激酶抑制剂的多晶型、含有该化合物的药物组合物、制备方法、及应用
1.当前揭示的主题属于医药晶体形式的领域,具体而言涉及激酶抑制剂的多晶型、含有此化合物的药物组合物、制备方法及其应用。
背景技术:
2.蛋白酪胺酸激酶(protein tyrosine kinase;ptk)为用以将磷酸基转移至蛋白质酪胺酸残基,由此活化酚类羟基的主要酶家族。ptk在细胞增殖及恶性病的过程中起重要作用。ptk包括受体ptk(受体酪胺酸激酶,rtk)及非受体ptk(参见zhang,z.y.,“functional studies of protein tyrosine phosphatases with chemical approaches f biochim biophys acta,2005,1754(1-2):100-107)。
3.rtk家族为具有内源性ptk活性的单通道受体。迄今为止,已鉴别属于20子族的超过50种rtk,主要包括肝细胞生长因子受体、表皮生长因子受体、血小板衍生的生长因子受体、胰岛素及类胰岛素生长因子-1受体、神经生长因子受体、纤维母细胞生长因子受体及血管内皮生长因子受体。其在调节与细胞增殖、凋亡、分化及代谢相关的信号转导路径中起重要作用。rtk家族成员由三个组分组成,包括结合于配位体的胞外部分、跨膜部分以及具有ptk活性及下游信号蛋白质结合位点的胞内部分。
4.mer酪胺酸激酶(mer tk)属于tam(axl/tyro-3/mer)rtk家族。人类的mer基因于1994年由graham发现(参见graham,d.k.等人,「cloning and mrna expression analysis of a novel human proto-oncogene,c-mer」,cell growth differ.,1994,5(6):647-657)。该基因位于2q14.1处,具有130,755kb的全长且含有19个外显子,包括编码的999个胺基酸,及约109,000的相对分子量。mer蛋白由胞外域、跨膜域及胞内激酶域组成。胞外域由两个免疫球蛋白域及两个纤维黏连蛋白域组成。免疫球蛋白域为编码有生长遏制特定基因6(gas6)的蛋白质产物的组合域,而纤维黏连蛋白域在mer及gas 6的组合中起调节作用。受体ptk子族的胞内激酶域独特kwiaies序列具有三个视情况存在的酪胺酸残基(y-749、y-753及y-754),其可作为自体磷酸化位点调节mer且与下游信号分子(磷脂酰肌醇-3-激酶(pi3k)/蛋白激酶b(pkb)及促分裂原活化蛋白激酶(mapk))相互作用。mer激酶的活化需要完全磷酸化三个酪胺酸残基(参见ling,l.等人,「identification of the major autophosphorylation sites of nyk/mer,an ncam-related receptor tyrosine kinase」,biol.chem.,1996,271(31):18355-18362)。
5.mer不能出现在正常t及b淋巴球及粒细胞中,但已报导mer在诸如jurkat、peer、k562及raji的一些恶性血液学疾病中的异常表现(参见graham,d.k.等人,「cloning and mrna expression analysis of a novel human proto-oncogene,c-mer」,cell growth differ.,1994,5(6):647-657)。此指示mer与一些恶性血液疾病相关。mer tk亦增加淋巴母细胞的药物耐受性。已在关于人类急性淋巴母细胞性白血病(acute lymphoblastic leukemia;all)细胞株的研究发现mer tk的表现亦可增加药物在治疗all时的药物耐受性
(参见linger,r.m.等人,「mer receptor tyrosine kinase is a novel therapeutic target in pediatric b-cell acute lymphoblastic leukemia」,blood,2009,114(13):2678-2687)。活体内实验进一步证明mer tk的抑制可降低白血病细胞的存活率且与其他化学疗法具有协同效应(参见lee-sherick,a.b.等人,「targeting pediatric acute lymphoblastic leukemia:novel therapies currently in development」,bn j.haematol.,2010,151(4):295-311;nguyen,k.等人,「factors influencing survival after relapse from acute lymphoblastic leukemia:a children's oncology group study」,leukemia,2008,22(12):2142-2150;scott,r.s.等人,「phagocytosis and clearance of apopotic cells is mediated by mer」,nature,2001,411(6834):207-211)。因此,mer酪胺酸激酶抑制剂(mer tki)将为治疗all的有效药物。
6.fms样酪胺酸激酶(flt3)属于iii型受体ptk家族。flt3的基因位于染色体13q12中,具有约l00kb的全长,含有24个外显子,包括编码的993个胺基酸。相对分子量为130,000,其中其呈非糖基化形式的相对分子量为160,000。flt3由包括五个免疫球蛋白样域的n端胞外域、一个跨膜域及一个胞内激酶域(包括近膜域及激酶催化域)组成。胞内激酶域的激酶催化域由n端、含有活化环的c端及含有atp的结合位点的可挠性激酶插入域组成(参见liao,j.j.,「molecular recognition of protein kinase binding pockets for design of potent and selective kinase inhibitor」,j.med.chem.,2007,50(3):409-424)。
7.flt3突变为flt3的异常活化的主要原因。存在三种主要突变体形式:近膜域的内部串联重复(internal tandem duplication;itd)、tkd突变及近膜域点突变。flt3的异常活化可引起all、急性骨髓白血病(acute myeloid leukemia;aml)及慢性骨髓性白血病(chronic myelogenous leukemia;cml)(参见hatzimichael,e.等人,「profile of pacritinib and its potential in the treatment of hematologic disorder」,j.blood med.,2014,5:143-152)。研究已显示,超过70%的aml患者及all患者具有较高的flt3表现(参见smith,c.c.等人,「validation of itd mutations in flt3 as a therapeutic target in human an acute myeloid leukaemia」,nature,2012,485(7397):260-263)。因此,flt3可用于治疗作为靶标的白血病。
8.目前,已报导多种mer ptk抑制剂及flt3 ptk抑制剂。举例而言,由wang等人设计及研发的吡咯并嘧啶化合物mrx-2843为mer ptk及flt3 ptk的双重高效抑制剂,外加展示抑制其他家族成员,诸如tam ptk及其他相关ptk的axl/tyro-3。mrx-2843的结构已揭示于wo2013052417a1及cn103958510a中,且描绘于下文:
[0009][0010]
特定有机药物化合物的多晶型由于其独特的三维结构而具有不同的物理特性,诸
如溶解度、吸湿性及稳定性。然而,通常不可能预测特定有机药物化合物是否将形成不同的晶体形式,且不大可能预测晶体形式本身的结构及特性。探究医药学上可接受的化合物的新颖晶体或多晶型形式提供一种改良医药产品的总体效能同时扩增调配物科学家可获得的多种材料的机会。有利的系藉由发现适用化合物的新颖晶体形式来扩增多种调配物。
[0011]
发明概述
[0012]
本发明所揭示的发明涉及式i化合物:(反式)-4-((2-环丙基乙基)氨基)-5-(4-((4-甲基哌嗪-l-基)甲基)苯-7h-吡咯并[2,3-d]嘧啶-7-基)环己醇三盐酸盐。
[0013][0014]
本发明还涉及式i化合物、其水合物及/或其溶剂合物的多种实质上纯的晶体形式。
[0015]
在本发明中,式i化合物的化合物、水合物及/或溶剂合物以一或多种晶体形式存在。
[0016]
在一具体实例中,提供式i化合物、其水合物及/或其溶剂合物的晶体形式。在一具体实例中,x射线粉末衍射图具有衍射角2θ,其在以下处具有特征峰:5.7
±
0.2
°
、17.7
±
0.2
°
、19.7
±
0.2
°
、22.7
±
0.2
°
及23.2
±
0.2
°
。为方便起见,特征在于此xrpd图的晶体形式称为晶体形式1。
[0017]
在另一具体实例中,晶体形式1的x射线粉末衍射图具有衍射角2θ,其在以下处具有特征峰:5.7
±
0.2
°
、11.3
±
0.2
°
、17.1
±
0.2
°
、17.7
±
0.2
°
、19.7
±
0.2
°
、20.2
±
0.2
°
、22.7
±
0.2
°
及23.2
±
0.2
°
。
[0018]
在另一具体实例中,晶体形式1的x射线粉末衍射图具有衍射角2θ,其在以下处具有特征峰:5.7
±
0.2
°
、11.3
±
0.2
°
、12.3
±
0.2
°
、12.7
±
0.2
°
、17.1
±
0.2
°
、17.7
±
0.2
°
、19.7
±
0.2
°
、20.2
±
0.2
°
、21.9
±
0.2
°
、22.7
±
0.2
°
、23.2
±
0.2
°
及25.2
±
0.2
°
。
[0019]
在另一具体实例中,以上晶体形式1具有如图1中所展示的x射线粉末衍射图。
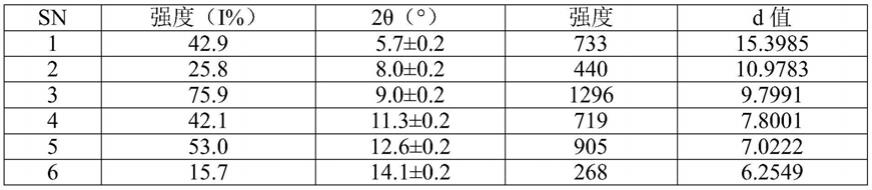
[0020]
当峰的相对强度大于10%时,晶体形式1的x射线粉末衍射光谱的特征峰展示于表1中。
[0021]
表1
[0022]
sn强度(i%)2θ(
°
)强度d值1100.05.7
±
0.2125515.5619213.59.5
±
0.21709.2757319.79.7
±
0.22479.0674
415.210.5
±
0.21918.4324529.511.3
±
0.23717.8445628.212.3
±
0.23547.1797723.812.7
±
0.22986.9681811.413.7
±
0.21446.4759913.814.1
±
0.21736.29821014.614.6
±
0.21836.07851111.915.8
±
0.21495.61191213.216.3
±
0.21655.42941330.617.1
±
0.23845.17001448.317.7
±
0.26074.99781548.919.7
±
0.26134.50191629.320.2
±
0.23674.40261711.020.8
±
0.21384.27481826.221.9
±
0.23294.06171941.222.7
±
0.25183.91442038.823.2
±
0.24873.82942116.224.0
±
0.22033.69902221.025.2
±
0.22633.52852314.625.9
±
0.21833.44182415.827.6
±
0.21993.22882510.628.1
±
0.21333.17212614.529.3
±
0.21823.0424
[0023]
在一具体实例中,此晶体形式的纯度≥85%。在另一具体实例中,此晶体形式的纯度≥95%。在另一具体实例中,此晶体形式的纯度≥99%。在另一具体实例中,此晶体形式的纯度≥99.5%。
[0024]
此外,晶体形式1具有如图3中所展示的差示扫描热量测定-热解重量分析(dsc-tga)图。
[0025]
在一替代具体实例中,本文所描述的发明进一步包含呈其水合物及/或其溶剂合物的另一晶体形式的式i化合物。在此具体实例中,晶体形式的x射线粉末衍射光谱具有衍射角2θ,其在以下处具有特征峰:9.0
±
0.2
°
、16.6
±
0.2
°
、24.2
±
0.2
°
、24.6
±
0.2
°
及24.8
±
0.2
°
。为方便起见,本发明称为晶体形式2。
[0026]
在一具体实例中,晶体形式2的x射线粉末衍射光谱具有衍射角2θ,其在下处具有特征峰:9.0
±
0.2
°
、12.6
±
0.2
°
、16.6
±
0.2
°
、16.9
±
0.2
°
、19.0
±
0.2
°
、24.2
±
0.2
°
、24.6
±
0.2
°
及24.8
±
0.2
°
。
[0027]
在另一具体实例中,晶体形式2的x射线粉末衍射光谱具有衍射角2θ,其在以下处具有特征峰:5.7
±
0.2
°
、9.0
±
0.2
°
、11.3
±
0.2
°
、12.6
±
0.2
°
、16.6
±
0.2
°
、16.9
±
0.2
°
、19.0
±
0.2
°
、20.0
±
0.2
°
、22.7
±
0.2
°
、24.2
±
0.2
°
、24.6
±
0.2
°
及24.8
±
0.2
°
。
[0028]
在另一具体实例中,晶体形式2具有如图2中所展示的x射线粉末衍射光谱。
[0029]
当峰的相对强度大于10%时,晶体形式2的x射线粉末衍射光谱的特征峰展示于表2中。
[0030]
表2
[0031][0032][0033]
以上晶体形式可仅使用主峰的概述特征化。此等主峰在
±
0.2的误差边际内可再现。
[0034]
如本文中所使用,术语“具有如图1中所展示的x射线粉末衍射图」或「具有如图2中所展示的x射线粉末衍射图”意谓x射线粉末衍射图的主峰展示于图1或图2中,其中与图1或图2中的最高峰相比(相对强度指定为100%),主峰意谓相对强度值大于10%且较佳地大于30%的彼等峰。
[0035]
在一具体实例中,晶体形式2的纯度≥85%。在另一具体实例中,晶体形式2的纯度≥95%。在另一具体实例中,晶体形式2的纯度≥99%。在另一具体实例中,晶体形式2的纯度≥99.5%。
[0036]
此外,晶体形式2具有如图4中所展示的差示热量-热解重量分析(dsc-tga)图。
[0037]
在一具体实例中,本发明亦提供一种药物组合物,其包含治疗有效量的如上文所描述的晶体形式1及/或晶体形式2。
[0038]
本发明亦提供如上文所描述的药物组合物的具体实例。
[0039]
在一具体实例中,所描述的药物组合物含有治疗有效量的晶体形式1或晶体形式
2,以及药学上可接受的赋形剂、佐剂或载体。
[0040]
在另一具体实例中,所描述的药物组合物含有治疗有效量的由本发明提供的晶体形式1及晶体形式2,以及药学上可接受的赋形剂、佐剂或载体。
[0041]
在另一具体实例中,药物组合物含有治疗有效量的晶体形式1或晶体形式2及至少一种其他治疗剂。
[0042]
在另一具体实例中,药物组合物含有治疗有效量的晶体形式1及晶体形式2及至少一种其他治疗剂。
[0043]
在一具体实例中,药物组合物为口服制剂。
[0044]
在一具体实例中,药物组合物为锭剂或胶囊。
[0045]
在一具体实例中,药物组合物包含5至150mg晶体形式1及/或晶体形式2,其与至少一种赋形剂、佐剂及/或载体一起调配,总量为约50mg至800mg。
[0046]
在一具体实例中,药物组合物中的赋形剂、佐剂及/或载体为微晶纤维素、预胶凝化淀粉、纤维素乳糖、交联聚维酮、微米化硅胶、硬脂酸镁及/或羟丙基纤维素。
[0047]
在一具体实例中,药物组合物含有0.01重量%至99重量%的晶体形式1或晶体形式2。在其他具体实例中,药物组合物含有0.01重量%至70重量%、1重量%至70重量%、1重量%至50重量%、1重量%至30重量%、或10重量%至30重量%的晶体形式1或晶体形式2。
[0048]
在一具体实例中,所描述的药物组合物含有0.01重量%至99重量%的本发明的晶体形式1及晶体形式2。在其他具体实例中,药物组合物含有0.01重量%至70重量%、1重量%至70重量%、1重量%至50重量%、1重量%至30重量%、或10重量%至30重量%的晶体形式1及晶体形式2。
[0049]
本发明亦提供所描述的晶体形式1及/或晶体形式2的用途,其用于生产用于治疗患者的疾病、症状或病况的医药品,其中疾病、症状或病况系由mer及/或flt3介导。
[0050]
本发明亦提供晶体形式1及/或晶体形式2在治疗疾病、症状或病况中的用途的具体实例。
[0051]
在一具体实例中,所描述的疾病、症状或病况为癌症及/或增殖性疾病。
[0052]
在一具体实例中,所描述的疾病、症状或病况为骨髓白血病、淋巴母细胞白血病、黑素瘤、乳癌、肺癌、结肠癌、肝癌、胃癌、肾癌、卵巢癌、子宫癌及/或脑癌。在另一具体实例中,所描述的疾病、症状或病况为骨髓白血病及/或淋巴母细胞白血病。
[0053]
在一具体实例中,本发明亦提供藉由向患者投予如本文所提供的具有晶体形式1及/或晶体形式2的式i化合物来治疗患者的疾病、症状或病况的方法。
[0054]
本发明进一步提供上文所提及的藉由使用所描述的晶体形式1及/或晶体形式2来治疗患者的疾病、症状或病况的方法的具体实例。
[0055]
在一具体实例中,疾病、症状或病况系由mer及/或flt3介导。
[0056]
在一具体实例中,所描述的疾病、症状或病况为癌症及/或增殖性疾病。
[0057]
在一具体实例中,所描述的疾病、症状或病况为骨髓白血病、淋巴母细胞白血病、黑素瘤、乳癌、肺癌、结肠癌、肝癌、胃癌、肾癌、卵巢癌、子宫癌及/或脑癌。在另一具体实例中,所描述的疾病、症状或病况为骨髓白血病及/或淋巴母细胞白血病。
[0058]
本发明进一步提供一种用于制备式i化合物的方法:
[0059][0060]
在一具体实例中,用于制备式i化合物的方法包括以下步骤:
[0061]
a)使化合物m-1与化合物m-2接触以得到化合物m-3;
[0062]
b)使化合物m-3与化合物m-4在存在钯催化剂的情况下接触以得到化合物m-5;及
[0063]
c)执行结晶方法以获得该式i化合物。
[0064]
本发明进一步提供制备式i化合物的中间产物。此等中间产物包括下文所描绘的m-3:
[0065][0066]
本发明进一步提供用于制备化合物、其水合物及/或其溶剂合物的晶体形式1及晶体形式2的方法。
[0067]
在一个具体实例中,晶体形式1的制备方法系如下:
[0068]
将m-5、hoac、活性炭及si-硫醇添加至反应瓶中,在室温下搅拌14小时(除钯以外)且随后用50g硅藻土过滤。将过滤物转移至反应瓶中以冷却,且在0℃
±
5℃下添加3n hcl cpme溶液。首先,添加3n hcl cpme的总体积的1/2,且将剩余1/2滴入混合物中直至存在固体沈淀。在完成添加的后,将mtbe添加至混合物中。将整个滴入过程的温度控制处于0℃
±
5℃。在逐滴添加mtbe的后,使用热绝缘以将温度维持处于0℃
±
5℃,搅拌混合物1小时且结晶。过滤所得固体,用mtbe冲洗滤饼,且将混合物在45℃下真空干燥。随后在室温下添加500ml乙醇,将混合物在45℃下过滤且干燥以获得晶体形式1。
[0069]
在一替代性方法中,将m-5及7l乙酸添加至反应釜中,于其中搅拌且溶解混合物。
在溶解的后,添加si-硫醇及活性炭。将混合物搅拌16小时,同时将整个过程的温度控制处于15至20℃。向此混合物中添加500g硅藻土,过滤混合物,且使用2.1l乙酸来冲洗滤饼。将过滤物转移至反应釜中且在15至20℃的温度下逐滴添加3n hcl cpme溶液。在完成添加的后,添加甲基第三丁基醚且搅拌混合物1小时。随后过滤结晶产物。用1l甲基第三丁基醚冲洗滤饼且藉由20℃至45℃的梯度加热将滤饼在真空下干燥15小时。将所获得的样品压碎且添加至反应釜中。在室温下添加15l乙酸乙酯,随后搅拌、拍打且过滤混合物。将滤饼在45℃下真空干燥。将以上样品添加至反应釜中,添加15l乙酸乙酯且在室温下搅拌,同时进行两次拍打及过滤。将滤饼在45℃下在真空下干燥3小时,随后加热至80℃且在真空下干燥15.5小时,以获得作为晶体形式1的黄色固体。
[0070]
在一具体实例中,晶体形式1的制备包含以下步骤:
[0071]
a)在反应瓶中混合m-5、hoac、活性炭及si-硫醇;
[0072]
b)在室温下掺合混合物;
[0073]
c)过滤步骤b的混合物;
[0074]
d)在0℃
±
5℃的温度下将3n hcl cpme添加至步骤c的过滤物中;及
[0075]
e)过滤步骤d的混合物以获得晶体形式1。
[0076]
在另一具体实例中,晶体形式1的制备包含以下步骤:
[0077]
a)将m-5及乙酸混合至反应容器中直至溶解;
[0078]
b)添加si-硫醇及活性炭;
[0079]
c)搅拌所得混合物持续一段时间,同时将温度控制处于15至20℃;
[0080]
d)过滤步骤c的混合物;
[0081]
e)将3n hcl cpme添加至步骤d的过滤物中;
[0082]
f)将甲基第三丁基醚添加至步骤e的混合物中;
[0083]
g)过滤步骤f的混合物以获得固体;
[0084]
h)将乙酸乙酯添加至步骤g中所获得的固体中;及
[0085]
i)过滤所得混合物以获得晶体形式1。
[0086]
晶体形式2可如下制备:
[0087]
在室温下用500ml乙醇拍打晶体形式1(约80g)持续2.5小时,过滤,且将滤饼在45℃下干燥,以获得75.01g固体样品。使用此样品以制造1500ml的7.5%水溶液。将混合物保持处于50℃,搅拌且拍打16.5小时。将混合物冷却至室温以用于过滤,且将滤饼在45℃下干燥以获得晶体形式2。
[0088]
在一替代性方法中,具有玻璃底部的80l夹套反应器配备有两个单信道图表记录器及一热控单元。将氮气施加至反应器持续15min。在干净玻璃瓶中制备7.5%v/v乙醇水溶液。输送管配备有串联过滤器(10微米)且将式i化合物添加于具有7.5%v/v乙醇水溶液(12.5l)的80l反应器中。将温度调节至50
±
5℃且将反应器内含物搅拌17小时。历经140min将批料温度升高至20
±
5℃且随后在20
±
5℃下搅拌1h。将批料材料过滤且用预过滤乙醇水溶液(2.1l)洗涤。将滤饼保持在氮气中持续1h且随后取样以用于hplc分析。历经约87小时将滤饼在45
±
5℃下在真空中干燥至恒重,且随后对残余etoh进行取样。将批料材料再干燥24小时以获得1967g晶体形式2。
[0089]
在一具体实例中,用于制备晶体形式2的方法包括以下步骤:
type)”或“晶体结构(crystal structure)”,而且“晶体形式”更应理解为“具有特定晶体结构的物质”或“具有特定晶体类型的晶体”。
[0114]
如本文中所使用,“晶体形式”可特征在于其代表性x射线衍射图。本领域中的彼等技术员可理解实验误差取决于仪器的条件、样品的制备及样品的纯度。在本领域中应了解,x射线衍射图通常随仪器的条件而改变。此外,峰角度的实验误差通常为5%或更小,且亦应考虑此等角度的误差,从而一般允许
±
0.2
°
的误差。由于诸如样品的高度的实验因素的影响,会发生峰角度的总体偏移,且通常允许一定偏移。因此,具有相同或类似特征峰的晶体形式在本发明的范围内。
[0115]
如本文所使用,术语“室温”系指习知室温,通常为10至30℃。
[0116]
如本文所使用,术语“药学上可接受”意谓其以不会有害地影响待投予的个体的形式或量存在。
[0117]
如本文所使用,术语“治疗有效量”系指一种化合物足以治疗疾病、病痛或疾病的症状的量。“治疗有效量”可随化合物,疾病,病痛,及/或疾病或病痛的症状,疾病、病况及/或疾病或病痛的症状严重度,待治疗患者的年龄,及/或待治疗患者的体重而改变。在任何特定病况中,适合量对于本领域中的彼等技术员而言可显而易见或可藉由常规实验来确定。在组合疗法的情况下,“治疗有效量”意谓有效治疗个体的疾病、病痛或症状的总量。
[0118]
本发明中的药物组合物的所有剂型可藉由医药领域中的习知方法来制备。举例而言,活性成分可与一或多种赋形剂混合且随后制备成所需剂型。
[0119]
如本文所使用,术语“药学上可接受的载体”意谓适用于所需医药制剂的熟知医药载体,例如稀释剂及赋形剂,诸如水、各种有机溶剂等;填充剂,诸如淀粉、预胶凝化淀粉、蔗糖、甘露糖醇、乳糖、喷雾干燥乳糖、微晶纤维素、硅化微晶纤维素及纤维素乳糖;黏合剂,诸如纤维素衍生物、海藻酸盐、明胶、羟丙基纤维素及聚乙烯吡咯啶酮(pvp);湿润剂,诸如甘油;崩解剂,诸如琼脂、碳酸钙、交联聚维酮、交联羧甲纤维素钠、羧甲基淀粉钠及碳酸氢钠;吸收增强剂,诸如四级铵化合物;界面活性剂,诸如鲸蜡醇、月桂基硫酸钠及吐温(tween);吸收载体,诸如高岭土及膨润土;润滑剂,诸如滑石、硬脂酸、硬脂酸钙、硬脂酸镁、硬脂酰反丁烯二酸钠、微型粉末硅胶及聚乙二醇。亦可将其他医药学上可接受的赋形剂添加至药物组合物,诸如分散剂、稳定剂、增稠剂、错合剂、缓冲液、渗透增强剂、聚合物、芳香剂、甜味剂或染料。较佳使用适用于所需剂型及所需投药模式的赋形剂。
[0120]
如本文所使用,术语“疾病”、“病痛”或“症状”系指任何疾病、不适、病痛、症状或适应症。
[0121]
如本文所使用,术语“额外治疗剂”系指在待治疗的疾病、病痛或症状中引发治疗反应的药剂。额外治疗剂的非限制性实例包括化学治疗剂、抗炎剂、免疫调节剂、亲神经因子、用于治疗心血管疾病的药剂、用于治疗肝病的药剂、抗病毒剂、用于治疗血液病症的药剂、用于治疗糖尿病的药剂及用于治疗免疫缺乏病症的药剂。
实施例
[0122]
以下具体实例进一步说明本发明,该等实施例不意欲限制本发明的范围。除非另外说明,否则在本发明的特定具体实例中,技术或方法为本领域中的常规技术或方法。
[0123]
除非另外说明,否则在以下具体实例及效应具体实例中,关于待使用的测试仪器
及测试方法参数的信息系如下:
[0124]
(1)x射线粉末衍射仪(xrd)、bruker d8高级衍射仪;技术规范:ka辐射(40kv,40ma),铜目标波长为1.54a,θ-2θ测角器、mo单色器、lynxeye探针;标准材料:al2o3;获取软件:diffrac plus xrd commander;分析软件:mdi jade 6;方法参数:测试角度,3-40
°
2θ/3-30
°
2θ(热量表xrd);步长,0.02
°
2θ;速度,0.15s/步骤;测试样品量》2mg。
[0125]
(2)同步热分析仪(dsc-tga)、sta 449f3;样品托盘:铝坩埚;测试样品剂量:0.5-5mg;屏蔽气体:氮气;气体流速:20ml/min;吹扫气体:60ml/min;测试方法:加热速率10℃/min,在30℃下平衡且随后升温直至400℃。对于细节,参考gb/t 19267.122008,第12章,法医技术的痕量证据的物理及化学检查:热分析(physical and chemical examination of trace evidence of forensic technology:thermal analysis)。
[0126]
(3)核磁共振光谱仪(nmr),agilent dd2 600mhz;测试类型:质子nmr光谱及碳光谱;测试方法:根据jy/t 007-1996用于核磁共振光谱的超导脉冲傅立叶变换(fourier transformation)的一般原理。
[0127]
缩写:
[0128]
api:活性医药成分;
[0129]
13
c-nmr:
13
c核磁共振;
[0130]
cpme:环戊基甲醚;
[0131]
dipea:n,n-二异丙基乙胺;
[0132]
dsc-tga:差示热量-热解重量分析;
[0133]
etoh:乙醇;
[0134]
h或hours:小时;
[0135]1h-nmr:1h核磁共振;
[0136]
hoac:乙酸;
[0137]
hplc:高效液相层析;
[0138]
ipa:异丙醇:
[0139]
lc-ms:液相层析-质谱法;
[0140]
min或mins:分钟;
[0141]
mtbe:甲基第三丁基醚;
[0142]
pd(ph3p)4:四(三苯基膦)钯;
[0143]
po:口服(peros);
[0144]
rh:相对湿度;
[0145]
xrd:x射线粉末衍射。
[0146]
实施案例1:式i化合物的合成。
[0147][0148]
化合物m-3的合成:
[0149]
具有玻璃底部的80l夹套反应器配备有两个单信道图表记录器及一个热控单元。将氮气施加至反应器持续75min,且随后将异丙醇、m-1、m-2及二异丙基乙胺添加于反应器中。将批料温度调节至97
±
5℃且将反应器内含物搅拌85小时。将批料材料进行取样以用于hplc分析。将批料温度调节至20
±
5℃且将批料材料搅拌隔夜。历经约30min将纯水添加于反应器中。当批料温度处于20
±
5℃时,将甲基第三丁基醚添加至反应器中且将内含物搅拌25min。分离各层且用mtbe萃取水相。将有机层用纯水洗涤且随后在真空下蒸馏至5.5l体积,同时保持批料温度《40℃。在将夹套温度调节至20
±
5℃的后,将乙腈添加至反应器中。将批料温度调节至0
±
5℃且将反应器内含物搅拌隔夜。将过滤物用作辅助过滤批料且用乙腈洗涤滤饼。将滤饼保持在氮气下持续4小时且随后取样以用于hplc分析。将滤饼在40
±
5℃下在真空烘箱中干燥直至恒重持续约21小时。
[0150]
lc-ms[m+h
+
]:379.1。
[0151]
化合物m-5(mrx-2843自由碱)的合成:
[0152]
具有玻璃底部的80l夹套反应器配备有两个单信道图表记录器及一个热控单元。将反应器保持在氮气流下隔夜。将m-3、m-4及碳酸钾添加至具有乙醇及水的混合物的反应器中且将氮气鼓泡至混合物中1小时。
[0153]
添加四(三苯基膦)钯,且将批料温度调节至40℃。将反应器内含物在40
±
5℃下搅拌2小时。将批料温度升高至58
±
5℃且将混合物搅拌18小时。随后采用批料取样以用于hplc分析且未侦测到m-3。将批料材料在真空下蒸馏至10l体积,同时保持批料温度≤60℃。将批料温度调节至58
±
5℃且保持处于此温度,同时将乙酸乙酯添加至反应器中。将混合物搅拌29min。分离各层且在58
±
5℃下用纯水洗涤有机层。将批料温度调节至20
±
5℃,将m-5晶种添加至反应器中且将内含物搅拌隔夜。将批料材料在真空下蒸馏至8l的体积,同时保
持夹套温度≤40℃。在将夹套温度调节至20
±
5℃的后,将乙腈添加至反应器中。将混合物冷却至10
±
5℃且搅拌约1.5小时。将过滤物用作辅助过滤批料且用乙酸乙酯洗涤滤饼。将滤饼保持在氮气下1h且在30
±
5℃下在真空烘箱中干燥至恒重。
[0154]
lc-ms[m+h
+
]:489.3。
[0155]
式i化合物的合成:
[0156]
具有玻璃底部的22l夹套反应器配备有两个单信道图表记录器及一个热控单元。将氮气施加至反应器中持续1h。将m-5及冰乙酸添加于反应器中并搅拌直至发现溶解。将批料温度调节至25
±
5℃。将活性炭及si-硫醇添加至反应器中且将内含物在25
±
5℃下搅拌19小时。使混合物穿过过滤垫且使任何松散的在冰乙酸及串联过滤器中成浆。将滤饼用冰乙酸洗涤且经由串联过滤器将过滤物转移至配备有两个单信道图表记录器及一个热控单元的具有玻璃底部的80公升夹套反应器中。将批料温度调节至5
±
5℃且保持处于此温度,同时将3n hcl添加于cpme中。将批料温度调节至0
±
5℃,且在此温度下历经40min经由串联过滤器添加mtbe。将反应器内含物在0
±
5℃下搅拌1h,过滤且用预过滤mtbe洗涤。将滤饼在氮气下干燥14.5小时且在45
±
5℃下在真空烘箱中干燥至恒重。此产生27,279g粗产物。将粗产物及乙醇添加至80l反应器中且将内含物在20
±
5℃下搅拌18.5小时。将批料材料过滤,且用预过滤乙醇洗涤滤饼。将滤饼保持在氮气下持续3.5小时且在45
±
5℃下在真空下干燥至恒重。1h nmr及
13
c nmr图与图5及图6中所展示的彼等一致。
[0157]
lc-ms[m+h
+
]:597.3。
[0158]
具体实例2:晶体形式1的制备方法
[0159]
方法1
[0160]
将m-5、hoac、活性炭及si-硫醇添加至反应瓶中,在室温下搅拌14小时(除钯以外)且随后用50g硅藻土过滤。将过滤物转移至反应瓶中以冷却,且在0℃
±
5℃下添加3n hcl cpme溶液。首先,添加总体积的1/2,持续添加剩余1/2,直至存在固体沈淀。在完成添加的后,将mtbe添加至系统中且将整个滴入过程温度控制处于0℃
±
5℃。在添加mtbe的后,在0℃
±
5℃下采用热绝缘。将混合物搅拌且伴随过滤结晶1小时并用mtbe冲洗滤饼。在45℃下真空干燥滤饼的后,在室温下拍打500ml乙醇,将混合物过滤,且在45℃下干燥滤饼,以获得晶体形式1。1h nmr及
13
c nmr图与图5及图6中所展示的彼等一致。
[0161]
方法2
[0162]
将m-5及7l乙酸添加至反应釜中以搅拌且溶解。在溶解的后,添加si-硫醇及活性炭并搅拌混合物16小时,同时将整个过程的温度控制处于15至20℃。添加500g硅藻土作为助滤剂,将2.1l乙酸用于冲洗滤饼,且将过滤物转移至反应釜中。随后在15至20℃的温度下添加3n hcl cpme溶液。在完成添加的后,添加甲基第三丁基醚并搅拌混合物,结晶l h并过滤。用1l甲基第三丁基醚冲洗滤饼且藉由20至45℃的梯度加热真空干燥滤饼15小时。将所获得的样品压碎且添加至反应釜中。在室温下添加15l乙酸乙酯。将混合物搅拌、拍打并过滤,且随后在45℃下真空干燥滤饼。将以上样品添加至反应釜中,在室温下添加15l乙酸乙酯并搅拌,同时进行两次拍打及过滤。将滤饼在45℃下真空干燥3小时,随后加热达至80℃且真空干燥15.5小时,以获得黄色固体,亦即晶体形式1。1h nmr及
13
c nmr图与图5及图6中所展示的彼等一致。
[0163]
具体实例3:晶体形式2的制备方法
[0164]
方法1
[0165]
在室温下用500ml乙醇拍打晶体形式1(约80g)持续2.5小时,过滤,且将滤饼在45℃下干燥,以获得75.01g固体样品。将所得物样品添加于1500ml的7.5%水溶液中,保持处于50℃,搅拌且拍打16.5小时。将混合物冷却至室温以用于过滤且在45℃下干燥滤饼,以获得晶体形式2。1h nmr及
13
c nmr图与图5及图6中所展示的彼等一致。
[0166]
方法2
[0167]
具有玻璃底部的80l夹套反应器配备有两个单信道图表记录器及一个热控单元。将氮气施加至反应器持续15min。在干净玻璃瓶中制备7.5%v/v乙醇水溶液。输送管配备有串联过滤器(10微米)且将式i化合物添加于具有7.5%v/v乙醇水溶液(12.5l)的80l反应器中。将批料温度调节至50
±
5℃且将反应器内含物搅拌17小时。历经140min将批料温度升高至20
±
5℃且随后在20
±
5℃下搅拌1小时。批料材料过滤且用预过滤乙醇水溶液(2.1l)洗涤。将滤饼保持在氮气下持续1h且随后取样以用于hplc分析。将滤饼在45
±
5℃下在真空中干燥至恒重持续87小时,且随后对残余etoh进行取样。将批料材料再干燥24小时以获得1967g晶体形式2。1h nmr及
13
c nmr图与图5及图6中所展示的彼等一致。
[0168]
具体实例4:晶体形式稳定性的测定
[0169]
将晶体形式1储存于25℃/60%rh及40℃/75%rh下持续1、3及6个月。在此等条件下,晶体形式保持不变。
[0170]
将晶体形式2储存于5℃下持续18及24个月,且晶体形式保持不变。将其储存于25℃/60%rh下持续1、3、6、9、12、18及24个月,且晶体形式保持不变。将其储存于40℃/75%rh下1、3及6个月,晶体形式不变。具体实例5:饱和溶解度的测定
[0171]
将适量的化合物样品在ph为1-8的水中过饱和,且在37℃下用振荡器振荡4小时。化合物经取样、离心,随后上清液经稀释且藉由hplc测试。将计算值与外部标准进行比较。
[0172]
式i化合物的晶体形式1及晶体形式2的化合物及mrx-2843自由碱在37℃下在不同ph值下的饱和溶解度展示于表3中。
[0173]
表3
[0174][0175]
具体实例实施例6.胶囊调配物及制备
[0176]
胶囊调配物展示于表4中。
[0177]
表4
[0178][0179]
a:根据理论盐度调节晶体形式1的活性医药成分(api)的单位含量。实际剂量系基于api含量。
[0180]
胶囊的制造制程系如下:
[0181]
1)api压碎/筛选
[0182]
将原料药压碎且用20目筛网筛分一次。
[0183]
2)混合
[0184]
将微晶纤维素ph-102、预胶凝化淀粉1500及经筛分原料药添加于v型混合器中持续20min。
[0185]
3)40mg大小的胶囊填充
[0186]
获取30%混合物且用其填充4号白色圆锥形明胶胶囊。
[0187]
4)100mg大小的胶囊填充
[0188]
获取30%混合物且用其填充1号白色圆锥形明胶胶囊。
[0189]
5)10mg大小的胶囊填充
[0190]
获取50%比例的微晶纤维素ph102的混合物及10%比例的预胶凝化淀粉1500的混合物及剩余材料以混合20min。用其填充第5个白色圆锥形明胶胶囊。
[0191]
6)最终封装
[0192]
将胶囊填充至100ml高密度聚乙烯瓶中,且添加干燥剂,且使用箔片型感应密封垫圈及儿童安全封盖进行密封。
[0193]
具体实例7:锭剂的调配及制备
[0194]
锭剂配方展示于表5中:
[0195]
表5
[0196][0197]
a:根据理论盐度调节晶体形式1的活性医药成分(api)的单位含量。实际剂量系基于api含量。
[0198]
b:在处理期间移除。
[0199]
锭剂的制造制程系如下:
[0200]
1)api压碎/筛选
[0201]
将晶体形式1压碎且用80目筛网筛分一次。
[0202]
2)预混合
[0203]
将纤维素乳糖添加至料斗混合器中且随后将原料药压碎并筛分,添加交联聚维酮及羟丙基纤维素,且最后添加预胶凝化淀粉并混合10分钟。
[0204]
3)材料处理
[0205]
使混合材料通过摆动粒化机(24目筛)两次。
[0206]
4)中间混合
[0207]
将材料转移至料斗混合器,使微型粉末硅胶通过40目筛且将其添加至混合器中持续30分钟。
[0208]
5)总体混合
[0209]
硬脂酸镁经由40目筛来筛分且与混合物混合5分钟。
[0210]
6)制锭
[0211]
将最终混合产物压制成卵形(100mg)或圆形核(10mg、40mg),且在制程中检查锭剂的重量、厚度及硬度。
[0212]
7)涂覆
[0213]
用高效涂覆盘涂覆且干燥核,并将锭剂温度控制处于38℃至42℃。将松散锭剂封装于双层拉链袋中,密封且置放于铝塑料复合膜袋中。将25g干燥剂置放于夹层的间,且在标记的后在封装制程中储存标记物。
[0214]
8)最终封装
[0215]
将锭剂封装于双重铝封装机器中且用代码喷雾标记。封装材料为聚酰胺/铝/聚氯乙烯及医药铝箔的冷冲压固体状医药复合硬质锭剂。
[0216]
具体实例8:药代动力学资料
[0217]
将九只雄性米格鲁犬(beagle dog)划分成三个组,每组三只。其各自分别地投予式i化合物的晶体形式1、晶体形式2及自由碱的5mg/kg口服胶囊(po)(根据自由碱的剂量来计算剂量)。在投药的前及投药的后15分钟、30分钟、1小时、2小时、4小时、6小时、8小时、10小时及24小时自颈静脉收集血液,且分离血浆并储存于-80℃下的冷冻机中。lc-ms随后用于分析。
[0218]
实验资料展示于表6中:
[0219]
表6
[0220][0221]
根据以上结果,表明式i化合物的晶体形式1及晶体形式2在体内比式i化合物的自由碱更好的吸收。
[0222]
尽管当前揭示的主题已经由各种方法及参考图的实施方案充分描述,但值得注意的系变化及修改对于本领域中的彼等技术员而言将为显而易见的。此等变化及修改应在本发明的随附申请专利范围的范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1