聚酰胺酸树脂、聚酰亚胺树脂及含有这些的树脂组合物的制作方法
1.本发明涉及新颖结构的聚酰胺酸树脂、该聚酰胺酸树脂的亚胺化物的聚酰亚胺树脂、含有这些的树脂组合物、及该树脂组合物的硬化物。
背景技术:
2.智能型手机或平板电脑等移动型通讯机器或通讯基地局装置、电脑或汽车导航等电子机器的不可或缺的构件可举出印刷配线板。在印刷配线板中使用与金属箔的密合性、耐热性及柔软性等特性优异的各种树脂材料。
3.另外,近年来进行高速、大容量的新一代高频无线用印刷配线板的开发,除了上述各特性以外也要求树脂材料要有低传送损失,也就是要求低介电/低介电损耗正切。
4.耐热性、阻燃性、柔软性、电气特性及耐药品性等特性优异的聚酰亚胺树脂广泛地使用于电性/电子零件、半导体、通讯机器及其电路零件、周边机器等。另一方面,已知石油或天然油等烃系化合物显示高绝缘性及低介电常数,专利文献1中揭示有发挥高绝缘性及低介电常数而于聚酰亚胺树脂中导入属于长链烷基的二聚物二胺的骨架的例子。
5.但是,专利文献1的聚酰亚胺树脂在低介电损耗正切方面虽为优异,但接着性及机械特性较差。
6.[现有技术文献]
[0007]
[专利文献]
[0008]
专利文献1:日本特开2018
‑
168369号公报。
技术实现要素:
[0009]
[发明所欲解决的技术问题]
[0010]
本发明的目的为提供可适用于印刷配线板的新颖结构的聚酰胺酸树脂及树脂组合物,该树脂组合物含有该聚酰胺酸树脂,且其硬化物的介电损耗正切低且接着性、耐热性及机械特性优异。
[0011]
[用以解决技术问题的手段]
[0012]
本发明人等专心致志进行检讨的结果,发现特定结构的新颖聚酰胺酸树脂的亚胺化物的聚酰亚胺树脂、或含有使用该聚酰亚胺树脂而得的末端改质聚酰亚胺树脂的树脂组合物可解决上述技术问题,从而完成本发明。
[0013]
也就是本发明涉及下述(1)至(13):
[0014]
(1)一种聚酰胺酸树脂,是胺基酚化合物(a)、脂肪族二胺基化合物(b)、四元酸二酐(c)及芳香族二胺基化合物(d)的反应物,且两末端具有胺基。
[0015]
(2)如前项(1)所记载的聚酰胺酸树脂,其中,胺基酚化合物(a)为下述式(1)所示的化合物,
[0016][0017]
(式(1)中,r1独立地表示氢原子、甲基或乙基,x表示
‑
c(ch3)2‑
、
‑
c(cf3)2‑
、
‑
so2‑
或下述式(2)所示的二价连结基、氧原子或直接键。)
[0018][0019]
(3)如前项(1)或(2)所记载的聚酰胺酸树脂,其中,脂肪族二胺基化合物(b)为具有二个胺基的碳数6至36的脂肪族烃。
[0020]
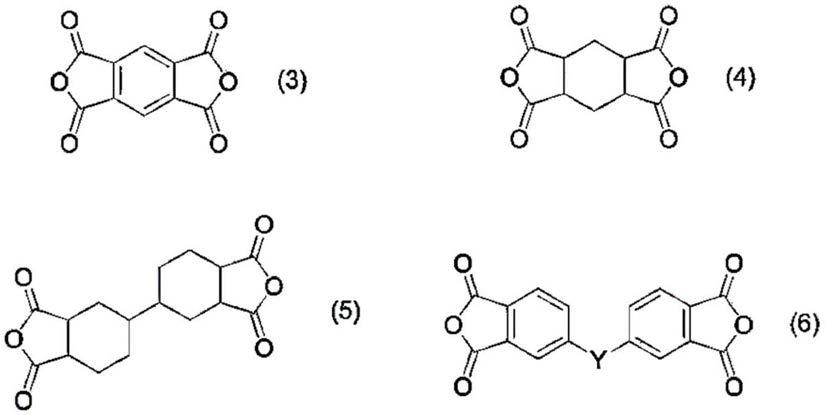
(4)如前项(1)至(3)中任一项所记载的聚酰胺酸树脂,其中,四元酸二酐(c)是选自由下述式(3)至(6)所成组中的化合物,
[0021][0022]
(式(6)中,y表示
‑
c(cf3)2‑
、
‑
so2‑
、
‑
co
‑
、或下述式(2)所示的二价连结基、氧原子或直接键。)
[0023][0024]
(5)如前项(1)至(4)中任一项所记载的聚酰胺酸树脂,其中,芳香族二胺基化合物(d)是选自由下述式(7)至(11)所成组中的化合物,
[0025][0026]
(式(9)中,r2独立地表示甲基或三氟甲基,式(10)中,r3独立地表示氢原子、甲基或乙基,式(11)中,z表示
‑
ch(ch3)
‑
、
‑
so2‑
、
‑
ch2‑
、
‑
o
‑
c6h4‑
o
‑
或下述式(2)所示的二价连结基、氧原子或直接键,r4独立地表示氢原子、甲基、乙基或三氟甲基。)
[0027][0028]
(6)如前项(1)至(5)中任一项所记载的聚酰胺酸树脂,其中,胺基酚化合物(a)的摩尔数ma、脂肪族二胺基化合物(b)的摩尔数mb、四元酸二酐(c)的摩尔数mc及芳香族二胺基化合物(d)的摩尔数md满足1.0<(ma+mb+md)/mc<1.5的关系。
[0029]
(7)一种聚酰亚胺树脂,是前项(1)至(6)中任一项所记载的聚酰胺酸树脂的亚胺化物。
[0030]
(8)一种末端改质聚酰亚胺树脂,是前项(7)所记载的聚酰亚胺树脂与马来酸酐的反应物。
[0031]
(9)一种树脂组合物,含有前项(7)所记载的聚酰亚胺树脂或前项(8)所记载的末端改质聚酰亚胺树脂、以及可与胺基和/或马来酰亚胺基反应的化合物。
[0032]
(10)如前项(9)所记载的树脂组合物,其中,可与胺基和/或马来酰亚胺基反应的化合物为马来酰亚胺树脂。
[0033]
(11)如前项(9)所记载的树脂组合物,其中,可与胺基和/或马来酰亚胺基反应的化合物为环氧树脂。
[0034]
(12)一种硬化物,是前项(9)至(11)中任一项所记载的树脂组合物的硬化物。
[0035]
(13)一种基材,具有前项(12)所记载的硬化物。
[0036]
[发明的功效]
[0037]
使用使用本发明的特定结构的聚酰亚胺树脂或该聚酰亚胺树脂所得的末端改质聚酰亚胺树脂,据此可提供耐热性、机械特性、低介电性及接着性等特性优异的印刷配线板等。
具体实施方式
[0038]
本发明的聚酰胺酸树脂为胺基酚化合物(a)(以下也仅称为“(a)成分”)、脂肪族二胺基化合物(b)(以下也仅称为“(b)成分”)、四元酸二酐(c)(以下也仅称为“(c)成分”)及芳香族二胺基化合物(d)(以下也仅称为“(d)成分”)的反应物,且两末端具有胺基。本发明的聚酰亚胺树脂为前述聚酰胺酸树脂的亚胺化物。
[0039]
(a)至(d)成分的反应为(a)、(b)及(d)成分中的胺基与(c)成分中的酸酐基的共聚反应,通过使用(a)成分的摩尔数ma、(b)成分的摩尔数mb、(c)成分的摩尔数mc及(d)成分的摩尔数md满足ma+mb+md>mc的关系的量的(a)至(d)成分进行共聚反应,会有可得两末端为胺基的本发明的聚酰胺酸树脂及聚酰亚胺树脂的情形。此时,(ma+mb+md)/mc的值较优选为超出1.0且未达2.0的范围,更优选为超出1.0且未达1.5的范围。前述值为2.0以上时,除了聚酰胺酸树脂及聚酰亚胺树脂的高分子量化有可能不充分以外,未反应原料的残存率提高而使树脂组合物(后述)在硬化后的耐热性或可挠性等各特性降低。
[0040]
上述共聚反应中,较优选为使用聚酰亚胺树脂的酚性羟基当量成为1,500至25,000g/eq.的范围的量的(a)成分。酚性羟基当量低于1,500g/eq.时,聚酰亚胺树脂的极性会变高,故会使含有聚酰亚胺树脂的树脂组合物的硬化物的介电损耗正切提高,酚性羟基当量高于25,000g/eq.高于时,会使含有聚酰亚胺树脂的树脂组合物的硬化物的接着强度及机械特性降低。
[0041]
另外,本说明书中的酚性羟基当量为根据jis k
‑
0070的方法测定的值。
[0042]
上述共聚反应中,较优选为使用成为从(a)成分、(b)成分、(c)成分及(d)成分的总质量减去(c)成分摩尔数的2倍摩尔数的水(脱水缩合反应所生成的水)的质量后的质量(生成的聚酰亚胺树脂质量)的10至50质量%的范围的量的(b)成分。(b)成分的量若低于前述范围,则聚酰亚胺树脂中源自(b)成分的脂肪族链过少,会提高介电损耗正切,若高于前述范围,则聚酰亚胺树脂中源自(b)成分的脂肪族链过多,会降低硬化物的耐热性。
[0043]
本发明的聚酰亚胺树脂为通过本发明的聚酰胺酸树脂的酰亚胺化反应,也就是通过脱水缩合所行环化反应而得。因此,为了合成具有所求羟基当量及脂肪族链量的两末端为胺基的聚酰亚胺树脂,所需的(a)成分、(b)成分、(c)成分及(d)成分的量(比率)可由共聚反应中使用的(a)至(d)成分各自的分子量及(a)成分中的酚性羟基数而容易地计算。
[0044]
作为一例,例如本发明的实施例1的作为聚酰亚胺树脂原料使用的bapp(2,2
‑
双[4
‑
(4
‑
胺基苯氧基)苯基]丙烷,分子量410.52g/mol)、priamine1075(分子量534.38g/mol)、abps(3,3
’‑
二胺基
‑
4,4
’‑
二羟基二苯基砜、分子量280.30g/mol)及odpa(氧双邻苯二甲酸酐,分子量310.22g/mol)的组合中,为了使聚酰亚胺树脂末端成为胺,相对于1摩尔的odpa,较优选为使bapp、priamine1075及abps的使用量总和为超出1摩尔的量(以下也以“1摩尔+α”表示),为了降低未反应而残存的原料成分,相对于1摩尔的odpa,较优选为使bapp、priamine1075及abps的使用量总和为2摩尔以下。
[0045]
另外,此时为了使聚酰亚胺树脂的酚性羟基当量成为1,500至25,000g/eq.的范围,例如相对于1摩尔的odpa而bapp、priamine1075及abps的使用量总和约为1摩尔(1摩尔+α)时,可使abps的使用量大致为0.02摩尔以上,相对于1摩尔的odpa而bapp、priamine1075及abps的使用量总和为1.5摩尔时,可使abps的使用量大致为0.03摩尔以上,相对于1摩尔的odpa而bapp、priamine1075及abps的使用量总和为2摩尔时,可使abps的使用量大致为0.04摩尔以上。
[0046]
另外,此时为了使生成的聚酰亚胺树脂的10至50质量%成为源自(b)成分的脂肪族链,例如相对于1摩尔的odpa而bapp、priamine1075及abps的使用量总和约为1摩尔(1摩尔+α)时,可使priamine1075的使用量大致为0.13摩尔以上,相对于1摩尔的odpa而bapp、priamine1075及abps的使用量总和为1.5摩尔时,可使abps的使用量大致为0.19摩尔以上,相对于1摩尔的odpa而bapp、priamine1075及abps的使用量总和为2摩尔时,可使abps的使用量大致为0.25摩尔以上。
[0047]
本发明的聚酰胺酸树脂(及聚酰亚胺树脂)的合成所使用的(a)成分只要为一分子中具有至少二个胺基及至少一个酚性羟基的化合物,则无特别限定。(a)成分的具体例可举出3,3
’‑
二胺基
‑
4,4
’‑
二羟基二苯基砜、3,3
’‑
二胺基
‑
4,4
’‑
二羟基二苯基醚、3,3
’‑
二胺基
‑
4,4
’‑
二羟基联苯、3,3
’‑
二胺基
‑
4,4
’‑
二羟基二苯基酮、2,2
‑
双(3
‑
胺基
‑4‑
羟基苯基)甲烷、2,2
‑
双(3
‑
胺基
‑4‑
羟基苯基)乙烷、2,2
‑
双(3
‑
胺基
‑4‑
羟基苯基)丙烷、1,3
‑
六氟
‑
2,2
‑
双(3
‑
胺基
‑4‑
羟基苯基)丙烷及9,9
’‑
双(3
‑
胺基
‑4‑
羟基苯基)芴等。这些可使用1种或混合2种以上使用。
[0048]
本发明的聚酰胺酸树脂(及聚酰亚胺树脂)的合成所使用的(a)成分较优选为下述式(1)所示的化合物。
[0049][0050]
式(1)中,r1独立地表示氢原子、甲基或乙基,x表示
‑
c(ch3)2‑
、
‑
c(cf3)2‑
、
‑
so2‑
或下述式(2)所示的二价连结基、氧原子或直接键。另外,直接键指二个苯基部分不通过原子而直接键结的状态。
[0051][0052]
本发明的聚酰胺酸树脂(及聚酰亚胺树脂)的合成所使用的(b)成分只要为一分子中具有至少二个胺基的化合物,则无特别限定。(b)成分的具体例可举出六亚甲二胺、1,3
‑
双(胺基甲基)环己烷、二聚物二胺及二胺基聚硅氧烷等。这些可使用1种或混合2种以上使用。
[0053]
(b)成分的具体例处所记载的二聚物二胺指油酸等不饱和脂肪酸的二聚物的二聚酸所具有的二个羧基取代为伯胺基的二聚物二胺(参照日本特开平9
‑
12712号公报等)。二
聚物二胺的市面贩卖品的具体例可举出priamine1074以及priamine1075(皆为croda japan股份有限公司制),及versamine 551(cognis japan股份有限公司制)等。这些可使用1种或混合2种以上使用。以下表示二聚物二胺的非限定性通式(各式中,较优选为m+n=6至17,较优选为p+q=8至19,虚线部指碳
‑
碳单键或碳
‑
碳双键)。
[0054][0055]
本发明的聚酰胺酸树脂(及聚酰亚胺树脂)的合成所使用的(c)成分只要一分子中具有二个酸酐基,则无特别限定。(c)成分的具体例可举出焦蜜石酸酐、乙二醇
‑
双(脱水偏苯三酸酯)、甘油
‑
双(脱水偏苯三酸酯)单乙酸酯、1,2,3,4
‑
丁烷四羧酸二酐、3,3’,4,4
’‑
二苯基砜四羧酸二酐、3,3’,4,4
’‑
二苯基酮四羧酸二酐、3,3’,4,4
’‑
联苯基四羧酸二酐、3,
3’,4,4
’‑
二苯基醚四羧酸二酐、5
‑
(2,5
‑
二侧氧基四氢
‑3‑
呋喃基)
‑3‑
甲基环己烯
‑
1,2
‑
二羧酸酐、3a,4,5,9b
‑
四氢
‑5‑
(四氢
‑
2,5
‑
二侧氧基
‑3‑
呋喃基)
‑
萘并[1,2
‑
c]呋喃
‑
1,3
‑
二酮、1,2,4,5
‑
环己烷四羧酸二酐、双环(2,2,2)
‑
辛
‑7‑
烯
‑
2,3,5,6
‑
四羧酸二酐及双环[2.2.2]辛烷
‑
2,3,5,6
‑
四羧酸二酐、5,5
’‑
((丙烷
‑
2,2
‑
二基双(4,1
‑
苯撑基))双(氧基))双(异苯并呋喃
‑
1,3
‑
二酮)等。其中以溶剂溶解性、与基材的密合性及感光性方面来看,较优选为3,3’,4,4
’‑
二苯基砜四羧酸二酐、3,3’,4,4
’‑
二苯基酮四羧酸二酐、3,3’,4,4
’‑
联苯基四羧酸二酐或3,3’,4,4
’‑
二苯基醚四羧酸二酐。这些可使用1种或混合2种以上使用。
[0056]
本发明的聚酰胺酸树脂(及聚酰亚胺树脂)的合成所使用的(c)成分较优选为含有选自由下述式(3)至(6)所成组中的化合物。
[0057][0058]
式(6)中,y表示
‑
c(cf3)2‑
、
‑
so2‑
、
‑
co
‑
、或上述式(2)所示的二价连结基、氧原子或直接键。在此,y表示直接键指以y为中心而左右的两芳香族基不通过原子而直接键结的状态。
[0059]
本发明的聚酰胺酸树脂(及聚酰亚胺树脂)的合成所使用的(d)成分只要为前述(a)成分以外的化合物,且一分子中具有二个胺基的芳香族系的化合物,则无特别限定。(d)成分的具体例可举出间苯二胺、对苯二胺、间甲苯二胺、4,4
’‑
二胺基二苯基醚、3,3
’‑
二甲基
‑
4,4
’‑
二胺基二苯基醚、3,4
’‑
二胺基二苯基醚、4,4
’‑
二胺基二苯基硫醚、3,3
’‑
二甲基
‑
4,4
’‑
二胺基二苯基硫醚、3,3
’‑
二乙氧基
‑
4,4
’‑
二胺基二苯基硫醚、3,3
’‑
二胺基二苯基硫醚、4,4
’‑
二胺基二苯基酮、3,3
’‑
二甲基
‑
4,4
’‑
二胺基二苯基酮、3,3
’‑
二胺基二苯基甲烷、4,4
’‑
二胺基二苯基甲烷、3,4
’‑
二胺基二苯基甲烷、3,3
’‑
二甲氧基
‑
4,4
’‑
二胺基二苯基硫醚、2,2
’‑
双(3
‑
胺基苯基)丙烷、2,2
’‑
双(4
‑
胺基苯基)丙烷、4,4
’‑
二胺基二苯基亚砜、3,3
’‑
二胺基二苯基砜、4,4
’‑
二胺基二苯基砜、联苯胺、3,3
’‑
二甲基联苯胺、3,3
’‑
二甲氧基联苯胺、3,3
’‑
二胺基联苯、对二甲苯二胺、间二甲苯二胺、邻二甲苯二胺、2,2
’‑
双(3
‑
胺基苯氧基苯基)丙烷、2,2
’‑
双(4
‑
胺基苯氧基苯基)丙烷、1,3
‑
双(4
‑
胺基苯氧基苯基)苯、1,3
’‑
双(3
‑
胺基苯氧基苯基)丙烷、双(4
‑
胺基
‑3‑
甲基苯基)甲烷、双(4
‑
胺基
‑
3,5
‑
二甲基苯基)甲烷、双(4
‑
胺基
‑3‑
乙基苯基)甲烷、双(4
‑
胺基
‑
3,5
‑
二乙基苯基)甲烷、双(4
‑
胺基
‑3‑
丙基苯基)甲烷及双(4
‑
胺基
‑
3,5
‑
二丙基苯基)甲烷等。这些可使用1种或混合2种以上使用。
[0060]
本发明的聚酰胺酸树脂(及聚酰亚胺树脂)的合成所使用的(d)成分较优选为含有
选自由下述式(7)至(11)所成组中的化合物。
[0061][0062]
式(9)中,r2独立地表示甲基或三氟甲基,式(10)中,r3独立地表示氢原子、甲基或乙基,式(11)中,z表示
‑
ch(ch3)
‑
、
‑
so2‑
、
‑
ch2‑
、
‑
o
‑
c6h4‑
o
‑
或上述式(2)所示的二价连结基、氧原子或直接键,r4独立地表示氢原子、甲基、乙基或三氟甲基。另外,z表示直接键指以z为中心而左右的两芳香族基不通过原子而直接键结的状态。
[0063]
本发明的聚酰胺酸树脂及聚酰亚胺树脂可以公知方法合成。
[0064]
例如将合成所使用的(a)至(d)成分溶解于溶剂后,在氮等惰性气体环境下在10至80℃加热搅拌,据此产生二胺类与四元酸二酐类的共聚反应,而得本发明的聚酰胺酸树脂溶液。
[0065]
另外,可任选于前述所得的聚酰胺酸树脂溶液中添加脱水剂或催化剂,在100至300℃加热搅拌,据此产生亚胺化反应(伴随脱水的闭环反应),而得本发明的聚酰亚胺树脂溶液。脱水剂可举出甲苯及二甲苯等,催化剂可举出吡啶及三乙胺等。另外,合成聚酰胺酸树脂及聚酰亚胺树脂时的反应时间会受反应温度大幅影响,但较优选为伴随反应进行的粘度上升达成平衡,且进行反应至获得最大分子量为止,通常为数分钟至20小时。
[0066]
上述例为通过聚酰胺酸合成聚酰亚胺树脂的方法,但也可将合成所使用的(a)至(d)成分溶解于溶剂后,任选添加脱水剂或催化剂并在100至300℃加热搅拌,据此一起进行共聚反应及亚胺化反应,而可得到聚酰亚胺树脂。
[0067]
另外,将上述所得聚酰胺酸树脂溶液或聚酰亚胺树脂溶液投入甲醇及己烷等贫溶剂中且分离生成聚合物后,通过再沉淀法进行精制并去除副产物,据此可得高纯度的聚酰胺酸树脂或聚酰亚胺树脂。
[0068]
聚酰胺酸树脂或聚酰亚胺树脂的合成时可使用的溶剂可举出甲基乙基酮、甲基丙基酮、甲基异丙基酮、甲基丁基酮、甲基异丁基酮、甲基正己基酮、二乙基酮、二异丙基酮、二
异丁基酮、环戊酮、环己酮、甲基环己酮、乙酰基丙酮、γ
‑
丁内酯、二丙酮醇、环己烯
‑1‑
酮、二丙基醚、二异丙基醚、二丁基醚、四氢呋喃、四氢吡喃、乙基异戊基醚、乙基
‑
叔丁基醚、乙基苄基醚、甲酚基甲基醚、苯甲醚、苯乙醚、乙酸甲酯、乙酸乙酯、乙酸丙酯、乙酸异丙酯、乙酸丁酯、乙酸异丁酯、乙酸戊酯、乙酸异戊酯、乙酸2
‑
乙基己酯、乙酸环己酯、乙酸甲基环己酯、乙酸苄酯、乙酰乙酸甲酯、乙酰乙酸乙酯、丙酸甲酯、丙酸乙酯、丙酸丁酯、丙酸苄酯、丁酸甲酯、丁酸乙酯、丁酸异丙酯、丁酸丁酯、丁酸异戊酯、乳酸甲酯、乳酸乙酯、乳酸丁酯、异戊酸乙酯、异戊酸异戊酯、草酸二乙酯、草酸二丁酯、安息香酸甲酯、安息香酸乙酯、安息香酸丙酯、水杨酸甲酯、n
‑
甲基吡咯烷酮(methyl pyrrolidone)、n,n
‑
二甲基甲酰胺、n,n
‑
二甲基乙酰胺、二甲基亚砜等,但并不限定于这些。这些可使用1种或混合2种以上使用。
[0069]
本发明的末端改质聚酰亚胺树脂为本发明的聚酰亚胺树脂末端以马来酸酐改质的聚酰亚胺树脂。末端的改质为通过使本发明的聚酰亚胺树脂两末端所具有的胺基与马来酸酐的共聚反应及亚胺化反应而进行,据此使聚酰亚胺树脂的两末端成为马来酰亚胺基。
[0070]
末端改质聚酰亚胺树脂溶液为于上述所得聚酰亚胺树脂溶液中添加必要量的马来酸酐后,在150至250℃加热搅拌即可获得,但上述亚胺化反应时生成的水残存于反应系统内时,较优选为一边去除一边进行共聚反应。
[0071]
末端改质时,相对于聚酰亚胺树脂1摩尔,较优选为使用稍微超出2摩尔左右的马来酸酐。相对于聚酰亚胺树脂1摩尔而马来酸酐量为2摩尔以下或过多时,由于末端改质聚酰亚胺树脂具高分子量而难以操作。
[0072]
本发明的树脂组合物含有本发明的聚酰亚胺树脂或本发明的末端改质聚酰亚胺树脂、以及可与胺基和/或马来酰亚胺基反应的化合物(以下将可与胺基和/或马来酰亚胺基反应的化合物仅称为“反应性化合物”)。
[0073]
反应性化合物只要为一分子中具有两个以上可与胺基和/或马来酰亚胺基反应的反应性基的化合物(树脂),则无特别限定。
[0074]
反应性化合物的具体例可举出mir
‑
3000(日本化药股份有限公司制)、bmi
‑
70、bmi
‑
80(皆为k
·
i chemical industry股份有限公司制)、bmi
‑
1000、bmi
‑
3000(皆为大和化成工业股份有限公司制)、jer828(三菱化学股份有限公司制)、nc
‑
3000、xd
‑
1000(皆为日本化药股份有限公司制)、间苯二甲酸、对苯二甲酸、karenz mt pe1(昭和电工股份有限公司制)、六亚甲二胺、kayaradr
‑
115(日本化药股份有限公司制)等,较优选为马来酰亚胺树脂或环氧树脂。
[0075]
本发明的含有聚酰亚胺树脂的树脂组合物中,反应性化合物的含量较优选为反应性化合物的反应性基当量相对于聚酰亚胺树脂的两末端胺基的活性氢1当量为0.1至500当量的量。另外,反应性化合物的反应性基具有与酚性羟基的反应性时,可于反应性化合物的反应性基当量相对于前述活性氢1当量为0.1至500当量的反应性化合物的含量中追加反应性化合物,该反应性化合物的追加量为使反应性化合物的反应性基当量相对于聚酰亚胺树脂的酚羟基1当量成为0.1至500当量的量。
[0076]
本发明的含有末端改质聚酰亚胺树脂的树脂组合物中,反应性化合物的含量较优选为反应性化合物的反应性基当量相对于末端改质聚酰亚胺树脂的马来酰亚胺基1当量为0.1至500当量的量。另外,反应性化合物的反应性基具有与酚性羟基的反应性时,可于反应性化合物的反应性基当量相对于前述马来酰亚胺基1当量为0.1至500当量的反应性化合物
含量中追加反应性化合物,该反应性化合物的追加量为使反应性化合物的反应性基当量相对于聚酰亚胺树脂的酚性羟基1当量成为0.1至500当量的量。
[0077]
反应性化合物的马来酰亚胺树脂只要为一分子中具有二个以上的马来酰亚胺基的马来酰亚胺树脂,则无特别限定,以机械强度或阻燃性等特性优异来看,树脂组合物的硬化物较优选为具有苯环、联苯基环及萘环等芳香族环的马来酰亚胺树脂,其具体例可举出mir
‑
3000(日本化药股份有限公司制)等。
[0078]
马来酰亚胺树脂的添加目的为用以与聚酰亚胺树脂的末端胺基或末端改质聚酰亚胺树脂的末端马来酰亚胺基反应,据此增加硬化物的交联密度并提高对极性溶剂的耐性,且提高与基材的密合性及耐热性。
[0079]
含有马来酰亚胺树脂的树脂组合物的硬化温度较优选为150至250℃。硬化时间虽取决于硬化温度,但大致为数分钟至数小时左右。
[0080]
含有聚酰亚胺树脂与马来酰亚胺树脂的本发明的树脂组合物中,马来酰亚胺树脂的含量较优选为使马来酰亚胺树脂的马来酰亚胺基当量相对于聚酰亚胺树脂的两末端胺基的活性氢1当量成为0.1至500当量的量。
[0081]
含有末端改质聚酰亚胺树脂与马来酰亚胺树脂的本发明的树脂组合物中,马来酰亚胺树脂的含量较优选为使马来酰亚胺树脂的马来酰亚胺基当量相对于末端改质聚酰亚胺树脂的马来酰亚胺基1当量成为0.1至500当量的量。
[0082]
另外,在此所述当量为从合成聚酰亚胺树脂或末端改质聚酰亚胺树脂时各原料的使用量所计算的值。
[0083]
含有马来酰亚胺树脂的本发明的树脂组合物中,以促进马来酰亚胺树脂的硬化反应为目的,可任选添加各种自由基引发剂。自由基引发剂可举出过氧化二异丙基苯及二丁基过氧化物等过氧化物类、2,2
’‑
偶氮双(异丁腈)及2,2
’‑
偶氮双(2,4
‑
二甲基戊腈)等偶氮化合物类等。
[0084]
含有马来酰亚胺树脂的本发明的树脂组合物中,自由基引发剂的添加量相对于马来酰亚胺树脂为0.1至10质量%。
[0085]
反应性化合物的环氧树脂只要为一分子中具有二个以上环氧基的环氧树脂,则无特别限定,以树脂组合物的硬化物的机械强度及阻燃性等特性优异来看,较优选为具有苯环、联苯基环及萘环等芳香族环的环氧树脂,其具体例可举出jer828(三菱化学股份有限公司制)、nc
‑
3000、xd
‑
1000(皆为日本化药股份有限公司制)等。
[0086]
环氧树脂的添加目的为与聚酰亚胺树脂的末端胺基或末端改质聚酰亚胺树脂的末端马来酰亚胺基反应,据此增加硬化物的交联密度并提高对极性溶剂的耐性,且提高与基材的密合性及耐热性。
[0087]
含有环氧树脂的树脂组合物的硬化温度较优选为150至250℃。硬化时间虽取决于硬化温度,但大致为数分钟至数小时左右。
[0088]
含有聚酰亚胺树脂及环氧树脂的本发明的树脂组合物中,环氧树脂含量较优选为环氧树脂的环氧基当量相对于聚酰亚胺树脂的两末端胺基的活性氢1当量成为0.1至500当量的量。另外,环氧树脂所具有的环氧基具有与酚性羟基的反应性,故较优选实施例为任选追加环氧树脂,该环氧树脂追加量为使环氧树脂的环氧基当量相对于聚酰亚胺树脂的酚性羟基1当量为0.1至500当量的量。
[0089]
含有末端改质聚酰亚胺树脂及环氧树脂的本发明的树脂组合物中,环氧树脂含量较优选为环氧树脂的环氧基当量相对于使末端改质聚酰亚胺树脂的两末端马来酰亚胺基1当量成为0.1至500当量的量。另外,环氧树脂所具有的环氧基具有与酚性羟基的反应性,故较优选实施例为任选追加环氧树脂,该环氧树脂追加量为使环氧树脂的环氧基当量相对于末端改质聚酰亚胺树脂的酚性羟基1当量为0.1至500当量的量。
[0090]
另外,在此所述当量为从合成聚酰亚胺树脂或末端改质聚酰亚胺树脂时各原料的使用量所计算的值。
[0091]
以促进环氧树脂的硬化反应为目的,可于含有环氧树脂的本发明的树脂组合物中任选添加各种热硬化催化剂。热硬化催化剂可举出2
‑
甲基咪唑、2
‑
乙基咪唑、2
‑
乙基
‑4‑
甲基咪唑、2
‑
苯基
‑
4,5
‑
二羟基甲基咪唑及2
‑
苯基
‑4‑
甲基
‑5‑
羟基甲基咪唑等咪唑类;2
‑
(二甲胺基甲基)酚及1,8
‑
二氮杂
‑
双环(5,4,0)十一烯
‑
7等3级胺类;三苯基膦等膦类及辛酸锡等金属化合物等。
[0092]
含有环氧树脂的本发明的树脂组合物中,热硬化催化剂的添加量相对于环氧树脂为0.1至10质量%。
[0093]
可于本发明的树脂组合物中并用有机溶剂而形成清漆状组合物(以下仅称为清漆)。可使用的溶剂可举例如γ
‑
丁内酯类、n
‑
甲基吡咯烷酮(methyl pyrrolidone)、n,n
‑
二甲基甲酰胺、n,n
‑
二甲基乙酰胺及n,n
‑
二甲基咪唑啶酮等酰胺系溶剂、四亚甲基砜等砜类、二乙二醇二甲基醚、二乙二醇二乙基醚、丙二醇、丙二醇单甲基醚、丙二醇单甲基醚单乙酸酯及丙二醇单丁基醚等醚系溶剂、甲基乙基酮、甲基异丁基酮、环戊酮及环己酮等酮系溶剂、甲苯及二甲苯等芳香族系溶剂。
[0094]
就有机溶剂的使用范围而言,排除了清漆中的有机溶剂的固形分浓度一般为10至80质量%,较优选为20至70质量%。
[0095]
本发明的树脂组合物中可任选并用公知的添加剂。可并用的添加剂的具体例可举出环氧树脂用硬化剂、聚丁二烯或其改质物、丙烯腈共聚物的改质物、聚苯醚、聚苯乙烯、聚乙烯、聚酰亚胺、氟树脂、马来酰亚胺系化合物、氰酸酯系化合物、聚硅氧凝胶、聚硅氧油、以及二氧化硅、氧化铝、碳酸钙、石英粉、铝粉、石墨、滑石、粘土、氧化铁、氧化钛、氮化铝、石棉、云母、玻璃粉等无机充填材、如硅烷耦合剂的充填材的表面处理剂、脱模剂、炭黑、酞青素蓝、酞青素绿等着色剂。这些添加剂的掺配量相对于树脂组合物100质量份较优选为1,000质量份以下,更优选为700质量份以下的范围。
[0096]
本发明的树脂组合物的调制方法并无特别限定,可仅均一混合各成分、或可进行预聚化。例如可通过将本发明的聚酰亚胺树脂或末端改质聚酰亚胺树脂及反应性化合物在催化剂的存在下或不存在下、溶剂的存在下或不存在下加热进行预聚化。各成分的混合或预聚化中,在溶剂的不存在下例如使用挤出机、捏合机、辊等,在溶剂的存在下使用附搅拌装置的反应釜等。
[0097]
通过将本发明的树脂组合物加热熔融并低粘度化再含浸于玻璃纤维、碳纤维、聚酯纤维、聚酰胺纤维、氧化铝纤维等强化纤维中即可得到预浸体。另外,也可通过将前述清漆含浸于强化纤维并加热干燥而获得预浸体。
[0098]
将上述预浸体裁切为所求形状,并任选与铜箔等层叠后,一边以压制成型法或高压釜成型法、薄片卷绕成型法等对层叠物施加压力一边加热硬化树脂组合物,据此可得到
电性电子用层叠板(印刷配线板)或碳纤维强化材等本发明的基材。
[0099]
另外,可于铜箔涂布树脂组合物并干燥溶剂后,层叠聚酰亚胺膜或lcp(液晶聚合物),热压制后加热硬化,据此获得本发明的基材。视情况可于聚酰亚胺膜或lcp侧涂布树脂组合物并层叠铜箔,据此获得本发明的基材。
[0100]
(实施例)
[0101]
以下以实施例、比较例进一步详细说明本发明,但本发明并不限定于这些实施例。另外,实施例中的“份”为质量份、“%”为质量%。
[0102]
实施例1(本发明的聚酰亚胺树脂的合成)
[0103]
于装设有温度计、回流冷却器、迪安
‑
斯塔克装置、粉体导入口、氮气导入装置及搅拌装置的300ml反应器中加入bapp(2,2
‑
双[4
‑
(4
‑
胺基苯氧基)苯基]丙烷、和歌山精化工业股份有限公司制、分子量410.52g/mol)16.40份、priamine1075(croda japan股份有限公司制、分子量534.38g/mol)10.87份、abps(3,3
’‑
二胺基
‑
4,4
’‑
二羟基二苯基砜、日本化药股份有限公司制、分子量280.30g/mol)0.32份、及nmp(n
‑
甲基吡咯烷酮(methyl pyrrolidone))106.47份,加热至70℃。接着加入odpa(氧双邻苯二甲酸酐、manac incorporated股份有限公司制、分子量310.22g/mol)18.04份、吡啶0.92份、及甲苯23.77份,一边将随着酰胺酸的闭环生成的水与甲苯共沸而去除,一边于180℃反应4小时。水的生成停止后,接着在180℃去除残留的吡啶及甲苯,据此获得聚酰亚胺树脂溶液(a
‑
1)(不挥发分31.2%)。实施例1所使用的二胺成分((a)成分、(b)成分及(d)成分)与酸酐成分((c)成分)的摩尔比(二胺成分的摩尔数/酸酐成分的摩尔数)为1.05。
[0104]
实施例2(本发明的末端改质聚酰亚胺树脂的合成)
[0105]
于装设有温度计、回流冷却器、迪安
‑
斯塔克装置、粉体导入口、氮气导入装置及搅拌装置的300ml反应器中加入bapp(2,2
‑
双[4
‑
(4
‑
胺基苯氧基)苯基]丙烷、和歌山精化工业股份有限公司制、分子量410.52g/mol)16.40份、priamine1075(croda japan股份有限公司制、分子量534.38g/mol)10.87份、abps(3,3
’‑
二胺基
‑
4,4
’‑
二羟基二苯基砜、日本化药股份有限公司制、分子量280.30g/mol)0.32份及nmp(n
‑
甲基吡咯烷酮(methyl pyrrolidone))106.47份,加热至70℃。接着加入odpa(氧双邻苯二甲酸酐,manac incorporated股份有限公司制、分子量310.22g/mol)18.04份、吡啶0.92份、及甲苯23.77份,一边将随着酰胺酸的闭环生成的水与甲苯共沸而去除,一边于180℃反应4小时。水的生成停止后,加入马来酸酐(分子量98.1g/mol)0.57份,一边将随着酰胺酸闭环生成的水与甲苯共沸而去除,一边以180℃反应2小时,使聚酰亚胺树脂的末端胺基改质为马来酰亚胺基。水的生成停止后,接着在180℃去除残留的吡啶及甲苯,据此获得末端改质聚酰亚胺树脂溶液(a
‑
2)(不挥发分31.0%)。实施例2所使用的二胺成分与酸酐成分的摩尔比为1.05。
[0106]
比较例1(比较用聚酰亚胺树脂的合成)
[0107]
于具备搅拌机、分水器、温度计及氮气导入管的反应容器中加入9,9
’‑
双[4
‑
(3,4
‑
二羧基苯氧基)苯基]芴酸二酐(商品名“bpf
‑
pa”、jfe化学股份有限公司制)290.00份、环己酮980.20份、及甲基环己烷196.04份,加热至60℃。接着滴入市面贩卖的二聚物二胺(商品名“priamine1075”、croda japan股份有限公司制)236.60份后,在140℃以12小时进行亚胺化反应,据此获得比较用聚酰亚胺溶液(r
‑
1)(不挥发分31.6%)。比较例1所使用的二胺成分与酸酐成分的摩尔比为0.97。
[0108]
实施例3至6、比较例2及3
[0109]
将实施例1所得的聚酰亚胺树脂溶液(a
‑
1)、实施例2所得的末端改质聚酰亚胺树脂溶液(a
‑
2)、比较例1所得的比较用聚酰亚胺树脂溶液(r
‑
1)、作为马来酰亚胺树脂的日本化药股份有限公司制mir
‑
3000(含有联苯基骨架的马来酰亚胺树脂)、作为自由基引发剂的过氧化双异苯丙基(dcp)、作为环氧树脂的日本化药股份有限公司制nc
‑
3000(含有联苯基骨架的环氧树脂、环氧基当量277g/eq、软化点60℃)、作为环氧树脂硬化剂的日本化药股份有限公司制gph
‑
65(含有联苯基骨架的酚硬化剂,羟基当量198g/eq.)、及作为硬化促进剂的四国化成工业股份有限公司制c11z
‑
a以表1所示的掺配量(单位为“份”)混合,得到本发明的树脂组合物及比较用树脂组合物。
[0110]
[表1]
[0111]
表1树脂组合物的组成
[0112][0113]
(接着强度的评价)
[0114]
使用实施例1至6及比较例1至3所得(末端改质)聚酰亚胺树脂溶液及树脂组合物,评价聚酰亚胺树脂、末端改质聚酰亚胺树脂及树脂组合物对铜箔的接着强度。
[0115]
于福田金属箔粉工业股份有限公司制铜箔cf
‑
t9fz
‑
hte的亮面(以下称为“cu镜面”)、cf
‑
t9fz
‑
hte的雾面(以下称为“cu粗面”)或福田金属箔粉工业股份有限公司制铜箔cf
‑
t4x
‑
su
‑
18(以下记载为“t4x”)表面使用自动分配器分别涂布前述聚酰亚胺树脂溶液、末端改质聚酰亚胺树脂或树脂组合物,在130℃加热干燥10分钟。干燥后的涂膜的厚度为30μm。于如此所得铜箔上的涂膜重叠cu粗面合,树脂组合物在200℃,其它则在180℃以60分钟、3mpa的条件真空压制。将所得试验片裁切为10mm宽度,使用autograph ags
‑
x
‑
500n(岛津制作所股份有限公司制)测定铜箔间的90
°
剥离强度(剥离速度为50mm/min),评价铜箔的
接着强度。
[0116]
另外,以目视确认试验后的样品,于cu镜面及t4x表面形成涂膜的样品皆在cu镜面或t4x表面与涂膜的界面产生剥离,另外,于cu粗面形成涂膜的样品皆在cu粗面与涂膜的界面产生剥离。
[0117]
结果呈示于表2、表3及表4。
[0118]
(机械特性、热特性及介电损耗正切的评价)
[0119]
以与上述“接着强度的评价”相同方法分别于cu镜面上形成厚度30μm的涂膜。另外,除了变更自动分配器的涂布厚度以外,以与上述“接着强度的评价”相同方法于cu镜面分别形成厚度100μm的涂膜。将如此所得铜箔上的涂膜以含有马来酰亚胺树脂的树脂组合物在200℃,其它则在180℃下加热60分钟后,以液比重45波美度的氯化铁(iii)溶液蚀刻铜箔,以离子交换水洗涤后,在105℃干燥10分钟,据此分别获得膜状硬化物。膜状的硬化物使用autograph ags
‑
x
‑
500n(岛津制作所股份有限公司制)测定拉伸强度(破裂点应力及破裂点伸长率)及弹性模数,使用动态粘弹性测定装置exstar6000(seiko epson股份有限公司制)测定玻璃转移温度,另外,使用网络分析仪8719et(agilent technologies制)以孔腔共振法测定10ghz中的介电损耗正切。
[0120]
结果呈示于表2、表3及表4。
[0121]
[表2]
[0122]
表2聚酰亚胺树脂及末端改质聚酰亚胺树脂的评价结果
[0123][0124]
[表3]
[0125]
表3包含马来酰亚胺树脂的树脂组合物的评价结果
[0126][0127][0128]
[表4]
[0129]
表4包含环氧树脂的树脂组合物的评价结果
[0130][0131]
由表2至4的结果可知:本发明的聚酰亚胺树脂、末端改质聚酰亚胺树脂及树脂组合物在接着强度、机械特性、热特性及介电常数皆优异,相对于此,分别对应的比较例其机械特性极差,接着强度、热特性及介电损耗正切也比实施例低劣。
[0132]
实施例7(本发明的聚酰亚胺树脂的合成)
[0133]
于装设有温度计、回流冷却器、迪安
‑
斯塔克装置、粉体导入口、氮气导入装置及搅拌装置的300ml反应器中加入bapp(2,2
‑
双[4
‑
(4
‑
胺基苯氧基)苯基]丙烷、和歌山精化工业股份有限公司制、分子量410.52g/mol)11.01份、priamine1075(croda japan股份有限公司制、分子量534.38g/mol)20.49份、bafa(2,2
‑
双(3
‑
胺基
‑4‑
羟基苯基)六氟丙烷、日本化药股份有限公司制、分子量366.26g/mol)1.59份及环己酮119.35份,加热至70℃。接着加入odpa(氧双邻苯二甲酸酐、manac incorporated股份有限公司制、分子量310.22g/mol)20.53份、三乙胺1.34份及甲苯25.69份,一边将随着酰胺酸的闭环所生成的水与甲苯共沸而去除,一边在140℃反应4小时。水的生成停止后,接着在140℃去除残留的三乙胺及甲苯,据此获得聚酰亚胺树脂溶液(a
‑
7)(不挥发分30.0%)。实施例7所使用的二胺成分与酸酐成分的摩尔比为1.05。
[0134]
实施例8(本发明的聚酰亚胺树脂的合成)
[0135]
于装设有温度计、回流冷却器、迪安
‑
斯塔克装置、粉体导入口、氮气导入装置及搅拌装置的300ml反应器中加入bafl(9,9
‑
双(4
‑
胺基苯基)芴、jfe化学股份有限公司制、分子量348.45g/mol)9.88份、priamine1075(croda japan股份有限公司制、分子量534.38g/mol)20.42份、bafa(2,2
‑
双(3
‑
胺基
‑4‑
羟基苯基)六氟丙烷、日本化药股份有限公司制、分
子量366.26g/mol)1.67份及环己酮119.30份,加热至70℃。接着加入odpa(氧双邻苯二甲酸酐,manac incorporated股份有限公司制,分子量310.22g/mol)21.63份,三乙胺1.41份及甲苯25.69份,一边将随着酰胺酸的闭环生成的水与甲苯共沸而去除,一边在140℃反应4小时。水的生成停止后,接着在140℃去除残留的三乙胺及甲苯,据此获得聚酰亚胺树脂溶液(a
‑
8)(不挥发分30.0%)。实施例8所使用的二胺成分与酸酐成分的摩尔比为1.02。
[0136]
比较例4(比较用聚酰亚胺树脂的合成)
[0137]
于装设有温度计、回流冷却器、迪安
‑
斯塔克装置、粉体导入口、氮气导入装置及搅拌装置的300ml反应器中加入bafl(9,9
‑
双(4
‑
胺基苯基)芴、jfe化学股份有限公司制、分子量348.45g/mol)6.10份、priamine1075(croda japan股份有限公司制、分子量534.38g/mol)11.38份及环己酮119.30份,加热至70℃。接着加入odpa(氧双邻苯二甲酸酐、manac incorporated股份有限公司制、分子量310.22g/mol)12.41份、三乙胺0.81份及甲苯18.75份,一边将随着酰胺酸的闭环所生成的水与甲苯共沸而去除,一边在140℃反应4小时。水的生成停止后,接着在140℃去除残留的三乙胺及甲苯,据此获得聚酰亚胺树脂溶液(r
‑
2)(不挥发分30.0%)。比较例4所使用的二胺成分与酸酐成分的摩尔比为0.97。
[0138]
实施例9及10、比较例5
[0139]
将实施例7及8所得的聚酰亚胺树脂溶液(a
‑
7)及(a
‑
8)、比较例4所得的比较用聚酰亚胺树脂溶液(r
‑
2)、作为马来酰亚胺树脂的日本化药制mir
‑
3000(含有联苯基骨架的马来酰亚胺树脂)及作为自由基引发剂的过氧化双异苯丙基(dcp)以表5所示的掺配量(单位为“份”)混合,得到本发明的树脂组合物及比较用树脂组合物。
[0140]
[表5]
[0141]
表5树脂组合物的组成
[0142] 实施例9实施例10比较例5a
‑
7100
ꢀꢀ
a
‑
8 100 r
‑2ꢀꢀ
100mir
‑
300012.8612.8612.86dcp0.4290.4290.429
[0143]
(接着强度、机械特性、热特性及介电损耗正切的评价)
[0144]
使用实施例7至10、比较例4及5所得的聚酰亚胺树脂溶液及树脂组合物。以根据上述实施例1至6及比较例1至3所得的聚酰亚胺树脂溶液、末端改质聚酰亚胺树脂溶液及树脂组合物的评价的方法评价聚酰亚胺树脂及树脂组合物对铜箔的接着强度、机械特性、热特性及介电损耗正切。
[0145]
结果呈示于表6及7。
[0146]
[表6]
[0147]
表6聚酰亚胺树脂的评价结果
[0148][0149]
[表7]
[0150]
表7包含马来酰亚胺树脂的树脂组合物的评价结果
[0151][0152]
(产业上的可利用性)
[0153]
通过使用本发明的特定结构的聚酰亚胺树脂、使用该聚酰亚胺树脂所得的末端改质聚酰亚胺树脂,及使用利用这些的树脂组合物,可提供耐热性、机械特性、低介电性及接着性等特性优异的印刷配线板等。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1