。
[0207]
尤其,在需要提高固化物表面的硬度、耐磨性的情况下,相对于树脂组合物的总质量(在包含溶剂的情况下,除溶剂以外的固体成分质量),树脂组合物优选包含5~50质量%的在分子中具有3个以上的(甲基)丙烯酰基的多官能(甲基)丙烯酸酯单体,更优选包含10~45质量%,进一步优选包含25~40质量%。在近紫外光吸收性有机化合物不是聚合性化合物的情况下,优选为25~75质量%,更优选为35~65质量%,进一步优选为45~55质量%。
[0208]
[聚合物]
[0209]
本发明的树脂组合物除了上述的化合物以外,还可以包含聚合物。尤其,由于具有自由基聚合性基团的聚合物发挥提高树脂组合物的粘度的作用,因此也能够称为增稠剂或者增稠聚合物。为了调节树脂组合物的粘度而能够添加聚合物。但是,聚合物可以包含自由基聚合性基团。
[0210]
作为聚合物的例子,能够举出在侧链上具有下述自由基聚合性基团的聚合物、聚丙烯酸酯、聚氨酯低聚物、聚酯、聚亚烷基等。作为聚丙烯酸酯的例子,可举出聚丙烯酸甲酯、聚丙烯酸丁酯。并且,作为聚合物,还能够使用市售品的lir―30、50、290、310、390、700(kuraray co.,ltd.)等。
[0211]
(具有自由基聚合性基团的聚合物)
[0212]
具有自由基聚合性基团的聚合物可以为均聚物,也可以为共聚物。更优选为相对于聚丙烯酸酯、聚氨酯低聚物、聚酯、聚亚烷基在侧链导入了具有自由基聚合性基团的部位的聚合物。
[0213]
作为自由基聚合性基团,能够举出(甲基)丙烯酸酯基、乙烯基、苯乙烯基、烯丙基等。在侧链上具有自由基聚合性基团的聚合物中,优选包含5~100质量%的具有自由基聚合性基团的重复单元,更优选包含10~90质量%,进一步优选包含20~80质量%。
[0214]
以下,列举优选地用于本发明的具有自由基聚合性基团的聚合物的具体例,但具有自由基聚合性基团的聚合物并不限定于以下结构。
[0215]
[化学式20]
[0216][0217]
[化学式21]
[0218][0219]
并且,作为市售品的例子,可举出uc
‑
102m、203m(kuraray co.,ltd.)、aa
‑
6、as
‑
6s、ab
‑
6(toagosei co.,ltd.)、紫光系列(nippon synthetic chemical industry co.,ltd.)ebecryl270、8301r、8402、8465、8804(daicel
‑
allnex ltd.)。
[0220]
聚合物的分子量(重均分子量)优选为1,000~10,000,000,更优选为5,000~300,000,进一步优选为10,000~200,000。并且,聚合物的玻璃化转变温度优选为
‑
50~400℃,
更优选为
‑
30~350℃。
[0221]
相对于树脂组合物的总质量,聚合物的含量优选为40质量%以下,更优选为30质量%以下,进一步优选为25质量%以下。另外,聚合物的含量可以是0质量%,也优选不添加聚合物的方式。
[0222]
[聚合引发剂]
[0223]
包含由通式1表示的化合物的树脂组合物优选包含选自热自由基聚合引发剂及光自由基聚合引发剂中的至少一种。
[0224]
(热自由基聚合引发剂)
[0225]
树脂组合物优选包含热自由基聚合引发剂。根据该作用,能够通过将树脂组合物热聚合来成型耐热性高的固化物。
[0226]
作为热自由基聚合引发剂,具体而言,能够使用以下化合物。例如能够举出1,1
‑
二(叔己基过氧)环己烷、1,1
‑
二(叔丁基过氧)环己烷、2,2
‑
二(4,4
‑
二
‑
(叔丁基过氧)环己基)丙烷、叔己基过氧化异丙基单碳酸酯、叔丁基过氧
‑
3,5,5
‑
三甲基己酸酯、叔丁基过氧化月桂酸酯、过氧化二异丙苯、过氧化二叔丁基、叔丁基过氧
‑2‑
己酸乙酯、叔己基过氧
‑2‑
己酸乙酯、氢过氧化枯烯、叔丁基过氧化氢、叔丁基过氧
‑2‑
乙基己基、2,3
‑
二甲基
‑
2,3
‑
二苯基丁烷等。
[0227]
相对于树脂组合物的总质量,热自由基聚合引发剂的含量优选为0.01~10质量%,更优选为0.05~5.0质量%,进一步优选为0.05~2.0质量%。
[0228]
(光自由基聚合引发剂)
[0229]
树脂组合物优选包含光自由基聚合引发剂。作为光自由基聚合引发剂,具体而言,能够使用以下化合物。例如,能够举出双(2,6
‑
二甲氧基苯甲酰基)
‑
2,4,4
‑
三甲基戊基氧化膦、双(2,6
‑
二甲基苯甲酰基)
‑
2,4,4
‑
三甲基戊基氧化膦、双(2,4,6
‑
三甲基苯甲酰基)
‑
2,4,4
‑
三甲基戊基氧化膦、双(2,6
‑
二氯苯甲酰氯)
‑
2,4,4
‑
三甲基戊基氧化膦、1
‑
苯基
‑2‑
羟基
‑2‑
甲基丙烷
‑1‑
酮、1
‑
羟基环己基苯基甲酮、1
‑
(4
‑
异丙基苯基)
‑2‑
羟基
‑2‑
甲基丙烷
‑1‑
酮、1,2
‑
二苯乙烷二酮、苯基乙醛酸甲酯、1
‑
[4
‑
(2
‑
羟基乙氧基)
‑
苯基]
‑2‑
羟基
‑2‑
甲基
‑1‑
丙烷
‑1‑
酮、2
‑
羟基
‑1‑
{4
‑
[4
‑
(2
‑
羟基
‑2‑
甲基
‑
丙酰基)
‑
苯甲基]苯基}
‑2‑
甲基
‑
丙烷
‑1‑
酮、2,2
‑
二甲氧基
‑
1,2
‑
二苯乙烷
‑1‑
酮、2
‑
甲基
‑1‑
(4
‑
甲硫基苯基)
‑2‑
吗啉基丙烷
‑1‑
酮、2
‑
苯甲基
‑2‑
二甲基氨基
‑1‑
(4
‑
吗啉代苯基)
‑
丁酮
‑
1、2,4,6
‑
三甲基苯甲酰基
‑
二苯基
‑
氧化膦、双(2,4,6
‑
三甲基苯甲酰基)
‑
苯基氧化膦等。
[0230]
其中,在本发明中,作为光自由基聚合引发剂,能够优选使用basf公司制造的,irgacure 184(1
‑
羟基环己基苯基甲酮)、irgacure 819(双(2,4,6
‑
三甲基苯甲酰基)
‑
苯基氧化膦)、irgacure tpo(2,4,6
‑
三甲基苯甲酰基
‑
二苯基
‑
氧化膦)、irgacure 651(2,2
‑
二甲氧基
‑
1,2
‑
二苯乙烷
‑1‑
酮)、1
‑
[4
‑
(2
‑
羟基乙氧基)
‑
苯基]
‑2‑
羟基
‑2‑
甲基
‑1‑
丙烷
‑1‑
酮、2
‑
甲基
‑1‑
(4
‑
甲硫基苯基)
‑2‑
吗啉基丙烷
‑1‑
酮。
[0231]
相对于树脂性组合物的总质量,光自由基聚合引发剂的含量优选为0.01~5.0质量%,更优选为0.05~1.0质量%,进一步优选为0.05~0.5质量%。
[0232]
另外,树脂性组合物优选包含光自由基聚合引发剂与热自由基聚合引发剂这两者,在该情况下,相对于树脂性组合物的总质量,光自由基聚合引发剂与热自由基聚合引发剂的总含量优选为0.01~5质量%,更优选为0.05~1.0质量%,进一步优选为0.05~0.5质
量%。
[0233]
(其他添加剂等)
[0234]
只要不违背本发明的主旨,则包含近紫外光吸收性有机化合物的树脂组合物可以包含除上述成分以外的聚合物或单体、分散剂、增塑剂、热稳定剂、脱模剂等添加剂。
[0235]
<其他树脂组合物的性质等>
[0236]
本发明的树脂组合物的粘度优选为5000mpa
·
s以下,更优选为3000mpa
·
s以下,进一步优选为2500mpa
·
s以下,尤其优选为2000mpa
·
s以下。通过将树脂组合物的粘度设为上述范围内,能够提高成型固化物时的操作性,并形成优质的固化物。另外,本发明的固化性树脂组合物的粘度优选为50mpa
·
s以上,更优选为100mpa
·
s以上,进一步优选为200mpa
·
s以上,尤其优选为500mpa
·
s以上。
[0237]
<固化物>
[0238]
本发明的固化物由上述树脂组合物形成。固化物是通过聚合性化合物(由通式1表示的化合物、(甲基)丙烯酸酯单体等)聚合而获得,但是本发明的固化物可以包含未反应的单体。
[0239]
使本发明的树脂组合物固化而获得的固化物是透明的,阿贝数(νd)低,折射率(nf)低。
[0240]
例如,作为将上述固化物形成为6μm厚度的片材时的波长780nm的透射率,能够获得83%以上的值。在此,透射率是指利用分光光度计(例如jasco corporation制造的分光光度计“v
‑
670”)测定的值。
[0241]
在本说明书中,“折射率(nf)”为波长486.13nm处的折射率。并且,“阿贝数(νd)”是根据不同的波长下的折射率测定值通过下述式计算的值。
[0242]
νd=(nd
‑
1)/(nf
‑
nc)
[0243]
其中,nd表示波长587.56nm处的折射率,nf表示波长486.13nm处的折射率,nc表示波长656.27nm处的折射率。
[0244]
使本发明的树脂组合物固化而获得的固化物的阿贝数νd并无特别限定,但是优选为30以下,更优选为27以下,进一步优选为25以下,尤其优选为23以下。并且,上述固化物的阿贝数并无特别限定,但是优选为5以上,更优选为10以上,进一步优选为15以上,尤其优选为17以上。上述固化物的阿贝数优选为15以上且25以下。
[0245]
使本发明的树脂组合物固化而获得的固化物的折射率nf优选为1.42以上且1.60以下,更优选为1.45以上且1.58以下。
[0246]
本发明的树脂组合物的固化物的波长587nm处的双折射δn(在本说明书中,有时称为双折射δn(587nm))优选为0.00≤δn≤0.01。双折射δn(587nm)更优选为0.001以下,进一步优选为小于0.001。双折射δn(587nm)的下限值可以是0.00001或0.0001。
[0247]
固化物的双折射δn(587nm)能够通过以下方法来求出。制作薄膜状样品,使用双折射评价装置(例如,wpa
‑
100,photonic lattice,inc.制造)来测定包含样品的中心的直径10mm的圆内的双折射,并求出波长587nm处的双折射的平均值,由此能够获得双折射δn(587nm)。
[0248]
<<树脂组合物的用途>>
[0249]
本发明的树脂组合物的用途并无特别限定,但是优选用作衍射光学元件制作用材
料。尤其,可用作用于多层型衍射光学元件中的低阿贝数衍射光学元件的制作的材料,能够赋予优异的衍射效率。
[0250]
<衍射光学元件>
[0251]
使本发明的树脂组合物固化而形成的衍射光学元件的最大厚度优选为2μm~100μm。最大厚度更优选为2~50μm,尤其优选为2μm~30μm。并且,衍射光学元件的阶梯差优选为1μm~100μm,更优选为1μm~50μm。而且,衍射光学元件的间距只要在0.1mm~10mm之间即可,优选根据所需的光像差在同一个衍射光学元件内发生变化。
[0252]
衍射光学元件例如能够按照以下步骤来制造。
[0253]
将树脂组合物夹在具有加工成衍射光栅形状的表面的模具的上述表面与透明基板之间。然后,可以将树脂组合物进行加压并拉伸至所期望的范围。在夹持的状态下,从透明基板侧进行光照射,使树脂组合物固化。然后,将固化物从模具中脱模。脱模之后,还可以从与透明基板侧相反的一侧进行光照射。
[0254]
作为上述透明基板,可举出平板玻璃、平板的透明树脂((甲基)丙烯酸树脂、聚碳酸酯树脂、聚对苯二甲酸乙二酯等)。
[0255]
上述制造中所使用的透明基板可以直接包含在衍射光学元件中,也可以剥离。
[0256]
上述模具的加工成衍射光栅形状的表面优选实施了氮化铬处理。由此,能够获得良好的模具脱模性,且能够提高衍射光学元件的制造效率。
[0257]
作为氮化铬处理,例如能够举出在模具表面形成氮化铬膜的方法。作为在模具表面形成氮化铬膜的方法,例如有cvd(化学气相沉积(chemical vapor deposition))法和pvd(物理气相沉积(physical vapor deposition))法。cvd法为使含有铬的原料气体和含有氮的原料气体在高温下反应而在基体表面形成氮化铬膜的方法。并且,pvd法为利用电弧放电在基体表面形成氮化铬膜的方法(电弧式真空蒸镀法)。在该电弧式真空蒸镀法中,在真空容器内配置例如由铬制成的阴极(蒸发源),在阴极与真空容器的壁面之间通过触发产生电弧放电,蒸发阴极的同时,实现基于电弧等离子体的金属的离子化,对基体施加负电压,并且在真空容器中装入几十mtorr(1.33pa)左右的反应气体(例如氮气),由此使离子化的金属与反应气体在基体的表面进行反应来制作化合物的膜。
[0258]
用于树脂组合物的固化的光照射中所使用的光优选为紫外线或可见光线,更优选为紫外线。例如优选使用金属卤化物灯、低压汞灯、高压汞灯、超高压汞灯、杀菌灯、氙气灯、led(发光二极管,light emitting diode)光源灯等。用于树脂组合物的固化的光照射中所使用的紫外光的照度优选为1~100mw/cm2,更优选为1~75mw/cm2,进一步优选为5~50mw/cm2。可以照射多次照度不同的紫外光。紫外光的曝光量优选为0.4~10j/cm2,更优选为0.5~5j/cm2,进一步优选为1~3j/cm2。光照射时的气氛优选为空气气氛或用不活泼气体置换的气氛,更优选为用氮气置换空气直至氧浓度成为1%以下的气氛。
[0259]
<多层型衍射光学元件>
[0260]
优选将使本发明的树脂组合物固化而形成的衍射光学元件设为第1衍射光学元件,进而将由不同材料形成的第2衍射光学元件以彼此的格栅形状的面对置的方式重叠而制成多层型衍射光学元件。此时,优选彼此的格栅形状的面接触。
[0261]
通过相较于第1衍射光学元件由高折射率且高阿贝数的材料形成第2衍射光学元件来抑制光斑的产生等,从而能够充分地利用多层型衍射光学元件的色差减少作用。
[0262]
第2衍射光学元件的阿贝数νd并无特别限定,但是优选大于30,更优选为35以上,进一步优选为40以上。并且,第2衍射光学元件的阿贝数并无特别限定,但是优选为70以下,更优选为60以下,进一步优选为50以下。
[0263]
第2衍射光学元件的折射率nf优选为1.55以上且1.70以下,更优选为1.56以上且1.65以下。并且,第2衍射光学元件的折射率nf比在多层型衍射光学元件中同时使用的第1衍射光学元件的折射率nf大。
[0264]
作为用于形成第2衍射光学元件的材料,只要可获得高折射率且高阿贝数的固化物,则并无特别限定。例如,能够使用包含硫原子、卤原子或具有芳香族结构的(甲基)丙烯酸酯单体的树脂组合物、或者包含氧化锆及(甲基)丙烯酸酯单体的树脂组合物等。
[0265]
多层型衍射光学元件例如能够按照以下步骤来制造。
[0266]
将用于形成第2衍射光学元件的材料夹在使本发明的树脂组合物固化而形成的衍射光学元件的衍射光栅形状表面(上述脱模之后获得的面)与透明基板之间。然后,可以将材料进行加压并拉伸至所期望的范围。在夹持的状态下,从透明基板侧进行光照射,使上述材料固化。然后,将固化物从模具中脱模。
[0267]
作为上述透明基板,能够举出与在制造上述衍射光学元件(第1衍射光学元件)时使用的透明基板相同的例子。
[0268]
上述制造中所使用的透明基板可以直接包含在多层型衍射光学元件中,也可以剥离。
[0269]
多层型衍射光学元件的最大厚度优选为50μm~20mm。最大厚度更优选为50μm~10mm,尤其优选为50μm~3mm。
[0270]
<透镜>
[0271]
上述衍射光学元件及多层型衍射光学元件能够分别用作透镜。
[0272]
根据透镜的使用环境或用途,在透镜的表面或周围能够设置膜或部件。例如,在透镜的表面能够形成保护膜、防反射膜、硬涂膜等。并且,能够设为层叠于玻璃透镜、塑料透镜上的复合透镜。而且,还能够将透镜的周围嵌入到基材保持框等而进行固定。但是,这些膜或框架等为附加于透镜的部件,与在本说明书中指的透镜本身有区别。
[0273]
透镜优选用于移动电话或数码相机等摄像用透镜或电视机、摄像机等摄影透镜以及车载透镜。
[0274]
实施例
[0275]
以下,举出实施例及比较例对本发明的特征进一步具体地进行说明。以下的实施例中所示的材料、使用量、比例、处理内容、处理步骤等在不脱离本发明的主旨的范围内能够进行适当的变更。因此,本发明的范围不应被以下所示的具体例进行限定性解释。
[0276]
<近紫外光吸收性有机化合物的合成>
[0277]
[化学式22]
[0278]
(合成例1)
[0279][0280]
<化合物(i
‑
1d)的合成>
[0281]
利用“journal of chemical crystallography”(1997);27(9);p.515
‑
526.中所记载的方法进行了化合物(i
‑
1d)的合成。
[0282]
<化合物(i
‑
4a)的合成>
[0283]
化合物(i
‑
4a)按照日本特开2016
‑
81035号公报中所记载的化合物(i
‑
4a)的合成法进行了合成。
[0284]
<化合物(i
‑
4)的合成>
[0285]
将羧酸化合物(i
‑
4a)15.5g(67.4mmol)、乙酸乙酯185ml、n,n
‑
二甲基乙酰胺46ml及2,6
‑
二叔丁基
‑4‑
甲基苯酚60mg进行混合,并将内温冷却至0℃。在内温0~5℃下,向混合物中滴加了亚硫酰氯7.75g(65.1mmol)。在5℃下搅拌60分钟之后,在内温0~8℃下滴加了化合物(i
‑
1d)6.85g(27.6mmol)及thf(四氢呋喃)52ml的溶液。
[0286]
然后,在内温0~10℃下滴加了n,n
‑
二异丙基乙胺16.8g。在内温20~25℃下搅拌1小时之后,添加乙酸乙酯40ml、水165ml及浓盐酸14ml,进行了清洗。将有机层利用饱和食盐水140ml进行清洗、分液,接着,利用饱和食盐水100ml、7.5质量%碳酸氢钠水溶液10ml进行了清洗、分液。然后,进行浓缩,获得油状的组合物之后,利用柱色谱法提纯,获得了化合物(i
‑
4)(产率为85%)。
[0287]1h
‑
nmr(400mhz,cdcl3):δ(ppm)1.25
‑
1.35(d,6h)、2.78(t,4h)、2.95(t,4h)、4.10
‑
4.35(m,4h)、5.25(sext,2h)、5.83(d,2h)、6.05
‑
6.15(m,2h)、6.40(d,2h)、7.33(s,2h)
[0288]
[化学式23]
[0289]
(合成例2)
[0290][0291]
<化合物(iii
‑
3db)的合成>
[0292]
利用“journal of organic chemistry”(2004);69(6);p.2164
‑
2177.中所记载的方法进行了化合物(iii
‑
3db)的合成。
[0293]
<化合物(iii
‑
3d)的合成>
[0294]
将化合物(iii
‑
3db)5.0g(15.3mmol)、氰基乙酸甲酯1.66g(16.80mmol)、异丙醇25ml进行混合,并在加热回流下搅拌了3小时。然后,冷却至室温,在混合物中加入水50ml,过滤了析出的晶体。利用水
‑
异丙醇(10比1)的混合溶液、0.5n盐酸溶液清洗所获得的晶体之后,溶解于n,n
‑
二甲基乙酰胺中并进行了过滤。在所获得的滤液中加入水,过滤所析出的晶体,由此获得了化合物(iii
‑
3d)2.2g(7.82mmol)(产率为51%)。
[0295]
<化合物(iii
‑
3)的合成>
[0296]
将实施例1中所记载的化合物(i
‑
4)的合成法中的化合物(i
‑
1d)变更为化合物(iii
‑
3d),除此以外,通过与实施例1相同的方法获得了化合物(iii
‑
3)(产率为86%)。
[0297]1h
‑
nmr(400mhz,cdcl3):δ(ppm)1.25
‑
1.35(d,6h)、2.78(t,4h)、2.95(t,4h)、3.89(s,3h)、4.10
‑
4.35(m,4h)、5.25(sext,2h)、5.83(d,2h)、6.05
‑
6.15(m,2h)、6.40(d,2h)、7.28(s,2h)
[0298]
[化学式24]
[0299]
(合成例3)
[0300][0301]
<v
‑
3d的合成>
[0302]
参考日本特开2013
‑
71956号公报的0282段中所记载的化合物(11
‑
d)的合成法实施了化合物(v
‑
3d)的合成。
[0303]
<化合物(v
‑
3)的合成>
[0304]
将实施例1中所记载的化合物(i
‑
4)的合成法中的化合物(i
‑
1d)变更为化合物(v
‑
3d),除此以外,通过与实施例1相同的方法获得了化合物(v
‑
3)(产率为82%)。
[0305]1h
‑
nmr(400mhz,dmso
‑
d6):δ(ppm)1.25
‑
1.35(d,6h)、2.78(t,4h)、2.95(t,4h)、4.10
‑
4.35(m,4h)、5.25(sext,2h)、5.83(d,2h)、6.05
‑
6.15(m,2h)、6.40(d,2h)、7.03(s,1h)、7.35
‑
7.45(m,3h)、7.80(s,1h)
[0306]
[化学式25]
[0307]
(合成例4)
[0308][0309]
<化合物(i
‑
7)的合成>
[0310]
将karenz moi
‑
eg(i
‑
7a,showa denko k.k.制造)3.9g(19.5mmol)、化合物(i
‑
1d)2.7g(10.9mmol)、n,n
‑
二甲基乙酰胺2ml、氯仿20ml进行混合,并将内温加热至60℃。搅拌12小时之后,冷却至室温并进一步搅拌了12小时。接着,加入饱和碳酸氢钠水溶液并搅拌1小时之后,进行了分液。利用1n盐酸、饱和食盐水清洗所收集的有机层之后,利用无水硫酸钠进行干燥,利用旋转蒸发器去除溶剂,并通过硅胶色谱法进行提纯,由此获得了化合物(i
‑
7)5.7g(8.90mmol)(产率为82%)。
[0311]1h
‑
nmr(400mhz,cdcl3):δ(ppm)1.95(s,6h)、3.37(m,4h)、3.60
‑
3.70(m,8h)、4.20(t,4h)、5.15(br,s,2h)、5.58(s,2h)、6.13(s,2h)、7.32(s,2h)
[0312]
[化学式26]
[0313]
(合成例5)
[0314][0315]
<化合物(iv
‑
14a)的合成>
[0316]
将溴乙酸叔丁酯55.8g(285.9mmol)、化合物(iv
‑
1d)30g(114.4mmol)、碳酸铯111.8g(343.1mmol)、四丁基溴化铵3.7g(11.4mmol)、thf300ml、n,n
‑
二甲基乙酰胺150ml进行混合,并将内温加热至75℃。搅拌5小时之后,冷却至25℃,加入水750ml,过滤了析出的固体。利用水和甲醇进行清洗,由此获得了化合物(iv
‑
14a)(产率为92%)。
[0317]
<化合物(iv
‑
14b)的合成>
[0318]
将叔丁酯化合物(iv
‑
14a)50g(102mmol)、二氯甲烷500ml进行混合之后,添加三氟乙酸150ml并在25℃下搅拌了2小时。将内温冷却至5℃,过滤析出的晶体之后,利用二氯甲烷进行清洗,由此获得了化合物(iv
‑
14b)(产率为98%)。
[0319]
<化合物(iv
‑
14)的合成>
[0320]
将羧酸化合物(iv
‑
14b)33.0g(87.2mmol)、二氯甲烷500ml、丙烯酸羟丙酯26.1g(200.6mmol)、n,n
‑
二甲基氨基吡啶1.1g(8.7mmol)及盐酸1
‑
乙基
‑3‑
(3
‑
二甲基氨基丙基)碳二亚胺38.3g(200.6mmol,简称:edac)进行了混合。在40℃下搅拌2小时之后,加入1n盐酸水300ml,并进行了清洗、分液。通过进行基于硫酸镁的脱水、过滤及浓缩来获得油状组合物之后,利用柱色谱法进行了提纯(产率为60%)。
[0321]1h
‑
nmr(400mhz,cdcl3):δ(ppm)1.25
‑
1.35(d,6h)、2.36(s,3h)、4.10
‑
4.30(m,2h)、4.30
‑
4.45(m,2h)、4.52(d,2h)、4.72(d,2h)、5.20
‑
5.40(m,2h)、5.83(m,2h)、6.05
‑
6.15(m,2h)、6.40(d,2h)、6.65(d,1h)
[0322]
[化学式27]
[0323]
(合成例6)
[0324][0325]
<化合物(iv
‑
15a)的合成>
[0326]
将合成例11中所记载的化合物(iv
‑
14a)的合成法中的溴乙酸叔丁酯变更为4
‑
溴乙酸乙酯,除此以外,通过相同的方法获得了化合物(iv
‑
15a)(产率为75%)。
[0327]
<化合物(iv
‑
15b)的合成>
[0328]
将酯化合物(iv
‑
15a)2.5g(102mmol)、浓盐酸5ml、乙酸25ml进行混合之后,在60℃下搅拌了1小时。然后,添加水80ml并过滤了析出的固体。利用柱色谱法将所获得的固体进行提纯,获得了化合物(iv
‑
15b)(产率为80%)。
[0329]
<化合物(iv
‑
15)的合成>
[0330]
将羧酸化合物(iv
‑
15b)37.9g(87.2mmol)、二氯甲烷500ml、丙烯酸羟丙酯26.1g(200.6mmol)、n,n
‑
二甲基氨基吡啶1.1g(8.7mmol)及盐酸1
‑
乙基
‑3‑
(3
‑
二甲基氨基丙基)碳二亚胺38.3g(200.6mmol,简称:edac)进行了混合。在40℃下搅拌2小时之后,加入1n盐酸水300ml,并进行了清洗、分液。通过进行基于硫酸镁的脱水、过滤及浓缩来获得油状组合物之后,利用柱色谱法进行了提纯(产率为55%)。
[0331]1h
‑
nmr(400mhz,cdcl3):δ(ppm)1.20
‑
1.35(m,6h)、2.10
‑
2.20(m,4h)、2.32(s,3h)、2.60
‑
2.75(m,4h)、3.91(t,2h)、4.10
‑
4.30(m,6h)、5.24(sext,2h)、5.84(d,2h)、6.05
‑
6.15(m,2h)、6.40(d,2h)、6.70(s,1h)
[0332]
[化学式28]
[0333]
(合成例7)
[0334]
<化合物(iv
‑
16)的合成>
[0335][0336]
将合成例14中所记载的化合物(iv
‑
15)的合成法中的丙烯酸羟丙酯变更为甲基丙烯酸羟丙酯,除此以外,通过相同的方法获得了化合物(iv
‑
16)(产率为57%)。
[0337]1h
‑
nmr(400mhz,cdcl3):δ(ppm)1.20
‑
1.35(m,6h)、1.93(s,6h)、2.10
‑
2.20(m,4h)、2.32(s,3h)、2.60
‑
2.75(m,4h)、3.92(t,2h)、4.10
‑
4.30(m,6h)、5.15
‑
5.35(m,2h)、5.57(s,2h)、6.10(s,2h)、6.69(s,1h)
[0338]
按照以下步骤进行了上述制造的化合物及下述比较例中所使用的化合物c
‑
1的吸收光谱(吸光度)的测定。
[0339]
精确地称量50mg的各化合物,使用5ml容量瓶并利用四氢呋喃(thf)进行稀释之后,进一步利用thf进行稀释,以使溶液浓度成为1/500倍,从而获得了测定溶液。使用shimadzu corporation制造的uv
‑
2550进行了测定。首先,放置试样光路、对照光路两者均装有对照试样(thf)的角型石英比色池(比色池长10mm),将250~800nm的波长区域中的吸光度调节至零。接着,将试样光路侧比色池内的试样替换为近紫外光吸收性有机化合物的溶液,测定出250~800nm的吸收光谱。任一个化合物在波长410~800nm下实质上均不显示光吸收。将由测定结果获得的340nm~400nm处的最大吸光度abs(λmax)以及410nm及430nm
处的各吸光度和各式的值示于表1。并且,将化合物iv
‑
15及化合物c
‑
1的波长250~450nm处的吸收光谱示于图1。
[0340]
[表1]
[0341]
化合物v
‑
3i
‑4ⅲ‑
3i
‑
7iv
‑
14iv―15iv―16c
‑
1λmax/nm351353362355372374374310abs(λmax)0.8820.6890.7330.7480.7730.7270.7200.235abs(410nm)0.0000.0000.0000.0000.0030.0050.0050.000abs(430nm)0.0000.0000.0000.0000.0000.0010.0010.000pa
‑
11.001.001.001.001.000.990.991.00pa
‑
21.001.001.001.001.000.990.991.00p10.0150.0120.0150.0140.0200.0200.0200.002
[0342]
表中,pa
‑
1、pa
‑
2及p1如下。
[0343]
pa
‑
1=(abs(λmax)
‑
abs(410nm))/abs(λmax)
[0344]
pa
‑
2=(abs(λmax)
‑
abs(410nm))/(abs(λmax)
‑
abs(430nm))
[0345]
p1=(abs(λmax)
‑
abs(410nm))/(410
‑
λmax)
[0346]
<ito粒子(ito
‑
1)的合成>
[0347]
向烧瓶中投入了75ml的油酸(sigma
‑
aldrich co.llc.制造,technical grade,90%)、10.060g(34.5mmol)的乙酸铟(alfa aesar公司制造,99.99%)及1.079g(3.0mmol)的乙酸锡(iv)(alfa aesar公司制造)。将该烧瓶中的混合物在氮气流的环境中在160℃下加热1小时,从而获得了黄色透明的前体溶液。
[0348]
接着,将另一个烧瓶中的油醇(wako pure chemical corporation制造,65%up)90ml在氮气流中加热至290℃。使用注射泵,以1.75ml/min的速度将上述前体溶液滴加到所加热的油醇中。上述前体溶液的滴加结束之后,将所获得的反应溶液在290℃下保持120分钟,然后停止加热,冷却至室温。
[0349]
在所获得的反应溶液中加入乙醇之后,进行离心分离而使粒子沉淀。重复进行3次去除上清液并再分散于甲苯中的操作,获得了油酸配位的ito粒子(ito
‑
1)的甲苯分散液(ito固体成分为4.75质量%,表面处理表面修饰成分的固体成分为0.25质量%)。
[0350]
利用tem观察上述ito粒子(ito
‑
1)的结果,平均粒径为21nm。
[0351]
<树脂组合物1
‑
1的制备方法>
[0352]
在ito
‑
1的甲苯分散液58.9g中加入化合物(v
‑
3)3.1g、phosmer pp(uni
‑
chemical co.,ltd.制造)0.28g及1,6
‑
己二醇二丙烯酸酯(hdda,tokyo chemical industry co.,ltd.制造)3.82g而使其溶解。一边在约70℃的水浴中进行加热一边进行减压吸引而蒸馏除去了甲苯。在蒸馏除去之后获得的混合物中加入irgacure 819(basf公司制造)0.01g并使其溶解而获得了树脂组合物1
‑
1。
[0353]
[化学式29]
[0354][0355]
<树脂组合物1
‑
2~1
‑
6的制备方法>
[0356]
将树脂组合物1
‑
1的制备方法中所记载的化合物(v
‑
3)变更为表2中所记载的近紫外光吸收性有机化合物,除此以外,通过相同的方法获得了树脂组合物1
‑
2~1
‑
6。
[0357]
<树脂组合物1
‑
7的制备方法>
[0358]
在ito
‑
1的甲苯分散液58.9g中加入化合物(iv
‑
16)3.1g、phosmer pp(uni
‑
chemical co.,ltd.制造)0.28g及1,6
‑
己二醇二甲基丙烯酸酯(hddma,tokyo chemical industry co.,ltd.制造)3.82g而使其溶解。一边在约70℃的水浴中进行加热一边进行减压吸引而蒸馏除去了甲苯。在蒸馏除去之后获得的混合物中加入irgacure 819(basf公司制造)0.01g并使其溶解而获得了树脂组合物1
‑
7。
[0359]
<树脂组合物1
‑
8~1
‑
12的制备方法>
[0360]
按照表所记载的组成比率,加入ito
‑
1的甲苯分散液、化合物(iv
‑
15)、phosmer pp(uni
‑
chemical co.,ltd.制造)、1,6
‑
己二醇二丙烯酸酯(hdda,tokyo chemical industry co.,ltd.制造)而使其溶解。一边在约70℃的水浴中进行加热一边进行减压吸引而蒸馏除去了甲苯。在蒸馏除去之后获得的混合物中加入irgacure 819(basf公司制造)0.01g并使其溶解而获得了树脂组合物1
‑
8~1
‑
12。
[0361]
<树脂组合物a的制备方法>
[0362]
在ito
‑
1的甲苯分散液90.5g中加入phosmer pp(uni
‑
chemical co.,ltd.制造)0.43g及1,6
‑
己二醇二甲基丙烯酸酯(hddma,tokyo chemical industry co.,ltd.制造)
chemical co.,ltd.制造)并搅拌至均匀。一边在约70℃的水浴中进行加热一边进行减压吸引而蒸馏除去了甲醇及mek(甲基乙基酮)。在蒸馏除去之后获得的混合物中加入irgacure 651(0.40g,basf公司制造)并使其溶解而制备了树脂组合物2
‑
3。树脂组合物2
‑
3的光学特性为(nf=1.601,νd=48.2)。
[0375]
<树脂组合物2
‑
4的制备>
[0376]
在氧化锆分散液(szr
‑
k,sakai chemical industry co.,ltd.制造)58.6g中加入fa
‑
512as(18.3g,hitachi chemical co.,ltd.制造)和a9300
‑
1cl(1.1g,shin
‑
nakamura chemical co.,ltd.制造)并搅拌至均匀。一边在约70℃的水浴中进行加热一边进行减压吸引而蒸馏除去了甲醇及mek(甲基乙基酮)。在蒸馏除去之后获得的混合物中加入irgacure 651(0.40g,basf公司制造)并使其溶解而制备了树脂组合物2
‑
4。树脂组合物2
‑
4的光学特性为(nf=1.618,νd=49.1)。
[0377]
<树脂组合物1
‑
1的固化物制作>
[0378]
将树脂组合物1
‑
1夹在经疎水化处理的玻璃板之间,使用uv照射装置(execure 3000,hoya candeo optronicscorporation制造),以1.0j/cm2(30mw/cm2)进行uv照射之后,再次以1.0j/cm2(5mw/cm2)进行uv照射而制作了固化物。上述单膜的膜厚设为6μm。
[0379]
<树脂组合物1
‑
2~1
‑
12、树脂组合物a~c的固化物制作>
[0380]
通过与树脂组合物1
‑
1的固化物制作方法相同的方法,获得了树脂组合物1
‑
2~1
‑
12、树脂组合物a~c的固化物。利用多波长阿贝折射仪dr
‑
m2(atago co.,ltd.制造)测定波长587.56nm、486.13nm、656.27nm处的折射率,求出阿贝数νd。将波长486.13nm处的折射率和阿贝数νd示于表2。
[0381]
<树脂组合物2
‑
1的固化物制作>
[0382]
将树脂组合物2
‑
1夹在经疎水化处理的玻璃板之间,使用uv照射装置(execure 3000,hoya candeo optronicscorporation制造),以2.0j/cm2(5mw/cm2)进行uv照射而制作了固化物。上述单膜的膜厚设为6μm。
[0383]
<树脂组合物2
‑
2~2
‑
4的固化物制作>
[0384]
通过与树脂组合物2
‑
1的固化物制作方法相同的方法,获得了树脂组合物2
‑
2~2
‑
4的固化物。
[0385]
<透射率测定>
[0386]
使用在上述条件下制作的各树脂组合物的固化物,测定波长400至800nm的透射率,并以780nm的透射率为基准进行了评价。若为b以上,则具有实用性。将结果示于表2。
[0387]
a:透射率为86%以上
[0388]
b:透射率为83%以上且小于86%
[0389]
c:透射率小于83%
[0390]
<透镜的衍射效率评价>
[0391]
使用多波长阿贝折射仪dr
‑
m2(atago co.,ltd.制造)测定出树脂组合物1
‑
1~1
‑
12、树脂组合物a~c的固化物及树脂组合物2
‑
1、2
‑
2、2
‑
3、2
‑
4的固化物的波长486.13(486)nm处的折射率。使用表2中所记载的组合的树脂组合物的固化物,参考日本特开2008
‑
241734号公报的式23、式24,计算出波长486nm处的1次光的衍射效率。若为评价b以上,则可以说是良好。将结果示于表2。
[0392]
a:衍射效率为96%以上。
[0393]
b:衍射效率为93%以上且小于96%。
[0394]
c:衍射效率为90%以上且小于93%。
[0395]
d:衍射效率小于90%。
[0396]
<热循环试验用固化物的制作>
[0397]
将树脂组合物1
‑
1~1
‑
12、树脂组合物a~c分别注入直径为30mm的圆形模具中,以使固化物的厚度成为20μm,然后从上放置平板玻璃(bk
‑
7),在氧浓度为1%以下的气氛下使用uv照射装置(execure3000,hoya candeo optronics corporation.制造),以1.0j/cm2(30mw/cm2)进行了uv照射。将与bk
‑
7成为一体的固化物从模具中脱模,从树脂侧再次以1.0j/cm2(5mw/cm2)进行了uv照射。
[0398]
接着,在上述固化物上分别放置表2中所记载的树脂组合物2的任一种和平板玻璃之后,按压平板玻璃直至厚度成为20μm,使用上述uv照射装置以1.0j/cm2(5mw/cm2)进行uv照射,从而制作了热循环试验用固化物。
[0399]
<热循环试验>
[0400]
通过上述方法分别制作了10个热循环试验用固化物。将在100℃下加热12小时之后,恢复到室温,进一步在
‑
20℃下经过12小时之后恢复到室温的工序设为一个循环,反复进行了4次。利用keyence corporation.制造的数码显微镜及激光显微镜进行了试验样品的形态观察。将观察到变形、裂痕、界面剥离等形状变化的产品设为不合格品,将未发生上述形状变化的产品设为合格品。评价10个固化物,将其合格品的比例设为合格品率,并以下述基准进行了评价。将结果示于表2。
[0401]
a:合格品率为80%以上。
[0402]
b:合格品率为70%以上且小于80%。
[0403]
c:合格品率小于70%。
[0404]
根据表2可知,相对于比较例的c评价,在实施例中获得了a评价。能够推断为,通过加入了近紫外光吸收性有机化合物(由通式1表示的化合物),树脂组合物1的固化物容易追随树脂组合物2的固化物,减少了裂痕、界面剥离。
[0405]
[表2]
[0406]
[0407]
[0408][0409]
表中数值为质量%。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1