用光反应性基团官能化的支化嵌段共聚物光交联剂及其在使适用于医疗和组织工程应用的可降解光交联弹性体成形中的用途
1.本发明涉及用选自芳基叠氮基、(甲基)丙烯酸酯基或硫醇基的光反应性基团官能化的新型可降解支化嵌段共聚物(特别是星形共聚物),以及所述可降解支化嵌段共聚物作为光交联剂用于提供可降解光交联弹性体的用途,所述可降解光交联弹性体作为生物材料适用于医疗应用和组织工程应用。
背景技术:
2.聚合物材料广泛用于医疗器械。根据其用途,需要具有弹性性质的可降解聚合物,特别是在组织工程领域。这种(生物)可吸收性材料解决了与生物稳定性材料(其长期存在通常是有问题的)相关的问题,同时产生最小的长期炎症。
3.可生物降解的聚合物材料通常由热塑性聚合物或交联材料构成。特别地,交联弹性体目前由于包括以下性质的许多特定性质有益于软组织工程而备受关注:1)随着时间的推移可保持材料的力学性质和3d结构的线性降解;2)达到高杨氏模量的可能性;以及3)来自取向性较小的聚合物链的有限结晶度,其改善细胞定植并降低炎症反应。
4.通过所有化学交联弹性体,热交联(自由基和缩合反应)、点击化学(狄尔斯-阿尔德环加成、“硫醇-迈克尔加成”、硫醇-炔“点击”)和光交联是用于产生(生物)可吸收性三维(3d)网络的主要后处理。然而,目前可用的通过缩聚获得的可降解弹性体是从低分子量官能预聚物开始制备的,所述低分子量官能预聚物用于确保反应性,但导致弹性体不适合在照射前成形以及其弹性域通常较低(li等人,rsc advances,2,22(2012)8229)。
5.因此,光交联因其诸如以下的优点而受到特别关注:易于实施,产生较低的热能,以及快速处理从而促进工业发展。该方法需要至少两个可光交联的侧基或链端基(例如丙烯酸酯基)和光引发剂。
6.然而,目前可用的通过这种方法获得的可降解弹性体有几个缺点。它们是从低分子量官能预聚物开始制备的,所述低分子量官能预聚物用于确保反应性,但导致弹性体不适合大多数的热塑性成形加工以及弹性范围有限;或者它们是由线形高分子量官能预聚物制备的,所述高分子量官能预聚物的反应性有限,以及导致弹性体具有低的力学性质和减小的弹性范围(chen等人,progress in polymer science,38,3,(2013)584)。
7.因此,需要提供具有增强弹性域的弹性体,该弹性体可以容易地通过适当的加工(例如电纺丝或3d打印技术)成形,以提供在医疗领域中应用的生物材料,这些生物材料具有有利地与天然组织一致的性质。
8.此外,最近的研究重点是开发新的生物相容性光引发剂和光交联剂,以提高交联效率并调节弹性体的力学性质和降解性质。在这方面,rupp等人(green chemistry,12(2010)1796)报道了光交联弹性体的制备,由于使用双(芳基叠氮化物)2,6-双(叠氮基亚苄基)-4-甲基环己酮作为光交联剂,由非官能的(聚)羟基丁酸酯-共-羟基戊酸酯(phbv)生成
光交联弹性体。该策略依赖于芳基叠氮基的紫外线活化来生成高反应性氮烯物种,这些氮烯物种可以插入到聚合物主链的碳氢键中,从而使得通过胺基团进行交联。然而,所公开的光交联剂双(芳基叠氮化物)2,6-双(叠氮基亚苄基)-4-甲基环己酮导致低的交联效率,尤其是当目标材料的厚度超过10μm时。
9.因此,本发明寻求提供一种用光反应性基团官能化的可降解支化嵌段共聚物作为光交联剂,其尤其能够使自身、非官能化的可降解聚合物和/或官能化的高分子量预聚物以高交联率交联,用以制备易于成形为适用于医疗和软组织工程的生物材料的交联可降解弹性体。
技术实现要素:
10.为了解决该技术问题,本发明人设计了一种新型的可降解支化嵌段共聚物,特别是星形共聚物,其被选自叠氮基、(甲基)丙烯酸酯基或硫醇基的光反应性基团官能化。
11.支化嵌段共聚物(特别是星形共聚物)相对于线形共聚物的特性在于具有大量链端。实际上,本发明的星形嵌段共聚物的中央核心的每个臂在其末端带有光反应性基团。这种特征是星形共聚物上的官能团的良好可及性和反应性的原因,这些官能团使得可使用高分子量预聚物以及产生高交联率以及改善的缠结,从而提供具有超弹性性质的弹性体生物材料。
12.与现有技术相比,根据本发明的支化嵌段共聚物交联剂(特别是星形共聚物)使得所得弹性体的交联率增加,同时控制最终弹性体的降解速率。
13.特别地,当所述光反应性基团为芳基叠氮基时,待交联的聚合物不需要用可交联基团进行预官能化。相比于现有技术的双官能的(芳基叠氮化物)交联剂(参见rupp.等人),通过芳基叠氮基的光活化(例如紫外活化)产生的活性氮烯物种更有可能与待交联的聚合物接触并充当交联剂。此外,氮烯物种插入到待交联的聚合物的碳氢键中,使得经由胺基团在任何非官能化的聚合物之间交联(图1)。因此,这种通用交联剂能够使任何含有c-h基团的聚合物交联,而无需进行预官能化。
14.此外,当所述光反应性基团为(甲基)丙烯酸酯基或硫醇基时,支化嵌段共聚物交联剂可以具有高分子量,即高于10000g.mol-1
,同时提供高交联率。此外,由于多官能星形交联剂的高交联效率,待交联的官能聚合物不需要是低分子量,以及可以用较小反应性的高分子量官能预聚物获得令人满意的交联率。通过光活化(例如紫外活化)产生的光反应性物质确实更有可能与待交联的聚合物的反应性官能团接触,因此使得可制备性质得以改进并同时适合于热塑性成形加工(例如电纺丝、熔融沉积成型等)的弹性体。
15.因此,本发明的支化嵌段共聚物交联剂特别适合于制造可生物降解的弹性体材料,尤其是有利地用于软组织工程的弹性体纤维支架,通常通过电纺丝加工或3d打印技术来进行所述制造。特别地,本发明的支化嵌段共聚物具有足够高的分子量,即至少10000g/mol,优选至少20000g/mol,以用于电纺丝加工或3d打印技术方式的成形加工。
16.因此,在第一方面,本发明涉及可降解支化嵌段共聚物,特别是星形共聚物,其包含具有n个臂的聚醚中央核心和从聚醚中央核心的每个臂延伸的可降解聚合物链,所述聚醚中央核心为星形核心或线形核心,每个臂包含对应于聚醚的m个单体单元,每个可降解聚合物链由可降解聚合物的l个单体单元构成,其特征在于,每个可降解聚合物链是相同的并
且在其末端被光反应性基团官能化,所述光反应性基团选自芳基叠氮化物衍生物、(甲基)丙烯酸酯基和硫醇基。
17.在另一方面,本发明涉及如上限定的可降解支化嵌段共聚物作为交联剂,特别是作为光交联剂的用途。
18.在又一方面,本发明涉及一种用于制备可降解光交联聚合物,优选可降解光交联弹性体的方法,其包括以下步骤:
19.(a)制备包含如上限定的可降解支化嵌段共聚物和任选的预聚物的溶液或固体共混物,
20.(b)对由步骤(a)产生的溶液或固体共混物进行成形加工以提供成形物体,
21.(c)在光(例如紫外光)下照射由步骤(b)产生的成形物体,
22.(d)回收可降解光交联聚合物,优选可降解光交联弹性体。
23.定义
24.如在本文所用,“共聚物”理解为包含多种不同重复单元(即至少两种不同重复单元)的聚合物。共聚物可以是无规共聚物、嵌段共聚物或梯度共聚物。
25.如在本文所用,“嵌段共聚物”理解为包含一系列不同嵌段的共聚物,每个嵌段仅包含一种重复单元。嵌段共聚物是单一分子,因此每个嵌段通过共价键与下一个嵌段共价连接。例如重复单元a、b和c的嵌段共聚物可以具有以下结构:aaaaaaaaaaabbbbbbbbbcccccccccaaaaaaaaacccccccccbbbbbbbbb。
26.如在本文所用,“支化嵌段共聚物”理解为包含中央核心和经由共价键与该核心连接的线形链的支化嵌段共聚物。这些线形链的数量为至少三个,优选至少四个。核心可以是原子、分子,通常是环状分子,或聚合物(作为一种嵌段共聚物)。核心可以是线形聚合物,其中所述聚合物的单体单元一个接一个排列,或者所述聚合物的每个单体经由唯一的一个原子连接从而形成“星形聚合物”。在本发明的上下文中,核心是线形或星形的聚醚核心。从核心延伸的线形链,也称为“臂”,在此由至少一种可降解聚合物构成。
27.当中央核心是线形聚合物核心时,应理解它包含一个接一个排列的至少4个聚合物单体单元,核心通过包含末端官能团的官能团或原子(例如氧原子或nh基团)与线形聚合物链连接。在这种情况下,线形链(臂)的数量与末端官能团或原子的数量一样多。这样的共聚物被称为“超支化嵌段共聚物”。
28.如在本文所用,“星形共聚物”或“星形化共聚物”理解为支化共聚物,其中至少四个线形链经由共价键与星形中央核心连接。中央核心具有与唯一的一个原子连接以形成星形的至少四个单体。
29.如在本文所用,“交联剂”理解为促进聚合物之间交联的化学试剂。交联是通过大分子链的共价结合提供三维网络的化学反应。该反应通常由热、压力、ph值变化、照射引发,以及大多需要交联剂。
30.如在本文所用,“光交联剂”理解为如上定义的交联剂,其在光照射(例如紫外照射)下反应以促进光交联,所述光交联被定义为光诱导形成至少两个大分子链之间的共价键。
31.如在本文所用,“交联”材料理解为由至少一种聚合物在与如上定义的交联剂反应后形成的三维网络。单一聚合物可以交联,条件是它具有两个或多个臂。
32.根据本发明,当聚合物由支化嵌段共聚物交联剂交联,或支化嵌段共聚物自身进行交联时,应理解支化嵌段共聚物的芳基叠氮官能团在光(例如紫外光)下反应,以形成与聚合物或支化嵌段共聚物的另一分子共价结合的胺官能团。因此,一旦支化嵌段共聚物在光下反应并交联,支化嵌段共聚物的叠氮官能团不再存在,以及在所得交联聚合物中被胺官能团取代。
33.根据本发明,当聚合物由支化嵌段共聚物交联剂交联,或支化嵌段共聚物自身进行交联时,应理解支化嵌段共聚物的(甲基)丙烯酸酯官能团在光(例如紫外光)下反应,以形成使待交联的官能聚合物与支化嵌段共聚物共价结合或使支化嵌段共聚物与支化嵌段共聚物的另一分子共价结合的c-c键。因此,一旦支化嵌段共聚物在光下反应并交联,支化嵌段共聚物的(甲基)丙烯酸酯官能团不再存在,以及在所得交联聚合物中被c-c键取代。
34.根据本发明,当聚合物由支化嵌段共聚物交联剂交联,或支化嵌段共聚物自身进行交联时,应理解支化嵌段共聚物的硫醇官能团在光(例如紫外光)下反应,以形成使待交联的官能聚合物与支化嵌段共聚物共价结合或使支化嵌段共聚物与支化嵌段共聚物的另一分子共价结合的硫基键(例如硫醚)。因此,一旦支化嵌段共聚物在光下反应并交联,支化嵌段共聚物的硫醇官能团不再存在,以及在所得交联聚合物中被硫基键取代。
35.如在本文所用,在本发明中,“分子量”是指数均分子量。本发明的聚合物由nmr(核磁共振),而不是sec(尺寸排阻色谱)表征。
36.如在本文所用,“光反应性基团”理解为在光活化(例如紫外活化)下进行化学、结构和/或物理变化的化学基团。
37.如在本文所用,“可光交联基团”理解为用于在光活化下使聚合物交联的如上定义的光反应性基团。
38.如在本文所用,“芳基叠氮化物衍生物”理解为包含至少一个芳基叠氮官能团的官能团。例如,根据本发明的芳基叠氮化物衍生物为叠氮基苯甲酰基。
39.如在本文所用,“可降解聚合物”理解为能够尤其通过酶(酶促降解,通常使用微生物)或水(水解降解)或任何化学反应(例如氨解)分解成小分子(例如水、二氧化碳、甲烷)的聚合物。“可降解”和“可生物降解”在本说明书中可互换。
40.如在本文所用,“可降解弹性体”理解为具有弹性性质并包含可降解大分子链的聚合物。弹性体意指具有弹性性质,使得弹性体材料在被拉伸或压缩后趋于恢复其原始形状。
41.如在本文所用,“预聚物”理解为旨在通过与交联剂反应来进行交联以形成交联材料的聚合物。根据本发明,交联有利地为光交联。
42.如在本文所用,“非官能化的预聚物”理解为旨在进行交联的聚合物,其不包含能够与合适试剂反应以交联的可交联侧基或链端基。这种可交联基团为例如(甲基)丙烯酸酯。
43.如在本文所用,“生物材料”理解为与动物(包括人体)相容以及适用于医疗应用(特别是组织工程或医疗器械(导管、引流管、固定装置)或植入物)的聚合物材料,特别是弹性体材料。
44.如在本文所用,“成形物体”是经过成形加工(例如电纺丝、挤出或3d打印技术)以及通过例如照射被交联的聚合物或聚合物混合物,例如预聚物与本发明的支化嵌段共聚物的混合物。成形物体可以是例如薄膜、丝线、纤维、管、网或垫。
45.如在本文所用,“弹性体纤维支架”理解为以可被恰当使用的方式成形以及由弹性体纤维制成的支撑物。在本发明的上下文中,弹性体纤维通过如本文所述的星形共聚物进行交联以及通过包括电纺丝步骤的方法制得。本发明的弹性体纤维支架通常适用于制造医疗器械。
46.如在本文所用,“电纺丝”理解为用于制造聚合物纤维的成形加工,其使用电力将聚合物溶液的带电丝线拉伸至数百纳米量级的纤维直径。
47.如在本文所用,“挤出”理解为其中迫使压缩材料穿过具有待获得部件的横截面的模具的成形加工。可以获得不同形式的聚合物材料,例如小管、大管、板材、片材、膜。
48.如在本文所用,“组织工程”是指修复受损或患病的组织和器官的技术领域。特别地,由于人体大部分软组织通常由胶原纤维支撑以形成三维微结构,因此纤维聚合物支架具有模仿天然软组织的结构、力学和生物环境的优势,这有利于天然软组织的再生和重构。
49.在本发明中,peg代表聚乙二醇。术语(peg)
n臂
意指peg核心在星形共聚物中提供n个臂,每个臂被m个peg单体单元取代。
50.例如,(peg)
4臂
化合物由以下分子式反映:
[0051][0052]
(peg)
6臂
化合物由以下分子式反映:
[0053][0054]
(peg)
8臂
,也称为8臂聚(乙二醇)(三季戊四醇),由以下分子式反映:
[0055][0056]
在本发明的另一个实施方案中,支化嵌段共聚物的聚合物核心可以是线形核心。特别地,它可以是线形peg核心,其包含一个接一个排列以及在每一侧被官能团r取代的peg单体重复单元,每个官能团r提供n/2个末端官能团或原子,可降解聚合物链可以从这些末端官能团或原子延伸。这样的末端官能团或原子可以是氧原子或nh基团。
[0057]
例如,这样的线形核心可以由以下分子式反映:
[0058][0059]
在本发明中,pla代表聚(丙交酯)。pla
94
意指pla由94%的l-乳酸单元和6%的d-乳酸单元构成。
[0060]
在本发明中,pcl代表聚己内酯,也称为聚(ε-己内酯)。
[0061]
在本发明中,phb代表聚羟基丁酸酯。
[0062]
在本发明中,phbv代表聚羟基丁酸酯-共-羟基戊酸酯。
[0063]
在本发明中,pga代表聚乙醇酸。
[0064]
在本发明中,代表包含pla单元和泊洛沙姆(poloxamer)单元并具有以下结构的共聚物:
[0065][0066]
在以下实施例中,该共聚物也称为或pla
50
plu。
[0067]
如在本文所用,用语“(甲基)丙烯酸酯基”包括甲基丙烯酸酯基或丙烯酸酯基。
[0068]
详细描述
[0069]
在本发明的第一方面,提供一种可降解支化嵌段共聚物,其包含具有n个臂的聚醚中央核心和从聚醚中央核心的每个臂延伸的可降解聚合物链,所述聚醚中央核心为星形核心或线形核心,每个可降解聚合物链由可降解聚合物的l个单体单元构成,其特征在于,每个可降解聚合物链是相同的并且在其末端被光反应性基团官能化,所述光反应性基团选自芳基叠氮化物衍生物、(甲基)丙烯酸酯基或硫醇基,所述可降解支化嵌段共聚物通过以下结构说明:
[0070][0071]
其中
‑‑‑‑‑‑‑‑
是构成可降解聚合物链的可降解聚合物的单体单元,
[0072]
g是光反应性基团,
[0073]
n是至少为4的整数,以及
[0074]
是
[0075]
其中是星形聚醚中央核心,以及
[0076]
是对应于聚醚核心的单体单元,以及
[0077]
m介于4至400之间以及l介于4至1500之间,或者
[0078]
是
[0079]
其中是形成线形聚醚中央核心的单体单元,以及
[0080]
r是包含数量为n/2个的末端官能团或原子的多价支化官能团,所述末端官能团或原子选自氧原子或nh基团,每个所述末端官能团与一个聚合物链连接,以及
[0081]
m介于4至600个单元之间以及l介于2至400之间。
[0082]
尽管支化嵌段共聚物的分子量高,但至少为4的臂数n使得确保本发明的支化嵌段共聚物与聚合物或其自身之间的高交联率。实际上,选择本发明的共聚物中的n、m和l数值以获得共聚物本身的高分子量与共聚物链端的高反应性之间的最佳折衷。通过具有至少为4的臂数n,本发明的可降解支化嵌段共聚物可以具有对于上述应用足够高的分子量(即至少10000 g/mol,甚至至少20000 g/mol的分子量),同时具有通过m和l数值限定的聚合物链
长,所述聚合物链长较短,足以提供良好的反应性并因此提供高交联率。
[0083]
在一个优选的实施方案中,中央核心的聚醚选自聚乙二醇(peg)、泊洛沙姆和泊洛沙胺(poloxamine)。优选地,聚醚中央核心为peg中央核心。
[0084]
在另一个实施方案中,聚醚中央核心的每个臂被可降解聚合物链取代,所述可降解聚合物链由可降解聚合物的l个单元构成。聚合物链可以包含一种可降解聚合物,或至少两种可降解聚合物的混合物。聚合物链可以是例如嵌段共聚物或聚合物“ababababa”。
[0085]
优选地,可降解聚合物链的可降解聚合物选自聚酯、聚碳酸酯及其混合物。有利地,可降解聚合物链是疏水的。
[0086]
在一个优选的实施方案中,可降解聚合物链的可降解聚合物是聚酯,例如选自聚(丙交酯)(pla)、聚(ε-己内酯)(pcl)、聚羟基丁酸酯(phb)、聚羟基丁酸酯-共-羟基戊酸酯(phbv)、聚乙醇酸(pga)、聚(3-羟基戊酸酯)、聚二噁烷酮及其混合物,但不限于这些物质。更优选地,可降解聚酯为pla。
[0087]
根据本发明,支化嵌段共聚物的分子量有利地高于10000g/mol,优选高于15000g/mol,更优选高于20000g/mol,甚至更优选高于25000g/mol。
[0088]
在一个优选实施方案中,可降解支化嵌段共聚物的每个聚合物链在其末端被芳基叠氮化物衍生物,例如叠氮基苯甲酰基,更优选4-叠氮基苯甲酰基(bz-n3)官能化。
[0089]
芳基叠氮官能团的光活化(例如紫外光活化)引起高反应性氮烯物种的形成。氮烯是碳烯类似物,其中氮原子具有亲电子性以及例如能够插入到ch键中以形成胺键。
[0090]
根据该实施方案,可降解聚合物链的可降解聚酯优选为半结晶pla
94
,其由于结晶节点与3d网络的化学交联的组合而使得可增强所得交联弹性体的预期弹性体性能。
[0091]
在另一个优选的实施方案中,可降解支化嵌段共聚物的每个聚合物链在其末端被(甲基)丙烯酸酯基或硫醇基,优选(甲基)丙烯酸酯基,更优选甲基丙烯酸酯基官能化。
[0092]
根据本发明,(甲基)丙烯酸酯或硫醇官能化的可降解支化嵌段共聚物是非水溶性的。这种非水溶性性质由亲水性的聚醚核心与优选为疏水性的聚合物链之间的比值产生。在根据本发明的具有星形核心的支化嵌段共聚物中,所述比值由比值m/l表示,其严格大于0且小于或等于3。在根据本发明的具有线形核心的支化嵌段共聚物中,所述比值由比值m/(n*l)表示,其严格大于0且小于或等于1。
[0093]
根据该实施方案,可降解聚合物链的可降解聚酯优选为无定形pla
50
。
[0094]
在一个实施方案中,支化嵌段共聚物的核心是线形的,因此不同于星形中央核心,产生超支化嵌段共聚物。在根据该实施方案的所述超支化嵌段共聚物中,从线形核心延伸的臂数n为至少4,有利地介于4至32之间,优选地在4至16之间,更优选n等于4、8或16。根据该实施方案,具有线形中央核心的超支化嵌段共聚物可以通过以下结构说明:
[0095][0096]
其中是形成线形聚醚中央核心的单体单元,以及
[0097]
r是包含数量为n/2个的末端官能团或原子的多价支化官能团,所述末端官能团或原子选自氧原子或nh基团,每个所述末端官能团与一个聚合物链连接,
[0098]
‑‑‑‑‑‑‑‑
是构成可降解聚合物链的可降解聚合物的单体单元,
[0099]
g是光反应性基团,以及
[0100]
n是至少为4的整数,m介于4至600个单元之间以及l介于2至400之间。
[0101]
在本发明的具有线形中央核心的超支化嵌段共聚物中,每个末端官能团(其为氧原子或nh基团)被如上限定的一个可降解聚合物链取代。
[0102]
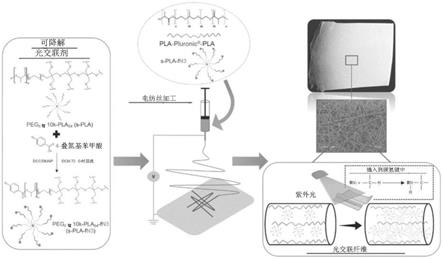
在一个优选的实施方案中,超支化形状的中央核心的聚醚选自聚乙二醇(peg)、泊洛沙姆或泊洛沙胺。优选地,聚醚中央核心为peg中央核心。
[0103]
根据本发明的具有线形中央核心的超支化嵌段共聚物有利地是对称的。线形中央核心在每一侧被相同的官能团r取代,每个基团r提供如上限定的数量为n/2个的相同末端官能团或原子,每个末端官能团被相同的聚合物链取代。
[0104]
根据一个优选的实施方案,本发明涉及一种星形共聚物,其包含具有n个臂的星形聚醚中央核心和从所述聚醚中央核心的每个臂延伸的聚合物链,n为至少为4的整数,每个臂包含对应于聚醚核心的m个单体单元,以及每个聚合物链由可降解聚合物的l个单元构成,其特征在于,每个聚合物链是相同的并且在其末端被光反应性基团官能化,所述光反应性基团选自叠氮基、(甲基)丙烯酸酯基或硫醇基,m介于4至400之间以及l介于4至1500之间。
[0105]
本发明的星形共聚物可以通过以下结构说明:
[0106][0107]
其中是星形聚醚中央核心,
[0108]
是对应于聚醚核心的单体单元,
[0109]
‑‑‑‑‑‑‑‑
是构成聚合物链的可降解聚合物的单体单元,以及
[0110]
g是光反应性基团。
[0111]
在本发明的另一实施方案中,本发明的可降解星形共聚物的臂数n介于4至12之间,优选介于4至8之间。有利地,本发明的星形嵌段共聚物具有4、6或8个臂。更优选地,聚醚中央核心具有8个臂。优选8臂星形嵌段共聚物以增加每个共聚物分子的反应性位点数量。
[0112]
在一个优选的实施方案中,星形中央核心的聚醚选自聚乙二醇(peg)、泊洛沙姆或泊洛沙胺。优选地,聚醚中央核心为peg中央核心。
[0113]
在一个优选实施方案中,可降解星形共聚物的每个可降解聚合物链在其末端被芳基叠氮化物衍生物,例如叠氮基苯甲酰基,更优选4-叠氮基苯甲酰基(bz-n3)官能化。
[0114]
在一个优选的实施方案中,根据本发明的(芳基叠氮)官能化的可降解星形共聚物由以下分子式反映:
[0115][0116]
其中是具有以下分子式的部分[peg-pla-bz-n3]:
[0117][0118]
m和l如上所限定。
[0119]
在以下实施例中,这种优选的星形嵌段共聚物也称为peg
8臂
10k-pla
94-fn3或s-pla-fn3。
[0120]
在另一个优选的实施方案中,可降解星形共聚物的每个聚合物链在其末端被(甲基)丙烯酸酯基或硫醇基,优选(甲基)丙烯酸酯基,更优选甲基丙烯酸酯基官能化。
[0121]
根据本发明,(甲基)丙烯酸酯或硫醇官能化的可降解星形共聚物有利地具有小于或等于100000 g/mol,优选小于或等于50000g/mol的分子量。高于100000 g/mol的分子量通常与由于(甲基)丙烯酸酯或硫醇官能团反应的可能性较低而引起的共聚物的反应性损失有关。
[0122]
根据本发明,(甲基)丙烯酸酯或硫醇官能化的可降解星形共聚物是非水溶性的。这种非水溶性性质由比值m/l产生,所述比值亦为亲水性的聚醚核心与优选为疏水性的聚合物链之间的比值,所述比值m/l严格大于0且小于或等于3。
[0123]
在一个优选的实施方案中,根据本发明的(甲基)丙烯酸酯官能化的可降解星形嵌段共聚物由以下分子式反映:
[0124][0125]
其中是具有以下分子式的部分[peg-pla-mc]:(在以下实施例中也称为peg
8臂
10k-pla
50-mc或s-pla-mc),m介于4至400之间,l介于4至1500之间。
[0126]
或者由以下分子式反映:
[0127][0128]
其中是具有以下分子式的部分[peg-pla-a]:(在以下实施例中也称为peg
8臂
10k-pla
50-a或s-pla-a),m介于4至400之间,l介于4至1500之间。
[0129]
根据另一方面,本发明涉及如上限定的可降解支化嵌段共聚物,优选星形共聚物作为光交联剂的用途。
[0130]
根据另一个实施方案,本发明涉及如上限定的支化嵌段共聚物,优选本发明的星形共聚物作为光交联剂以制备可降解聚合物生物材料,优选弹性体生物材料的用途。有利地,所述聚合物生物材料适合于医疗和软工程应用,例如导管、引流管、固定装置、敷料、膜、贴片、或医疗重建系统如植入物。
[0131]
根据进一步的实施方案,本发明涉及如上限定的可降解支化嵌段共聚物,优选星形共聚物用于使预聚物光交联的用途。这种光交联使得可制备光交联材料。
[0132]
旨在通过支化嵌段共聚物光交联剂,优选本发明的星形共聚物光交联剂进行光交联的预聚物可以由一种单体形式构成(=均聚物),或者为由至少两种不同单体形式构成的共聚物。
[0133]
有利地,旨在通过本发明的支化嵌段共聚物光交联剂,优选本发明的星形共聚物光交联剂进行光交联的预聚物为可降解聚合物。
[0134]
在一个优选的实施方案中,本发明涉及如上限定的(芳基叠氮)官能化的支化嵌段共聚物光交联剂,特别是如上限定的(芳基叠氮)官能化的星形共聚物光交联剂通过芳基叠氮基在光(例如紫外光)下的活化而用作通用光交联剂的用途。这种光交联剂有利地用于使包含ch键的非官能化预聚物光交联,其中由芳基叠氮官能团的紫外活化产生的氮烯物种插入以形成胺键。
[0135]
有利地,旨在通过(芳基叠氮)官能化的支化嵌段共聚物光交联剂,优选上述限定的(芳基叠氮)官能化的星形共聚物光交联剂进行光交联的非官能化预聚物选自聚酯、聚碳酸酯、聚乙烯类、聚醚、聚酰胺、聚烯烃、聚亚胺、聚(烷基硅氧烷)、聚((甲基)丙烯酸)及其混合物。
[0136]
优选地,旨在通过(芳基叠氮)官能化的支化嵌段共聚物光交联剂,优选上述限定的(芳基叠氮)官能化的星形共聚物光交联剂进行光交联的非官能化预聚物选自pla、泊洛沙姆及其混合物,但不限于这些物质。更优选地,旨在通过(芳基叠氮)官能化的支化嵌段共聚物光交联剂,优选(芳基叠氮)官能化的星形共聚物光交联剂进行光交联的非官能化预聚物为pla与泊洛沙姆的共聚物,特别是如上所述的三嵌段共聚物
[0137]
实际上,这种聚合物已显示出作为热塑性材料用于韧带组织工程的潜力,这是由于它们具有可调的降解速率以及在加工成编织/捻合的支架后接近天
然组织的力学性质,因此可适用于形成用于软组织工程应用的弹性体(pinese,c.等人,j.biomed.mater.res.b appl.biomater.105(2017)735-743)。
[0138]
在另一个优选的实施方案中,本发明涉及(甲基)丙烯酸酯或硫醇官能化的支化嵌段共聚物光交联剂,优选如上限定的(甲基)丙烯酸酯或硫醇官能化的星形共聚物光交联剂用于使由至少一个可光交联基团官能化的预聚物光交联的用途。所述可光交联基团为例如(甲基)丙烯酸酯基。
[0139]
有利地,旨在通过本发明的(甲基)丙烯酸酯或硫醇官能化的支化嵌段共聚物光交联剂,优选如上限定的(甲基)丙烯酸酯或硫醇官能化的星形共聚物光交联剂进行交联的官能化预聚物具有小于或等于50000g/mol的分子量。低分子量预聚物可以改进其反应性和可光交联链端基团的可用性。
[0140]
在一个实施方案中,旨在通过本发明的(甲基)丙烯酸酯或硫醇官能化的支化嵌段共聚物光交联剂,优选如上限定的(甲基)丙烯酸酯或硫醇官能化的星形共聚物光交联剂进行交联的官能化预聚物有利地选自聚酯、聚碳酸酯、聚乙烯类、聚醚、聚酰胺、聚烯烃、聚亚胺、聚(烷基硅氧烷)、聚丙烯酸、聚((甲基)丙烯酸)及其混合物,所述预聚物由可光交联基团官能化。
[0141]
优选地,旨在通过(甲基)丙烯酸酯或硫醇官能化的支化嵌段共聚物光交联剂,优选如上限定的(甲基)丙烯酸酯或硫醇官能化的星形共聚物光交联剂进行光交联的官能化预聚物选自pla、泊洛沙姆及其混合物,但不限于这些物质。更优选地,旨在通过(甲基)丙烯酸酯或硫醇官能化的支化嵌段共聚物光交联剂,优选如上限定的(甲基)丙烯酸酯或硫醇官能化的星形共聚物光交联剂进行光交联的官能化预聚物为pla与泊洛沙姆的共聚物,特别是如上所述的三嵌段共聚物
[0142]
根据本发明的另一个实施方案,用芳基叠氮化物衍生物或(甲基)丙烯酸酯或硫醇基团官能化的本发明的支化嵌段共聚物光交联剂(特别是本发明的星形共聚物光交联剂)能够与自身反应形成光交联材料。在这样的实施方案中,不需要预聚物,以及目标交联聚合物(优选交联弹性体)仅从本发明的支化嵌段共聚物,优选仅从本发明的星形共聚物开始获得。
[0143]
根据另一方面,本发明涉及一种用于制备可降解光交联聚合物,优选可降解光交联弹性体的方法,其包括以下步骤:
[0144]
(a)制备包含本发明的支化嵌段共聚物(优选如上限定的星形共聚物)和任选的预聚物的溶液或固体共混物,
[0145]
(b)对由步骤(a)产生的溶液或固体共混物进行成形加工,
[0146]
(c)在光(例如紫外光)下照射由步骤(b)产生的溶液或固体共混物,
[0147]
(d)回收可降解光交联聚合物,优选可降解光交联弹性体。
[0148]
在一个实施方案中,从(芳基叠氮)官能化的支化嵌段共聚物光交联剂(特别是(芳基叠氮)官能化的星形共聚物光交联剂)和具有至少一个ch键的非官能化预聚物开始进行上述方法。在另一个实施方案中,从(芳基叠氮)官能化的超支化嵌段共聚物光交联剂和具有至少一个ch键的非官能化预聚物开始进行上述方法。
[0149]
在又一个实施方案中,从(甲基)丙烯酸酯官能化或硫醇官能化的支化嵌段共聚物光交联剂(特别是(甲基)丙烯酸酯官能化或硫醇官能化的星形共聚物光交联剂)和用可光
交联基团官能化的预聚物开始进行上述方法。在另一个实施方案中,从(甲基)丙烯酸酯官能化或硫醇官能化的超支化嵌段共聚物光交联剂和用光可交联基团官能化的预聚物开始进行上述方法。
[0150]
在又一个实施方案中,仅从(芳基叠氮)官能化的支化嵌段共聚物光交联剂开始进行上述方法。优选地,仅从(芳基叠氮)官能化的星形共聚物光交联剂开始进行上述方法。在另一个实施方案中,仅从(芳基叠氮)官能化的超支化嵌段共聚物光交联剂开始进行上述方法。
[0151]
在又一个实施例中,仅从(甲基)丙烯酸酯官能化的支化嵌段共聚物光交联剂开始进行上述方法。优选地,仅从(甲基)丙烯酸酯官能化的星形共聚物光交联剂开始进行上述方法。在另一个实施方案中,仅从(甲基)丙烯酸酯官能化的超支化嵌段共聚物光交联剂开始进行上述方法。
[0152]
在又一个实施方案中,仅从硫醇官能化的支化嵌段共聚物光交联剂开始进行上述方法。优选地,仅从硫醇官能化的星形共聚物光交联剂开始进行上述方法。在另一个实施方案中,仅从硫醇官能化的超支化嵌段共聚物光交联剂开始进行上述方法。
[0153]
根据进一步的实施方案,步骤(b)的成形加工选自挤出、涂膜、喷膜、铸膜、电喷涂、电纺丝或3d打印技术,例如熔融沉积成型、多喷口打印、立体光刻、数字光处理、选择性激光烧结或连续液体界面生产。
[0154]
根据另一个实施方案,同时完成步骤(b)和步骤(c),特别是当成形加工是电纺丝时。
[0155]
有利地,可以在光引发剂例如2,2二甲氧基-2-苯基乙酰苯或曙红-y的存在下完成步骤(c),特别是当光源与紫外光不同时。
[0156]
在另一个实施方案中,用汞或金属卤化物灯(优选汞灯)或紫外led进行紫外照射。
[0157]
在又一个实施方案中,照射的紫外波长介于254至400nm之间。
[0158]
在又一个实施方案中,紫外照射进行介于1秒至10分钟之间,优选5秒至5分钟之间的时间,更优选进行1分钟。
[0159]
在一个实施方案中,在步骤(a)中,预聚物与支化嵌段共聚物以介于10/90至90/10之间,优选在50/50至90/10之间的(预聚物/支化嵌段共聚物)重量比混合,更优选以50/50的比例混合。
[0160]
根据又一实施方案,可通过上述方法获得的可降解光交联聚合物,优选可降解光交联弹性体成形为膜、丝线、纤维、管、网或垫。特别地,可降解的光交联弹性体纤维支架通过上述方法使用电纺丝加工来获得。
[0161]
在另一个实施方案中,可通过上述方法获得的可降解光交联聚合物,优选可降解光交联弹性体是适用于医疗和软工程应用的可降解聚合物生物材料,例如导管、引流管、固定装置、敷料、膜、贴片或医疗重建系统如植入物。
[0162]
本发明还涉及可通过上述方法获得的可降解光交联弹性体。根据进一步的实施方案,提供一种用于医疗和组织工程应用的可通过上述方法获得的可降解光交联弹性体。所述可降解光交联弹性体为例如导管、引流管、固定装置、敷料、膜、贴片或医疗重建系统如植入物。
[0163]
根据又一个实施方案,可降解光交联弹性体为膜或弹性体纤维支架。
[0164]
根据一个实施方案,从预聚物,优选经由电纺丝加工通过根据本发明的星形共聚物光交联剂进行光交联的可降解预聚物开始生成可降解的光交联弹性体纤维支架。
附图说明
[0165]
图1:基于本发明多官能的芳基叠氮化物星形嵌段共聚物光交联剂的弹性微纤维支架的设计方案。
[0166]
图2:(a)可降解共聚物光交联剂peg
8臂
10k-pla
94-fn3(s-pla-fn3)的合成方案;(b)peg
8臂
10k-pla
94-fn3(s-pla-fn3)的1h nmr波谱;(c)(1)peg
8臂
10k-pla
94
的sec分析和(2)peg
8臂
10k-pla
94-fn3的sec分析。紫外检测器(270nm-1
)。
[0167]
图3:通过凝胶分率分析评估的用于生成弹性体的紫外灯类型(mb相对于mhb)的影响。
[0168]
图4:在使用两种不同的紫外线灯泡以不同的时间进行紫外照射前后对支架的ftir分析:(a)金属卤化物灯和(b)汞灯。通过位于2110cm-1
的特征叠氮ir谱带的丢失,表明了芳族双(芳基叠氮化物)光活化生成氮烯。
[0169]
图5:通过凝胶分率评估的弹性体pla
50
plu(50-200)/s-pla-fn3的交联动力学。(a)pla
50
plu50/s-pla-fn3;(b)pla
50
plu100/s-pla-fn3;(c)pla
50
plu200/s-pla-fn3;(d)通过凝胶分率评估的弹性微纤维支架pla
50
plu(50-200)/s-pla-fn3的交联动力学。(数据表示为平均值
±
sd,对应于n=3的测量值)。
[0170]
图6:随着芳基叠氮化物光交联剂(10、25和50重量%的s-pla-fn3相对于5重量%的ba)的性质和共混物中芳基叠氮基的总含量(n(n3))变化的凝胶分率(10分钟照射时间)。
[0171]
图7:基于pla
50-plu200/s-pla和pla
50-plu200/s-pla-fn3的不同支架的sem图像和微纤维直径分布(紫外固化2分钟前后)。sem的比例尺为30μm。(a)pla
50-plu200/s-pla 90/10;(b)未固化的pla
50-plu200/s-pla-fn3 90/10;(c)紫外固化的pla
50-plu200/s-pla-fn3 90/10;(d)pla
50-plu200/s-pla 75/25;(e)未固化的pla
50-plu200/s-pla-fn3 75/25;(f)紫外固化的pla
50-plu200/s-pla-fn375/25;(g)pla
50-plu200/s-pla 50/50;(h)未固化的pla
50-plu200/s-pla-fn350/50;(i)紫外固化的pla
50-plu200/s-pla-fn3 50/50。
[0172]
图8:在37℃的水合和干燥状态下的基于pla
50-plu200/s-pla-fn3的紫外固化纤维支架的力学拉伸性质。
[0173]
图9:基于不同比例的pla
50-plu200/s-pla或pla
50-plu200/s-pla-fn3的纤维支架的吸水性。
[0174]
图10:支架降解的评估:(a)在37℃的pbs中纤维支架在降解期间的剩余质量,数据对应于三个重复的测量值;(b)pla
50-plu200/s-pla或pla
50-plu200/s-pla-fn3 50/50在37℃的pbs中随着降解时间推移的sem图像,放大倍数x5000。(数据表示为平均值
±
sd,对应于n=3的测量值)。
[0175]
图11:用基于不同比例的pla
50-plu200/s-pla或pla
50-plu200/s-pla-fn3的纤维支架的提取物处理24小时后评估的对l929细胞的细胞毒性。(数据表示为平均值
±
sd,对应于每种条件n=9的测量值)。
[0176]
图12:基于peg(s-pla-mc mn≈50000g.mol-1)的膜的应力-应变曲线:(a)在紫外照射后通过热熔压机制备的膜,在37℃以及在10%、50%和100%应变(10次循环)下进行分
析;(b)在紫外照射后通过热熔压机制备的膜与不进行紫外照射的情况下通过热熔压机制备的膜的比较,在37℃以及在100%应变(10次循环)下进行分析。
[0177]
图13:基于s-pla-mc(mn≈50000g.mol-1)的弹性体在存在或不存在光引发剂(pi,在这种情况下为2,2二甲氧基-2-苯基乙酰苯)时的应力-应变曲线(在37℃下)。
[0178]
图14:通过溶剂蒸发的基于s-pla-50-mc的膜的降解,涉及a)剩余重量、b)剩余交联和c)吸水性。
[0179]
图15:基于pla
50-plu200/s-pla-fn
3 75/25(左)或pla
50-plu200/s-pla-fn350/50(右)的纤维支架在加工中紫外固化或在加工后紫外固化的情况下的凝胶分率(%)。
[0180]
图16:基于(a)pla-pluronic-pla/s-pla-fn
3 75/25和(b)pla-pluronic-pla/s-pla-fn
3 50/50的纤维支架的15%伸长的循环拉伸测试,两者均是在后处理紫外固化之后获得。在37℃的温度下以10个循环进行分析。
[0181]
图17:基于s-pla-100-mc(100000g.mol-1)的纤维支架的sem图像。
[0182]
图18:通过3d打印从s-pla-50-mc(50kg.mol-1)获得的多孔材料的照片。
[0183]
图19:通过3d打印从s-pla-50-mc(50kg.mol-1)获得的材料的照片。
[0184]
图20:通过3d打印从s-pla-50-mc(50kg.mol-1)获得的立方体的照片。
具体实施方式
[0185]
通过以下实施例说明本发明。
[0186]
实施例
[0187]
1.材料和方法
[0188]
1.1材料
[0189]
d,l-丙交酯和l-丙交酯购自purac(里昂,法国)。8臂聚(乙二醇)(三季戊四醇)(peg
8臂
10k,mw=10000g.mol-1
)购自jenkem technology有限责任公司(北京,中国)。泊洛沙姆(f127,mw=12600g.mol-1
)、2-乙基己酸锡(ii)(sn(oct)2,95%)、二氯甲烷(dcm)、乙醚(et2o)、n,n-二环己基-碳二亚胺(dcc)、4-(二甲胺)吡啶(dmap)和n,n-二甲基甲酰胺(dmf)、四氢呋喃(thf)购自sigma-aldrich(st quentin fallavier,法国)。2,6-双(4-叠氮基亚苄基)-4-甲基环己酮(ba)和4-叠氮基苯甲酸购自tci(巴黎,欧洲)。除dcm和dcc外,所有化学品均无需进一步纯化即可使用。dcm用钙杂化物干燥并新鲜蒸馏后使用。dcc通过mgso4溶解在无水dcm中,搅拌6小时,然后过滤并干燥后使用。
[0190]
1.2表征
[0191]
ft-ir
[0192]
用perkin elmer spectrum 100光谱仪记录聚合物膜的ft-ir光谱。
[0193]
tga
[0194]
在氮气氛下用perkin elmer tga 6记录tga分析。将样品在30℃下保持1分钟,然后以10℃.min-1
的速率加热至300℃。
[0195]
sec
[0196]
通过尺寸排阻色谱(sec)shimadzu使用两个混合介质柱plgel 5μm mixed-c(300x7.8mm)、shimadzu ri检测器20-a和shimadzu紫外检测器spd-20a(370nm-1
)(40℃恒温分析池)来确定平均分子量和分散性四氢呋喃(thf)是流动相,在30℃(柱温)
下流速为1ml.min-1
。将聚合物溶解在thf中达到10mg.ml-1
的浓度;之后,在注射前通过0.45μm微孔过滤器过滤溶液。和根据使用聚苯乙烯标样的校准来表示。
[0197]
nmr波谱
[0198]
在室温下从以300mhz运行的amx brucker波谱仪记录1h nmr波谱。所用溶剂为氘代氯仿和dmso-d6。化学位移以相对于四甲基硅烷(tms)的ppm表示。
[0199]
用对不同的聚合物表征的perkin elmer instrument dsc 6000热分析仪通过差示扫描量热法(dsc)分析聚合物的热性质。其在氮气下进行。将样品加热至100℃(10℃.min-1
),然后冷却至-50℃(10℃.min-1
),而后第二次升温至120℃(5℃.min-1
)。将基于peg
8臂
10k-pla
94
的样品加热至180℃(10℃.min-1
),然后冷却-50℃(10℃.min-1
),而后第二次升温至180℃(5℃.min-1
)。在第二次升温时测量玻璃化转变温度(tg)。
[0200]
用hitachi s4800扫描电子显微镜(balard化学极的iem实验室的技术平台)以2kv的加速电压以及500、1000和5000倍的放大倍数(每个放大倍数下3张图像)检查样品的形态。
[0201]
1.3共聚物的合成
[0202]
如发明人先前工作中所述,通过开环聚合(rop)来合成三嵌段共聚物(预聚物pla
50
plu)、peg
8臂
10k-pla
50
(50%的l-乳酸单元和50%的d-乳酸单元)、peg
8臂
10k-pla
94
(94%的l-乳酸单元和6%的d-乳酸单元)(非官能化的星形共聚物,s-pla)(leroy,a.et al,mater.sci.eng.c.33(2013)4133-4139)。
[0203]
(预聚物)
[0204]
对于pla
50
plu,目标是三种分子量:50000、100000和200000g.mol-1
,相应的共聚物分别称为pla
50
plu50、pla
50
plu100和pla
50
plu200。
[0205]
为此,将确定量的d.l-丙交酯、l-丙交酯和f127引入烧瓶中,然后向其中加入sn(oct)2(相对于
d,l-丙交酯单元为0.1摩尔%)。在真空下密封烧瓶之前实施氩-真空循环。在130℃的烘箱中并在持续搅拌下进行rop,持续5天。然后,将混合物溶解在dcm中并在冷et2o中沉淀。在减压下将最终的三嵌段共聚物干燥至恒重。
[0206]1h nmr(300mhz;cdcl3):δ(ppm)=5.1(q,1h,co
–
ch-(ch3)
–
o),3.6(s,4h,ch2–
ch2–
o),3.5(m,2h,ch(ch3)
–
ch2–
o),3.4(m,1h,ch(ch3)
–
ch2–
o),1.5(m,3h,co
–
ch(ch3)
–
o),1.1(m,3h,ch(ch3)
–
ch2–
o)。
[0207]
使用方程式(1)和(2)确定共聚物分子量,其中乳酸单元的分子量确认为72g.mol-1
。
[0208]
(1)
[0209]
(2)
[0210]
peg
8臂
10k-pla
94 20000g/mol(s-pla-20)、peg
8臂
10k-pla
50 25 000g/mol(s-pla-25)、peg
8臂
10k-pla
50 50000g/mol(s-pla-50)和peg
8臂
10k-pla
50
100000g/mol(s-pla-100)(非官能化)
[0211]
对于peg
8臂
10k-pla
94
,目标总分子量为20000g.mol-1
。
[0212]
对于peg
8臂
10k-pla
50
,目标总分子量为25000g.mol-1
或50000g.mol-1
或
100000g.mol-1
。
[0213]
为此,将确定量的d.l-丙交酯、l-丙交酯和peg
8臂
10k引入烧瓶中,然后向其中加入sn(oct)2(相对于
d,l-丙交酯单元为0.1摩尔%)。在真空下密封烧瓶之前实施氩-真空循环。在130℃的烘箱中并在持续搅拌下进行rop,持续5天。然后,将混合物溶解在dcm中并在冷et2o中沉淀。在减压下将最终的星形共聚物干燥至恒重。通过sec分析确定了1.1的低分散性。
[0214]
peg
8臂
10k-pla
94
:
[0215]1h nmr(300mhz;cdcl3):δ(ppm)=5.1(q,1h,co
–
ch-(ch3)
–
o),4.3(m,2h,o-ch
2-c-ch
2-o),3.6(s,4h,ch2–
ch2–
o),3.3(m,2h,o-ch
2-c-ch
2-o),1.5(t,3h,co-ch-(ch3)-o)。
[0216]
使用方程式(1)和(3)确定星形共聚物分子量
[0217]
(3)
[0218]
1.4芳基叠氮官能化的peg
8臂
10k-pla
94
(s-pla-fn3)的合成(图2a)
[0219]
将8臂星形共聚物peg
8臂
10k-pla
94
溶解在新鲜蒸馏的dcm(20%w/v)中。加入确定量的4-叠氮基苯甲酸(2.5eq./oh基团)、dcc(2.5eq./oh基团)和dmap(2.5eq./oh基团)。在黑暗中并在搅拌下将混合物在45℃加热6天。过滤反应介质并用na2co3水溶液洗涤(3次),然后用mgso4干燥。在黑暗中并在冷乙醚中沉淀共聚物溶液。在减压下将芳基叠氮官能化peg
8臂
10k-pla
94
(s-pla-fn3)干燥至恒重。通过对在8.0处的芳基叠氮化物特征信号的积分和在4.2ppm处的质子共振的积分进行比较来确定官能化率。
[0220]1h nmr(300mhz;dmso-d6):δ(ppm)=8.0(d,2h芳族环,ch=ch-c-n3),7.3(d,2h芳族环,ch=ch-n3),5.1(q,1h,co
–
ch-(ch3)
–
o),4.3(m,2h,o-ch
2-c-ch
2-o),3.6(s,4h,ch2–
ch2–
o),3.3(m,2h,o-ch
2-c-ch
2-o),1.5(t,3h,co-ch-(ch3)-o)(图2b)。
[0221]
根据1h nmr波谱计算的实验分子量和通过sec分析确定的d=1.1的分散性表明在合成过程中没有发生s-pla共聚物降解。
[0222]
sec分析进一步证实了4-叠氮基苯甲酸接枝到s-pla链端。官能化后,芳基叠氮基团(270nm-1
)的特征紫外信号在对应于星形共聚物折射率信号的保留时间是可见的(图2-c-2)。起始s-pla共聚物的情况并非如此(图2-c-1)。
[0223]
这些结果证实了由芳基叠氮部分成功地在链端官能化s-pla,产生了预期的多(芳基叠氮化物)大分子光交联剂s-pla-fn3。
[0224]
1.5甲基丙烯酸酯官能化的peg
8臂
10k-pla
50 25000(s-pla-25-mc)、peg
8臂
10k-pla
50 50000(s-pla-50-mc)和peg
8臂
10k-pla
50 100000(s-pla-100-mc)的合成
[0225]
将8臂星形共聚物peg
8臂
10k-pla
50
peg
8臂
10k-pla
50
或peg
8臂
10k-pla
50
溶解在新鲜蒸馏的dcm(20%w/v)中。加入三乙胺(5eq./oh基团)并将所得混合物冷至0℃。在0℃下并在搅拌下,用浇铸安瓿加入甲基丙烯酰氯(5eq./oh基团)。一旦添加完成,将混合物在黑暗中并在室温下搅拌72小时。然后,过滤产物,之后在冷乙醚中沉淀。在黑暗中,将甲基丙烯酸酯官能化的peg
8臂
10k-pla
50
(s-pla-25-mc)、peg
8臂
10k-pla
50
(s-pla-50-mc)和peg
8臂
10k-pla
50
(s-pla-100-mc)溶解在dcm中并用碱性水相洗涤。在真空压力下浓缩有机层,得到浓缩溶液,将浓缩
溶液在冷乙醚中沉淀。然后在减压下干燥回收的产物。
[0226]
由nmr确定官能化率(95%官能化)。
[0227]
s-pla-25-mc:
[0228]1h nmr(300mhz;cdcl3)δ(ppm)=6.2(d,1h,co-c(ch3)=ch2),5.6(d,1h,co-c(ch3)=ch2),5.1(q,1h,co
–
ch-(ch3)
–
o),4.3(m,2h,c-ch
2-o),3.6(s,4h,ch2–
ch2–
o),3.3(o-ch
2-c-ch
2-o),2.0(s,3h,,co-c(ch3)=ch2),1.5(t,3h,co-ch-(ch3)-o)。
[0229]
1.6丙烯酸酯官能化的peg
8臂
10k-pla
50
(s-pla-a)的合成
[0230]
将8臂星形共聚物peg
8臂
10k-pla
50
溶解在新鲜蒸馏的dcm(20%w/v)中。加入三乙胺(15eq./oh基团)并将所得混合物冷至0℃。在0℃下并在搅拌下,用浇铸安瓿加入丙烯酰氯(15eq./oh基团)。一旦添加完成,将混合物在黑暗中并在45℃下加热72小时。然后,过滤产物,之后在冷乙醚中沉淀。在黑暗中,将丙烯酸酯官能化的peg
8臂
10k-pla
50
(s-pla-a)溶解在dcm中并用碱性水相洗涤。在黑暗中并在室温下,在真空压力下浓缩有机层,得到浓缩溶液,将浓缩溶液在冷乙醚中沉淀。然后在减压下干燥回收的产物。
[0231]
1.7聚合物的成形和光交联
[0232]
通过溶剂蒸发的膜
[0233]
对于用2,6-双(4-叠氮基亚苄基)-4-甲基环己酮(ba)交联的弹性体,将具有限定分子量的pla
50
plu共聚物在dcm中与2,6-双(4-叠氮基亚苄基)-4-甲基环己酮(ba)(聚合物的2至5重量%)一起搅拌。
[0234]
对于用s-pla-fn3交联的弹性体,将具有限定分子量的pla
50
plu共聚物与不同重量比(10、25和50重量%)的s-pla-fn3混合并在dcm中搅拌。
[0235]
对于对照,遵循相同的程序,其中将s-pla-fn3替换为非官能的s-pla。
[0236]
对于仅从s-pla-50-mc开始(没有其它预聚物)获得的弹性体,将s-pla-50-mc溶解在dcm中并进行搅拌。可以加入光引发剂2,2-二甲氧基-2-苯基乙酰苯(pi)(共聚物的2重量%)。
[0237]
在铝模具中干燥溶液,从而获得薄膜。将膜在暗处保存24小时。将所得膜在真空下进一步干燥24小时。
[0238]
通过压制的膜
[0239]
在155℃下加热压机。然后,将粉末形式的共聚物s-pla-50-mc沉积在聚四氟乙烯纸上并加热至155℃,施加5至6巴的压力10分钟。在此步骤后,将几微米的膜放入冰箱中5分钟。
[0240]
通过电纺丝加工的基于微纤维的组织
[0241]-电纺聚合物溶液
[0242]
将聚合物共混物pla
50
plu和s-pla-fn3或s-pla(90/10、75/25和50/50w/w在本文的其余部分中分别称为90/10、75/25和50/50)溶于dcm/dmf(50/50v/v)[40]。选择共混浓度以制备无珠粒的纤维(90/10:14重量%,75/25:18重量%,50/50:22重量%)。将所有混合物在室温下力学搅拌过夜,直至完全溶解。
[0243]
将共聚物、s-pla-50-mc或s-pla-100mc溶解在dcm/dmf溶液(70/30v/v)中,s-pla-100-mc的浓度为35重量%以及s-pla-50-mc为40重量%。将聚合物溶液在室温下力学混合过夜,直至完全溶解。
[0244]-电纺丝加工
[0245]
用水平注射泵装置进行电纺丝加工。高压电源设置为12至15kv。聚合物溶液装满具有21标准针头(内径为0.82mm)的10ml注射器。由注射泵(fresenius vial program 2iec)控制进料速率(s-pla-50-mc和s-pla-100-mc为1.8ml/h,其它聚合物为2.1ml/h)。收集器是方形铝箔,位于距针尖15cm处。在室温下进行实验。电纺丝40分钟后收集纤维支架。在进一步实验之前将其干燥过夜。
[0246]
任选地,在电纺丝加工期间使用紫外led完成纤维的紫外固化步骤。在整个电纺丝加工中进行紫外固化。
[0247]
dymax qx4控制器的led(365和385nm)位于距收集器8cm处。led具有在14w.cm-2
至19w.cm-2
之间的强度。accu-cal 50-led辐射仪用于测量样品接收到的紫外剂量。
[0248]
也可以在电纺丝加工之后完成纤维的所述紫外固化步骤,时间也为2分钟。
[0249]
薄膜的光交联
[0250]
使用dymax pc-2000系统(75mw.cm-2)在惰性气氛下将膜在紫外光(汞灯或金属卤化物灯)下照射不同的时间(1分钟《t《20分钟)。为清楚起见,在本文的其余部分,10分钟照射时间对应于膜每侧照射5分钟。灯与样品之间测得的距离为13.5cm。使用accu-cal
tm 50系统评估照射剂量强度。随后,将弹性体膜切割、称重并放入dcm(10ml)中。洗涤三次后,将不溶性交联部分从dcm中移出并真空干燥24小时。最后,对样品进行称重以根据下面的方程式(4)确定凝胶分率。
[0251]
纤维支架的光交联
[0252]
为了保证外壳内低温并保持纤维的形态,在紫外光(汞灯)和惰性气氛下以0.5hz的频率照射纤维支架2秒。使用dymax pc-2000系统(75mw.cm-2)在确定的时间段内施加连续闪光。使用针对膜描述的程序来测量距离、照射强度和凝胶分率。
[0253]
通过立体光刻的3d材料
[0254]
共聚物溶液的合成
[0255]
将共聚物s-pla-50-mc以400g/l的浓度溶解在乳酸乙酯中。将光引发剂omnirad rpo-l以2重量%的浓度加入到该溶液中。然后将所得混合物力学搅拌24小时。
[0256]
成形加工
[0257]
所需的结构由onshape软件建模,然后使用phrozen shuffle 3d打印机打印。使用405nm(50瓦)led逐层照射聚合物溶液(50μm需2分钟)。
[0258]
在打印过程结束时,使用formlab-form cure对物体进行后固化步骤:波长405nm,两侧照射5分钟,45℃。
[0259]
凝胶分率(=交联率)
[0260]
(4)凝胶分率(%)=(不溶性交联部分的重量/初始样品的重量)*100
[0261]
凝胶分率的百分比值使得可评估测试的光交联剂的效率。凝胶分率值越高,光交联剂越有效。
[0262]
1.8力学性质
[0263]
对微纤维支架样品进行拉伸力学测试。切割样品(30x10mm)并用测微计测量厚度。在37℃下,使用instron 3344以10mm/min的变形速率对支架(干燥和水合状态)进行三次重复分析。杨氏模量(e,mpa)、屈服应力(σy,mpa)、屈服应变(εy,%)、断裂应力(σ
断裂
,mpa)、断裂
应变(ε
断裂
,%)表示为三次测量的平均值。
[0264]
1.9纤维材料的降解研究
[0265]
将纤维测试样品切割(10x10mm)、称重(wi=初始重量)并在搅拌下置于恒温(37℃)的5ml磷酸盐缓冲盐水(pbs)(ph 7.4)中。在不同的时间点,从pbs中移出纤维材料,进行称重(ww=湿样品的重量),然后干燥至恒重(wx=在pbs中x时间后干燥的重量)。根据方程式(5)计算样品的剩余质量。
[0266]
(5)剩余质量(%)=(1-((wi-wx)/wi))*100
[0267]
根据方程式(6)确定吸水性
[0268]
(6)吸水性(%)=((ww-wi)/wi)*100。
[0269]
1.10膜的降解研究
[0270]
对基于s-pla-50-mc的膜的降解进行了一个月的研究。将膜(l=2mm和l=0.5mm)称重(wi=初始质量),然后引入到pbs溶液(ph=7.4)中并在37℃下搅拌。在不同的时间(3、8、15、15、22和30天),回收膜,称重(ww=湿质量)并干燥24小时。然后对膜再次称重(wd=干燥质量)并引入到dcm溶液中。洗涤三次后,将样品干燥过夜并称重(wcd=交联的干燥质量)。因此,在该降解过程中,根据以下相应的方程式来评估材料质量的保持、吸水性和保持的化学桥:
[0271]
(5)剩余质量(%)=(1-((wi-wd)/wi))*100
[0272]
(6)吸水性(%)=((ww-wi)/wi)*100
[0273]
(7)剩余化学桥(%)=(1-(凝胶分率(i)-(凝胶分率(m))/凝胶分率(i))*100
[0274]
其中凝胶分率(i)是初始凝胶分率,凝胶分率(m)是在不同时间的分率。作为提醒,
[0275]
(8)
[0276]
1.11细胞毒性试验
[0277]
根据iso 10993-5指南选择细胞和对照聚合物膜。将小鼠成纤维细胞l929细胞(ecacc 85011425)维持在补充有5%胎牛血清(fbs)、2mm l-谷氨酰胺和1%青霉素/链霉素的dmem高葡萄糖中,并在37℃和5%co2下培养。测试细胞不含支原体。阴性(rm-c高密度聚乙烯被称为c-)对照膜和阳性(rm-b 0.25%二丁基二硫代氨基甲酸锌(zdbc)聚氨酯被称为c+)对照膜购自hatano research institute(ochiai 729-5,hadanoshi,kanagawa 257,日本)。对提取物评估细胞毒性。首先,按照iso 10993-12建议,在37℃下并在无菌条件下,在完全生长培养基上以0.1g/ml提取72小时。将l929细胞以每孔15.103个细胞接种在96孔板中,并使其贴壁过夜。然后将培养基从培养物中去除并丢弃,而后将纤维提取物的等分试样加入到每个孔中。将空白、阴性对照和阳性对照的等分试样加入到额外的重复孔中(n=9)。在适当气氛下孵育24小时后,根据制造商的说明,通过乳酸脱氢酶(ldh)试验(pierce)评估提取物的细胞毒性。简而言之,将孔中的培养基转移至新板并与ldh反应混合物混合。在室温下孵育30分钟后,使用酶标仪(bmg labtech's)测量490nm和680nm处的吸光度以确定ldh活性。
[0278]
根据方程式(7)计算细胞毒性百分比
[0279]
(9)细胞毒性(%)=(((样品ldh活性)—ldh-)/(ldh+
″
—
″
ldh-))*100
[0280]
其中“ldh
‑”
表示自发ldh释放控制(水处理),“ldh+”表示细胞裂解后获得的最大
ldh释放控制活性。
[0281]
2.结果和讨论
[0282]
2.1现有技术中双(芳基叠氮化物)作为光交联剂的评价
[0283]
为了从非官能的聚酯开始制备可降解弹性生物材料,我们首先关注三嵌段(pla
50
plu)。根据1h nmr波谱,目标分子量和实验分子量(50000、100000和200000g.mol-1
)是一致的。通过sec分析确定了分散性在1.5至1.8之间,这与高分子量聚酯的rop通常获得的值一致。
[0284]
这些共聚物进一步用于评估现有技术中2,6-双(叠氮基亚苄基)-4-甲基环己酮(ba)作为光交联剂的实际潜力。
[0285]
三种不同的三嵌段共聚物pla
50
plu50、pla
50
plu100和pla
50
plu200与ba在不同的ba浓度(2重量%和5重量%)下混合。凝胶分率结果(图3)显示使用ba作为光交联剂的交联效率低(凝胶分率《15%),虽然已证明芳基叠氮化物被活化。这通过在2100nm-1
处的谱带(其是叠氮基的特征)消失得到证明(图4)。
[0286]
尽管芳基叠氮化物被光活化,但交联不足,这种不足归因于偶氮二聚体的形成和不允许交联的终止反应。此外,与所用紫外灯的性质(金属卤化物灯相对于汞灯)和ba浓度相比,预聚物pla
50
plu共聚物的分子量不会显著影响交联效率。如所预期的,凝胶分率随着汞灯和更高的ba浓度(5重量%)增加。
[0287]
考虑到这些结果,我们推测ba(2个芳基叠氮基团)的与该有机小分子上反应性基团的直接接近有关的有限官能性可以解释基于ba的交联的不良结果。
[0288]
2.2通过s-pla-fn3光交联的可降解弹性体成形为膜
[0289]
pla
50
plu预聚物分子量和s-pla-fn3含量对交联效率的影响
[0290]
为了评估s-pla-fn3用于制备可降解弹性体生物材料的潜力,我们首先关注pla
50
plu分子量和s-pla-fn3含量对交联效率的影响。基于对双(芳基叠氮化物)光交联剂进行的研究,由pla
50
plu(50-200)/s-pla-fn3共混物以各种组成(90/10、75/25和50/50w/w)制备20μm厚度的膜,然后在紫外光下照射10分钟的时间(每侧5分钟)。结果汇总在图5中。
[0291]
如所预期的,混合物中s-pla-fn3的初始含量对交联效率有很大的影响,其中在s-pla-fn3比例以10重量%、25重量%、50重量%变化时凝胶分率分别为约15%、35%和55%。相反,pla
50
plu的分子量对交联效率没有显示出任何显著影响。对于限定重量比的pla
50
plu(50-200)/s-pla-fn3,无论pla
50
plu的分子量如何,凝胶分率都是相似的。在紫外交联温度下,与pla
50
plu
200
相比,pla
50
plu
50
的链迁移率更高,但这种更高的迁移率似乎不会显著影响交联效率。与pla
50
plu
100
或pla
50
plu
200
相比,仅在50/50的比例下,pla
50
plu
50
所获得的凝胶分率略低。这一结果可能是由于较低的链缠结加上较高的链迁移率,这部分地阻止了活性氮烯物种与聚合物链之间的反应。
[0292]
光交联的动力学
[0293]
然后在10分钟的时间内追踪光交联的动力学(图5)。紫外照射2分钟后,大多数pla
50
plu/s-pla-fn3共混物已达到最大凝胶分率,这证实了芳基叠氮化物光交联是一个非常快的过程,而不论pla
50
plu共聚物的分子量如何。
[0294]
ba(现有技术)和s-pla-fn3作为光交联剂的效率比较
[0295]
最后,针对于共混物中芳基叠氮基总浓度,比较了分子双(芳基叠氮化物)光交联
剂ba和大分子多(芳基叠氮化物)光交联剂s-pla-fn3的交联效率(图6)。
[0296]
应注意的是,pla
50
plu(50-200)-ba5混合物(5重量%的ba,11μmol)中的芳基叠氮基的浓度高于所有pla
50
plu/s-pla-fn3共混物,即使在使用最高浓度的s-pla-fn3(50重量%,8μmol)时亦是如此。然而,使用大分子8支链星形光交联剂获得的凝胶分率比ba高,即使对于最低含量的s-pla-fn3(10重量%,约2μmol)也比ba高,所述最低含量的s-pla-fn3对应于与5重量%的ba相比少5.5倍的光反应性部分。
[0297]
如所预期的,相比于双官能ba,在s-pla-fn3星形大分子光交联剂上存在8个芳基叠氮基的情况下,活性氮烯物种更有可能与pla
50
plu聚合物链接触并充当交联剂。此外,由于其大分子性质和预期的链缠结而使得交联剂的迁移率降低也可以解释这种提高的交联效率。
[0298]
2.3使用芳基叠氮化物星形s-pla-fn3作为光交联剂的微型支架
[0299]
基于在膜pla
50
plu(50-200)上获得的证实s-pla-fn3作为光交联剂的高潜力的结果,下一步是评估这种方法向电纺丝加工的可转移性,以制备基于光交联纤维的弹性体可降解支架。已经表明pla
50
plu共聚物的分子量不会影响结果,所述下一项研究限于被证明易于电纺的pla
50
plu200。如实验部分所述,制备相同比例的pla
50-plu200/s-pla-fn3(90/10、75/25和50/50)。所得支架的厚度接近250μm。为了保证外壳内低温(更多细节参见实验部分和图6)并保持纤维的形态,在紫外光(汞灯)和惰性气氛下以0.5hz的频率照射纤维支架2秒。已经研究了各种参数,并在以下部分中进行讨论。
[0300]
纤维形态
[0301]
通过sem分析了纤维形态,典型的图像示于图7中。对于限定的比例,注意到,即使在紫外固化后,基于s-pla或s-pla-fn3的纤维之间的纤维直径分布也没有差异。简而言之,所有纤维直径都在1至2μm的范围内。最低的纤维直径(1.2μm)由pla
50-plu200/s-pla-fn3 90/10共混物获得以及随着s-pla-fn3的含量增加,75/25共混物和50/50共混物的纤维直径分别为1.65至1.98μm和1.74至2.13μm。然而,后者的纤维分布更加不均匀。这可能是由于非全部溶剂蒸发导致扁平纤维,从而导致互连纤维。
[0302]
原位光交联评价
[0303]
为了确定获得弹性微纤维支架的最佳紫外固化时间,进行了交联研究。在紫外光(汞灯)和惰性气氛下以0.5hz的频率照射基于pla
50
plu200/s-pla-fn3的纤维支架2秒。紫外照射2分钟后,获得的凝胶分率最大(20至25%)(图5-d)。因此,将该照射时间选择用于其余的研究。纤维支架的凝胶分率值(20至25%)比20μm的膜(15至65%)低。这可能是由于高度多孔支架的约250μm的厚度和不透明性质会将紫外穿入限制在支架表面的几微米内。考虑到芳基叠氮化物的紫外阻隔性质加上s-pla-fn3聚合物结晶度,紫外光会使得纤维仅在表面(几微米)上光固化。因此,可注意到,无论s-pla-fn3浓度如何,纤维支架之间都没有显著差异。
[0304]
力学性质
[0305]
致力于软组织重建的合成可吸收材料领域的一个主要挑战是确保生物材料/宿主组织复合体在降解和愈合过程中保持力学性质。因此,在37℃的干燥和水合状态下评估pla
50-plu200/s-pla-fn3的力学性能(表1)。
[0306]
表1:在37℃的干燥和水合状态下的弹性微纤维支架(fs)的力学性质:杨氏模量
(e)、极限应力(σ
断裂
)、极限应变(ε
断裂
)和弹性极限(εy)。(数据表示为平均值
±
sd,对应于n=3的测量值)。
[0307][0308]
在37℃的干燥状态下,基于pla
50
plu200/s-pla(比例为75/25)的非紫外固化的纤维支架具有较低的变形性、高杨氏模量(e=29.3mpa)和低弹性极限(εy=1.3%)。相反,50/50比例是最可变形的材料(e=2.4mpa和εy7.3%)。观察到的范围内的纤维直径(约1至2μm)不影响力学性质。另一方面,在37℃的干燥状态下,基于pla
50
plu200/s-pla-fn3的紫外固化纤维支架显示出比非紫外固化的纤维支架(e=2.44至29.3mpa和εy=1.3至7.3%)高的弹性性质(e=0.22至0.68mpa和εy=12至182%)。由于纤维以拟线性应力-应变曲线交联,因此获得显著增加的弹性极限(图7)。它产生的支架具有较低的极限应力(交联fs的0.58至1.38mpa相对于非交联的1.01至2.01mpa)和高得多的极限应变(174至333%相对于120至146%)。如所预期的,fs pla
50
plu200/s-pla-fn3 75/25和50/50显示出比90/10(e=0.68mpa;εy=12%)高的弹性性质(e=0.22至0.34mpa;εy=115至182%),确认了使用星形大分子s-pla-fn3光交联剂的益处。应注意的是,对于类似的交联效率(图5-d),fs pla
50
plu200/s-pla-fn3 75/25达到了最高的弹性和极限应力。这可以通过有效交联加上长聚合物链与短聚合物链之间的良好平衡来解释。
[0309]
在37℃的水合状态下,纤维支架的杨氏模量和极限应力始终高于干燥状态,而弹性极限和极限应变低于干燥状态(图8)。考虑到众所周知的水的增塑作用,这可能看起来违背常理。然而,这些结果可以归因于最近在文献中报道的pla-b-peg-b-pla共聚物的微相分离现象。更详细地,peg嵌段(更柔韧、更低的转变温度)对共混物起增塑剂的初始作用,但peg链段在吸水时易于迁移,这使得微相分离和硬化。在我们的情况中,由于交联纤维的核壳结构(交联的壳,未交联的核,参加降解),这种现象可能弱化了水合状态下交联的影响。
[0310]
降解
[0311]
追踪支架降解1个月(图10-a)。如所预期的,与交联的对应物(剩余质量90%至95%)相比,非交联的纤维支架显示出更快的降解(剩余质量65%至85%)。只有pla
50-plu200(90/10)含量高的fs表现出相似的降解曲线,1个月内几乎没有降解(重量损失《2%)。还观察到星形共聚物(s-pla和s-pla-fn3)含量越高,重量损失越快。这是由于peg的
亲水链段有利于fs(150至300%)的吸水(图9),从而促进它们的水解降解。有趣的是,如针对化学交联弹性体所预期的,所有交联纤维的降解曲线都是拟线性的。交联纤维与非交联纤维之间的另一个区别是后者观察到的额外侵蚀。这种现象由图10b中呈现的sem图片来说明。尽管重量损失(1个月后10%),fs pla
50-plu200/s-pla-fn3 50/50在降解时却没有侵蚀,这在一定程度上证实了核壳结构。事实上,已知交联网络在降解过程中保持其3d形状,这在此处有观察到。虽然非交联或较少交联的核心链降解,但它们扩散通过交联壳会受到阻碍,这使得重量损失较慢。因此,电纺纤维支架的紫外固化使得可调节降解曲线,以及可有助于在软组织工程应用的框架中适合支架的性质。
[0312]
细胞相容性研究
[0313]
最后,在对纤维支架进行力学和降解研究之后,验证它们与细胞一起使用的潜力的最后一个强制性步骤是验证它们的细胞相容性。不同的共聚物pla、pluronicf127和peg已经获得fda的批准。然而,纤维内部残留的未反应的s-pla-fn3可能会从纤维中渗出。出于该原因,根据iso 10993-12建议对提取物评估了支架的细胞毒性。在接种到孔中的l929成纤维细胞上添加来自支架、c-和c+的提取物,并在24小时的时间内评估细胞毒性。
[0314]
只有来自阳性对照膜(c+)的提取物对l929细胞产生约45至50%的细胞毒性。结果(汇总在图11中)显示,即使在来自含有最高s-pla-fn3浓度(50/50)的支架的提取物的情况下,与l929细胞接触的提取物也不存在细胞毒性。因此,该初步试验证实了所提出的可降解弹性体生物材料在细胞接触应用中的潜力,其细胞相容性将在未来的专门工作中进一步研究。
[0315]
2.4s-pla-fn3作为光交联剂的多功能性
[0316]
为了突出所提策略的广泛适用性和多(芳基叠氮化物)s-pla-fn3作为交联剂的多功能性,具有高分子量的非官能聚合物选自包括聚酯(pla50)、聚醚(peo)和聚(甲基丙烯酸酯)(pmma)的各种族类。45%至70%范围内的凝胶分率(表2)证实,无论聚合物性质如何以及即使分子量高,都可以获得交联。
[0317]
表2:通过凝胶分率分析评估的聚合物性质对交联效率的影响(s-pla-fn3用作交联剂,20μm厚的膜,汞灯,每侧5分钟紫外照射)。(数据表示为平均值
±
sd,对应于n=3的测量值)。
[0318][0319]
2.5通过s-pla-ma光交联的可降解弹性体成形为膜
[0320]
根据上述方法,使用压制或通过溶剂蒸发将甲基丙烯酸酯官能化的星形共聚物s-pla-ma成形为膜。然后如第1.7点所述用紫外光照射膜。
[0321]
凝胶分率
[0322]
交联弹性体膜的根据方程式(4)计算的凝胶分率汇总在表3中(还参见图12和图13)。
[0323]
表3:交联弹性体膜的凝胶分率
[0324][0325]
降解
[0326]
通过溶剂蒸发制成的基于s-pla-50-mc(50000g/mol)的膜的降解示于图14。
[0327]
非官能嵌段共聚物s-pla的剩余质量在水解降解1个月后减少并达到80%(图14a)-虚线)。相反,就剩余重量和交联而言,s-pla-50-mc在1个月后没有发生降解(图14.a)(实线)-b))。因此,通过在聚合物基质内引入共价键来减慢降解过程。
[0328]
此外,s-pla-50-mc显示出部分吸水性(80至85%),其材料结构在水中得以保存(图14c))。
[0329]
2.6通过电纺丝加工的基于微纤维的组织
[0330]
2.6.1使用芳基叠氮化物星形s-pla-fn3(作为光交联剂)和pla
50-plu200的基于微纤维的组织
[0331]
紫外固化步骤
[0332]
与在加工后紫外固化相比,在加工中紫外固化使得纤维支架的凝胶分率增加,对于纤维支架pla
50-plu/s-pla-fn
3 75/25是从23%增加到52%,纤维支架pla
50-plu/s-pla-fn
3 50/50是从22%增加到77%(参见图15)。纤维支架在厚度上的紫外固化阻止了芳基叠氮反应性基团的紫外阻隔,使得在纤维支架内形成更高的共价键。
[0333]
力学性质
[0334]
从力学研究来看,只有基于比例为75/25和50/50的pla
50-plu/pegs
8-pla-fn3的纤维支架表现出类似橡胶的性能。因此,已经通过循环应力-应变曲线研究了这些弹性体纤维支架在不损失能量的情况下可逆变形的能力(参见图16)。
[0335]
两种纤维支架的光交联fs pla
50-plu200/pegs
8-pla-fn3均显示出在15%变形下相对于循环载荷的力学保持。
[0336]
2.6.2使用s-pla-mc 100的基于微纤维的组织
[0337]
纤维形态
[0338]
基于s-pla-mc(100 000g.mol-1)的纤维支架具有适用于组织工程应用的微米级纤维(2.8
±
0.3μm)(参见图17)。
[0339]
2.7通过立体光刻的3d材料
[0340]
使用s-pla-50-mc聚合物通过立体光刻加工获得不同的材料,并汇总于图18至图20中。根据我们的研究,我们能够制备具有各种多孔直径的材料(d=1mm
–
图18|d=4mm和d=7mm
–
图19)。如在图20中所示,可以使用多(甲基丙烯酸酯)嵌段共聚物s-pla-50-mc获得毫米级的3d材料。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1