用于纳米孔测序的易位控制元件、报告子代码和用于易位控制的其他手段的制作方法
1.本发明总体上涉及新的合成报告子构建体,更具体地涉及新的无核苷酸、基于亚磷酰胺的易位控制元件、报告子代码和在穿过纳米孔时产生独特信号的其他特征,及其制造和利用方法,特别是在基于纳米孔的聚合物测序方法中。
背景技术:
2.生物分子的测量是现代医学的基础,广泛用于医学研究,更具体地用于诊断和治疗,以及药物开发。核酸编码生命体活动和繁殖所必需的信息,本质上是生命的蓝图。确定此类蓝图在纯研究和应用科学中都有用。在医学中,测序可用于诊断和研发用于各种病症的治疗方法,包括癌症、心脏病、自身免疫性疾病、多发性硬化症和肥胖症。在工业中,测序可用于设计改进的酶促过程或合成生物。例如,在生物学中,该工具可用于研究生态系统的健康,因此具有广泛的用途。类似地,蛋白质和其他生物分子的测量提供了标志物和对疾病和病原传播的了解。
3.个体独特的dna序列提供了有关他们对某些疾病的易感性的宝贵信息。它还为患者提供了筛查以早期发现和/或接受预防性治疗的机会。此外,根据患者的个人蓝图,临床医生将能够施用个性化疗法,以最大限度地提高药物疗效和/或最大限度地降低药物不良反应的风险。类似地,确定病原生物的蓝图可以为传染病提供新的治疗和更稳健的病原体监测。低成本的全基因组dna测序将为现代医学奠定基础。为了实现该目标,测序技术必须在通量、准确性和读段长度方面不断进步。
4.在过去的十年中,大量的下一代dna测序技术已经可商购,已大大降低了全基因组测序的成本。这些包括边合成边测序(
″
sbs
″
)平台(illumina,inc.,454 life sciences,ion torrent,pacific biosciences)和基于类似连接的平台(complete genomics,life technologies corporation)。许多其他技术正在开发中,这些技术利用了各种各样的样品处理和检测方法。例如,gnubio公司(cambridge,mass.)使用皮升反应容器来控制数百万个谨慎的探针测序反应,而halcyon molecular(redwood city,calif.)正在尝试开发使用透射电子显微镜进行直接dna测量的技术。
5.基于纳米孔的核酸测序是一种已被广泛研究的引人注目的方法。kasianowicz等人(proc.natl.acad.sci.usa 93:13770-13773,1996)在单链多核苷酸电易位通过嵌入脂双层中的α溶血素纳米孔时,表征了单链多核苷酸。已经证明,在多核苷酸易位期间,可以测量纳米孔孔口的部分堵塞作为离子电流降低的量度。然而,纳米孔中的多核苷酸测序因必须解析紧密间隔的碱基(0.34nm)而承受沉浸在显著背景噪音中的小信号差异。由于观察到多核苷酸的快速易位速率,通常大约为每微秒1个碱基,因此纳米孔中单碱基分辨率的测量挑战变得更加苛刻。可以通过调整运行参数(例如电压、盐成分、ph、温度和粘度等)来降低易位速度。然而,这种调整无法将易位速度降低到允许单碱基分辨率的水平。
6.stratos genomics开发了一种称为扩展测序(“sbx”)的方法,该方法使用生化过
程将dna序列转录到称为“xpandomer”的可测量聚合物上(kokoris等人,美国专利号7,939,259
″
high throughput nucleic acid sequencing by expansion
″
)。转录的序列沿xpandomer主链在高信噪比报告子中编码,所述报告子相隔约10nm,专为高信噪比、分化良好的反应而设计。这些差异在xpandomer相对于天然dna的序列读段效率和准确性方面提供了显著的性能增强。xpandomer可以实现多种下一代dna测序检测技术,并且非常适合纳米孔测序。
7.纳米孔已被证明是强大的放大器,就像它们被高度开发的前身库尔特计数器一样。然而,任务是dna的碱基识别的当代有机纳米孔(诸如溶血素和mspa)是在自然界中不与dna相互作用的跨膜蛋白。它们没有控制dna易位的天然功能。这是一个公认的缺点,一些人试图通过在纳米孔附近添加蛋白质马达的功能来纠正。例如,akeson的团队在α-溶血素纳米孔附近添加了聚合酶,以便ss-dna可以以受控的速率被送入孔中(参见g.m.cherf等人
″
automated forward and reverse ratcheting of dna in a nanopore at 5-a precision,
″
nat biotech,vol.advance online publication,february 2012)。这种方法使测定复杂化,并强制α溶血素中的测量区域与聚合酶中的位置控制分离,这可能会给测量带来额外的噪声和序列依赖性变化。
8.在另一种称为杂交易位控制(tch)的方法中,通过使用杂交产生的结构暂停纳米孔易位事件,所述结构解离以进行易位(参见,例如,授予mcruer和kokoris的美国专利号10,457,979)。akeson等人(美国专利号6,465,193)首先通过用顺序发夹双链区暂停dna易位来证明这一点。易位在双链体处停止,因为它大于α-溶血素纳米孔的孔口。当双链体因随机热波动而释放时,易位进行到下一个双链体。在每次暂停期间,可以测量和识别位于纳米孔中(与双链体相邻)的dna区域。当应用于纳米孔测序时,这种用于易位控制的双链方法存在局限性,包括双链体形成不完全或杂交填充率以及双链体解离的随机性,这可能会导致缺失或插入事件。无法本地化的插入和缺失会严重降低数据质量。
9.虽然在此领域取得了重大进展,但是商业上可行的易位控制的实施,例如使用xpandomer,将受益于克服由双链引起的限制的改进。本发明满足这些需要并提供如下文所述的进一步的相关优点。
10.背景部分中讨论的所有主题都不一定是现有技术,不应仅仅因为其在背景部分中的讨论而被假定为现有技术。沿着这些思路,除非明确说明是现有技术,否则对背景部分中讨论的或与此类主题相关的现有技术中的问题的任何认识不应被视为现有技术。相反,背景部分中对任何主题的讨论都应被视为发明人解决特定问题的方法的一部分,其本身也可能具有创造性。
技术实现要素:
11.简而言之,公开了用于改进聚合物分析物(例如,xpandomer)的纳米孔测序(例如,产生具有更高读取长度、准确度和/或通量的序列)的化合物(例如,xntp)和方法,所述化合物包括由一系列新颖亚磷酰胺单体单元合成的聚合物报告子和连接基构建体。
12.在一些实施例中,聚合物构建体可被设计成完全没有核苷酸。
13.在一个方面,本公开提供具有以下结构的化合物(即,xntp):
[0014][0015]
其中r是oh或h;核碱基是腺嘌呤、胞嘧啶、鸟嘌呤、胸腺嘧啶、尿嘧啶或核碱基类似物;报告子构建体是具有第一末端和第二末端的聚合物,并且包括从第一末端到第二末端串联的第一报告子代码、带有易位控制元件的对称化学分支和第二报告子代码;连接基a将α氨基磷酸酯的氧原子接合到报告子构建体的第一末端;连接基b将核碱基接合到报告子构建体的第二末端;并且其中易位控制元件是如下所述的聚合物。
[0016]
在一个实施例中,所述易位控制元件是包含两个或更多个选自以下的重复单元的聚合物:1,3-o-双(磷酸二酯)-2s-o-mpeg4-丙烷(化合物12b)、1,3-o-双(磷酸二酯)-2-(4-me-o-peg3)-1-(et-o-ac)-1,2,3-三唑)-丙烷(化合物35b)、1,3-o-双(磷酸二酯)-2s-o-mpeg6-丙烷(化合物12c)、1,2-o-双(磷酸二酯)-3-o-mpeg2-丙烷(化合物16)、2,3-o-双(磷酸二酯)-1-(5-苯并呋喃)-丙烷(化合物20i)、1,2-o-双(磷酸二酯)-3-(4-甲基哌嗪-1-基)-丙烷(化合物20j)、2,3-o-双(磷酸二酯)-1-(n1-(2-me-5-硝基吲哚)-丙烷(化合物20g)、1,8-o-双(磷酸二酯)-n,n-二乙基哌嗪(化合物26h)、1,2-o-双(磷酸二酯)-3-(4-(me-o-peg3-o-bz)-1-(1,2,3-三唑))-丙烷(化合物31d)、1,3-o-双(磷酸二酯)-2s-o-(4-(me-o-peg2)-1-(et-obz)-1,2,3-三唑)-丙烷(化合物35a)、1,3-o-双(磷酸二酯-2-(4-(me-o-peg5)-1-(et-o-ac)-1,2,3-三唑)-丙烷(化合物35c)、1,3-o-双(磷酸二酯-2s-o-(4-(me-o-peg7)-1-(et-obz)-1,2,3-三唑)-丙烷(化合物35d)、1,3-o-双(磷酸二酯-2s-o-(4-(me-o-peg3)-1-(me-乙酸酯)-1,2,3-三唑)-丙烷(化合物35e)、1,3-o-双(磷酸二酯-2s-o-(4-(me-o-peg3)-1-(et-2,2,2-三-(me-o-bz))-1,2,3-三唑)-丙烷(化合物37a)、1,3-o-双(磷酸二酯-2-(4-(me-o-peg5)-1-(et-2,2,2-三-(me-o-ac))-1,2,3-三唑)-丙烷(化合物37b)、1,3-o-双(磷酸二酯-2s-o-(peg4-o-bz)-丙烷(化合物38b)、1,3-o-双(磷酸二酯-2,2-双(me-o-mpeg2)-丙烷(化合物45b)、1,3-o-双(磷酸二酯-2,2-双(4-(me-o-peg2-o-me)-1-(et-o-bz)-1,2,3-三唑)-丙烷(化合物47f)、1,3-o-双(磷酸二酯-2,2-双(4-(me-o-peg3-o-me)-1-(et-o-bz)-1,2,3-三唑)-丙烷(化合物47gg)、1,3-o-双(磷酸二酯-2,2-双(4-(me-o-peg3-o-me)-1-(et-2,2,2-三-(me-o-bz))-1,2,3-三唑)-丙烷(化合物47i)或1,3-o-双(磷酸二酯-2,2-双(1-me-4-(me-o-peg2-o-bz)-1,2,3-三唑)-丙烷(化合物52)。
[0017]
在一些实施例中,r是oh。
[0018]
在一些实施例中,r是h。
[0019]
在其他实施例中,核碱基是腺嘌呤、胞嘧啶、鸟嘌呤、胸腺嘧啶或尿嘧啶。
[0020]
在其他实施例中,核碱基是核碱基类似物。
[0021]
在其他实施例中,所述对称化学分支是1,2,3-o-三-(磷酸二酯)-丙烷、1,3-双-(5-o-磷酸二酯-戊基酰胺基)-2-o-磷酸二酯-丙烷或1,4,7-o-三-(磷酸二酯)-庚烷。
[0022]
在其他实施例中,对称化学支化剂是1,2,3-o-三-(磷酸二酯)-丙烷。
[0023]
在其他实施例中,所述易位控制元件是包含两个或更多个选自表1a的重复单元的聚合物。
[0024]
在其他实施例中,所述易位控制元件是包含两个或更多个选自以下的重复单元的聚合物:1,3-o-双(磷酸二酯)-2s-o-mpeg4-丙烷(化合物12b)和1,3-o-双(磷酸二酯)-2-(4-me-o-peg3)-1-(et-o-ac)-1,2,3-三唑)-丙烷(化合物35b)。
[0025]
在再其他实施例中,所述易位控制元件是包含以下序列的聚合物:[(1,3-o-双(磷酸二酯)-2s-o-mpeg4-丙烷(化合物12b))]n1[(1,3-o-双(磷酸二酯)-2-(4-me-o-peg3)-1-(et-o-ac)-1,2,3-三唑)-丙烷(化合物35b))]n2,其中n1为0至6且n2为6至10。
[0026]
在其他实施例中,第一报告子代码和第二报告子代码是相同的。
[0027]
在进一步的实施例中,所述第一报告子代码和所述第二报告子代码是包含两个或更多个选自以下的重复单元的聚合物:六乙二醇(d)、乙烷(l)、三乙二醇(x)、1,3-o-双(磷酸二酯)-2s-o-mpeg4-丙烷(化合物12b)、1,3-o-双(磷酸二酯)-2-(4-me-o-peg3)-1-(et-o-ac)-1,2,3-三唑)-丙烷(化合物35b)、1,3-o-双(磷酸二酯-2,2-双(me-o-mpeg2)-丙烷(化合物45b)、1,3-o-双(磷酸二酯-2s-o-(peg4-o-bz)-丙烷(化合物38b)、1,3-o-双(磷酸二酯)-2s-o-mpeg6-丙烷(化合物12c)、1,3-o-双(磷酸二酯-2s-o-(4-(me-o-peg3)-1-(et-2,2,2-三-(me-o-bz))-1,2,3-三唑)-丙烷(化合物37a)、1,3-o-双(磷酸二酯-2s-o-(4-(me-o-peg3)-1-(me-乙酸酯)-1,2,3-三唑)-丙烷(化合物35e)、1,3-o-双(磷酸二酯)-2s-o-(4-(me-o-peg2)-1-(et-obz)-1,2,3-三唑)-丙烷(化合物35a)、1,3-o-双(磷酸二酯)-2-(4-et-1-(et-o-mpegl)-1,2,3-三唑)-丙烷(化合物31a)、2,3-o-双(磷酸二酯)-1-(1-二甲氧基喹唑啉二酮)-丙烷(化合物20c)、2,3-o-双(磷酸二酯)-1-(n9-(3,6-二甲氧基咔唑)-丙烷(化合物20e)、1,1
′‑
o-双(磷酸二酯)-2,2
′‑
(磺酰基双(苄-4-基))-二乙醇(化合物26d)、1,1
′‑
o-双(磷酸二酯)-2,2
′‑
联吡啶-4,4
′‑
基)-二甲醇(化合物26a)、2,3-o-双(磷酸二酯)-1-(n1-(4,6-二甲氧基-3-me-吲哚)-丙烷(化合物20b)、3-(1,2-o-双(磷酸二酯)-丙基)-8,8-二甲基六氢-3h-3a,6-甲桥苯并[c]异噻唑2,2-二氧化物(化合物20d)、2,3-o-双(磷酸二酯)-1-(n1-(6-氮杂胸腺嘧啶))-丙烷(化合物20f)、1,5-o-双(磷酸二酯)-六氢呋喃并[2,6]呋喃(化合物23)、1,1
′‑
o-双(磷酸二酯)-八氢-2,6-二甲基-3,8∶4,7-二甲桥-2,6-萘啶-4,8-二基)-二甲醇(化合物26e)、2,3-o-双(磷酸二酯)-1-(n1-(2-me-5-硝基吲哚)-丙烷(化合物20h)、2,3-o-双(磷酸二酯)-1-(n1-(2-me-5-硝基吲哚)-丙烷(化合物20g)、2,3-o-双(磷酸二酯)-1-(5-苯并呋喃)-丙烷(化合物20i)、1,2-o-双(磷酸二酯)-3-o-mpeg2-丙烷(化合物5b)、1,3-o-双(磷酸二酯)-2-(4-et-1-(et-o-mpeg3)-1,2,3-三唑)-丙烷(化合物31b)和1,3-o-双(磷酸二酯)-3-o-mpeg4-丙烷(化合物5a)。
[0028]
在其他实施例中,所述第一报告子代码和所述第二报告子代码是包含两个或更多个选自六乙二醇、乙烷、三乙二醇和表1a中所列任何化合物的重复单元的聚合物。
[0029]
在进一步的实施例中,第一报告子代码和所述第二报告子代码是包含两个或更多个选自六乙二醇、乙烷、三乙二醇和1,3-o-双(磷酸二酯)-2s-o-mpeg4-丙烷(化合物12b)的
1-(et-2,2,2-三-(me-o-ac))-1,2,3-三唑)-丙烷(化合物37b))4(六乙二醇)2]。
[0036]
在其他实施例中,连接基a通过包含三唑的键合接合到α氨基磷酸酯的氧原子,并且连接基b通过包含三唑的键合接合到核碱基。
[0037]
在另一方面,本发明提供一种报告子构建体,其包含具有第一末端和第二末端的聚合物,并且包含从所述第一末端到所述第二末端串联的第一报告子代码、带有易位控制元件的对称化学分支和第二报告子代码;并且其中所述易位控制元件是包含两个或更多个选自以下的重复单元的聚合物:1,3-o-双(磷酸二酯)-2s-o-mpeg4-丙烷(化合物12b)、1,3-o-双(磷酸二酯)-2-(4-me-o-peg3)-1-(et-o-ac)-1,2,3-三唑)-丙烷(化合物35b)、1,3-o-双(磷酸二酯)-2s-o-mpeg6-丙烷(化合物12c)、1,2-o-双(磷酸二酯)-3-o-mpeg2-丙烷(化合物16)、2,3-o-双(磷酸二酯)-1-(5-苯并呋喃)-丙烷(化合物20i)、1,2-o-双(磷酸二酯)-3-(4-甲基哌嗪-1-基)-丙烷(化合物20j)、2,3-o-双(磷酸二酯)-1-(n1-(2-me-5-硝基吲哚)-丙烷(化合物20g)、1,8-o-双(磷酸二酯)-n,n-二乙基哌嗪(化合物26h)、1,2-o-双(磷酸二酯)-3-(4-(me-o-peg3-o-bz)-1-(1,2,3-三唑))-丙烷(化合物31d)、1,3-o-双(磷酸二酯)-2s-o-(4-(me-o-peg2)-1-(et-obz)-1,2,3-三唑)-丙烷(化合物35a)、1,3-o-双(磷酸二酯-2-(4-(me-o-peg5)-1-(et-o-ac)-1,2,3-三唑)-丙烷(化合物35c)、1,3-o-双(磷酸二酯-2s-o-(4-(me-o-peg7)-1-(et-obz)-1,2,3-三唑)-丙烷(化合物35d)、1,3-o-双(磷酸二酯-2s-o-(4-(me-o-peg3)-1-(me-乙酸酯)-1,2,3-三唑)-丙烷(化合物35e)、1,3-o-双(磷酸二酯-2s-o-(4-(me-o-peg3)-1-(et-2,2,2-三-(me-o-bz))-1,2,3-三唑)-丙烷(化合物37a)、1,3-o-双(磷酸二酯-2-(4-(me-o-peg5)-1-(et-2,2,2-三-(me-o-ac))-1,2,3-三唑)-丙烷(化合物37b)、1,3-o-双(磷酸二酯-2s-o-(peg4-o-bz)-丙烷(化合物38b)、1,3-o-双(磷酸二酯-2,2-双(me-o-mpeg2)-丙烷(化合物45b)、1,3-o-双(磷酸二酯-2,2-双(4-(me-o-peg2-o-me)-1-(et-o-bz)-1,2,3-三唑)-丙烷(化合物47f)、1,3-o-双(磷酸二酯-2,2-双(4-(me-o-peg3-o-me)-1-(et-o-bz)-1,2,3-三唑)-丙烷(化合物47gg)、1,3-o-双(磷酸二酯-2,2-双(4-(me-o-peg3-o-me)-1-(et-2,2,2-三-(me-o-bz))-1,2,3-三唑)-丙烷(化合物47i)或1,3-o-双(磷酸二酯-2,2-双(1-me-4-(me-o-peg2-o-bz)-1,2,3-三唑)-丙烷(化合物52)。
[0038]
在一些实施例中,所述对称化学分支是1,2,3-o-三-(磷酸二酯)-丙烷、1,3-双-(5-o-磷酸二酯-戊基酰胺基)-2-o-磷酸二酯-丙烷或1,4,7-o-三-(磷酸二酯)-庚烷。
[0039]
在另一实施例中,对称化学支化剂是1,2,3-o-三-(磷酸二酯)-丙烷。
[0040]
在其他实施例中,所述易位控制元件是包含两个或更多个选自表1a的重复单元的聚合物。
[0041]
在再其他实施例中,所述易位控制元件是包含两个或更多个选自以下的重复单元的聚合物:1,3-o-双(磷酸二酯)-2s-o-mpeg4-丙烷(化合物12b)和1,3-o-双(磷酸二酯)-2-(4-me-o-peg3)-1-(et-o-ac)-1,2,3-三唑)-丙烷(化合物35b)。
[0042]
在进一步的实施例中,所述易位控制元件是包含以下序列的聚合物:[(1,3-o-双(磷酸二酯)-2s-o-mpeg4-丙烷(化合物12b))]n1[(1,3-o-双(磷酸二酯)-2-(4-me-o-peg3)-1-(et-o-ac)-1,2,3-三唑)-丙烷(化合物35b))]n2,其中n1为0至6且n2为6至10。
[0043]
在其他实施例中,第一报告子代码和第二报告子代码是相同的。
[0044]
在一些实施例中,所述第一报告子代码和所述第二报告子代码是包含两个或更多
个选自以下的重复单元的聚合物:六乙二醇(d)、乙烷(l)、三乙二醇(x)、1,3-o-双(磷酸二酯)-2s-o-mpeg4-丙烷(化合物12b)、1,3-o-双(磷酸二酯)-2-(4-me-o-peg3)-1-(et-o-ac)-1,2,3-三唑)-丙烷(化合物35b)、1,3-o-双(磷酸二酯-2,2-双(me-o-mpeg2)-丙烷(化合物45b)、1,3-o-双(磷酸二酯-2s-o-(peg4-o-bz)-丙烷(化合物38b)、1,3-o-双(磷酸二酯)-2s-o-mpeg6-丙烷(化合物12c)、1,3-o-双(磷酸二酯-2s-o-(4-(me-o-peg3)-1-(et-2,2,2-三-(me-o-bz))-1,2,3-三唑)-丙烷(化合物37a)、1,3-o-双(磷酸二酯-2s-o-(4-(me-o-peg3)-1-(me-乙酸酯)-1,2,3-三唑)-丙烷(化合物35e)、1,3-o-双(磷酸二酯)-2s-o-(4-(me-o-peg2)-1-(et-obz)-1,2,3-三唑)-丙烷(化合物35a)、1,3-o-双(磷酸二酯)-2-(4-et-1-(et-o-mpeg1)-1,2,3-三唑)-丙烷(化合物31a)、2,3-o-双(磷酸二酯)-1-(1-二甲氧基喹唑啉二酮)-丙烷(化合物20c)、2,3-o-双(磷酸二酯)-1-(n9-(3,6-二甲氧基咔唑)-丙烷(化合物20e)、1,1
′‑
o-双(磷酸二酯)-2,2
′‑
(磺酰基双(苄-4-基))-二乙醇(化合物26d)、1,1
′‑
o-双(磷酸二酯)-2,2
′‑
联吡啶-4,4
′‑
基)-二甲醇(化合物26a)、2,3-o-双(磷酸二酯)-1-(n1-(4,6-二甲氧基-3-me-吲哚)-丙烷(化合物20b)、3-(1,2-o-双(磷酸二酯)-丙基)-8,8-二甲基六氢-3h-3a,6-甲桥苯并[c]异噻唑2,2-二氧化物(化合物20d)、2,3-o-双(磷酸二酯)-1-(n1-(6-氮杂胸腺嘧啶))-丙烷(化合物20f)、1,5-o-双(磷酸二酯)-六氢呋喃并[2,6]呋喃(化合物23)、1,1
′‑
o-双(磷酸二酯)-八氢-2,6-二甲基-3,8:4,7-二甲桥-2,6-萘啶-4,8-二基)-二甲醇(化合物26e)、2,3-o-双(磷酸二酯)-1-(n1-(2-me-5-硝基吲哚)-丙烷(化合物20h)、2,3-o-双(磷酸二酯)-1-(n1-(2-me-5-硝基吲哚)-丙烷(化合物20g)、2,3-o-双(磷酸二酯)-1-(5-苯并呋喃)-丙烷(化合物20i)、1,2-o-双(磷酸二酯)-3-o-mpeg2-丙烷(化合物5b)、1,3-o-双(磷酸二酯)-2-(4-et-1-(et-o-mpeg3)-1,2,3-三唑)-丙烷(化合物31b)和1,3-o-双(磷酸二酯)-3-o-mpeg4-丙烷(化合物5a)。
[0045]
在其他实施例中,所述第一报告子代码和所述第二报告子代码是包含两个或更多个选自六乙二醇、乙烷、三乙二醇和表1a中所列任何化合物的重复单元的聚合物。
[0046]
在进一步的实施例中,第一报告子代码和所述第二报告子代码是包含两个或更多个选自六乙二醇、乙烷、三乙二醇和1,3-o-双(磷酸二酯)-2s-o-mpeg4-丙烷(化合物12b)的重复单元的聚合物。
[0047]
在进一步的实施例中,第一报告子代码和第二报告子代码是包含选自以下序列的聚合物:(i)[(六乙二醇)2(乙烷)3(六乙二醇)(三乙二醇)],(ii)[(六乙二醇)2(1,3-o-双(磷酸二酯)-2s-o-mpeg4-丙烷(化合物12b))2(乙烷)(三乙二醇)3],(iii)[(六乙二醇)2(1,3-o-双(磷酸二酯)-2s-o-mpeg4-丙烷(化合物12b))3(乙烷)2(六乙二醇)(三乙二醇)]和(iv)[(三乙二醇)2(乙烷)(1,3-o-双(磷酸二酯)-2s-o-mpeg4-丙烷(化合物12b))6(乙烷)7]。
[0048]
在另一方面,本发明提供一种对称合成的报告子系链(ssrt),其中所述对称合成的报告子系链是具有第一末端和第二末端的聚合物,并且包含从所述第一末端到所述第二末端串联的第一连接基、根据上述报告子构建体中任一者的报告子构建体以及第二连接基,其中所述第一连接基和所述第二连接基是相同的并且是包含两个或更多个选自以下的重复单元的聚合物:精胺(q)、六乙二醇(d)、2-((4-((3-(苯甲酰氧基)-2-(((1-(3-(苯甲酰氧基)-2-((苯甲酰氧基)甲基)-2-((磷酸二酯-氧基)甲基)丙基)-1h-1,2,3-三唑-4-基)甲氧基)甲基)-2-((苯甲酰氧基)甲基)丙氧基)甲基)-1h-1,2,3-三唑-1-基)甲基)-2-o-磷酸
二酯-丙烷-1,3-二基二苯甲酸酯(化合物62)、1,3-o-双(磷酸二酯-2,2-双(1-me-4-(me-o-peg2-o-bz)-1,2,3-三唑)-丙烷(化合物52)、1,3-o-双(磷酸二酯-2-(4-(me-o-peg5)-1-(et-o-ac)-1,2,3-三唑)-丙烷(化合物35c)、1,3-o-双(磷酸二酯-2s-o-(4-(me-o-peg7)-1-(et-obz)-1,2,3-三唑)-丙烷(化合物35d)、1,3-o-双(磷酸二酯-2s-o-(4-(me-o-peg3)-1-(et-2,2,2-三-(me-o-bz))-1,2,3-三唑)-丙烷(化合物37a)、1,3-o-双(磷酸二酯-2-(4-(me-o-peg5)-1-(et-2,2,2-三-(me-o-ac))-1,2,3-三唑)-丙烷(化合物37b)、1,2-o-双(磷酸二酯)-3-(4-(me-o-peg3-o-bz)-1-(1,2,3-三唑))-丙烷(化合物31d)、1,3-o-双(磷酸二酯-2,2-双(4-(me-o-peg2-o-me)-1-(et-o-bz)-1,2,3-三唑)-丙烷(化合物47f)、1,3-o-双(磷酸二酯-2,2-双(4-(me-o-peg3-o-me)-1-(et-2,2,2-三-(me-o-bz))-1,2,3-三唑)-丙烷(化合物47i)、1,2-o-双(磷酸二酯)-3-(4-甲基哌嗪-1-基)-丙烷(化合物20j)、1,3-o-双(磷酸二酯-2,2-双(4-(me-o-peg3-o-me)-1-(et-o-bz)-1,2,3-三唑)-丙烷(化合物47g)和1,1
′‑
o-双(磷酸二酯)-n(对甲苯基)-二乙醇胺(化合物26b)。
[0049]
在一些实施例中,对称合成的报道系链(ssrt)包括含有两个精胺重复单元的聚合酶增强区。
[0050]
在其他实施例中,所述对称合成的报告子系链(ssrt)包含含有两个或更多个选自以下的重复单元的易位减速区:1,3-o-双(磷酸二酯-2-(4-(me-o-peg5)-1-(et-o-ac)-1,2,3-三唑)-丙烷(化合物35c)、1,3-o-双(磷酸二酯-2s-o-(4-(me-o-peg7)-1-(et-obz)-1,2,3-三唑)-丙烷(化合物35d)、1,3-o-双(磷酸二酯-2s-o-(4-(me-o-peg3)-1-(et-2,2,2-三-(me-o-bz))-1,2,3-三唑)-丙烷(化合物37a)和1,3-o-双(磷酸二酯-2-(4-(me-o-peg5)-1-(et-2,2,2-三-(me-o-ac))-1,2,3-三唑)-丙烷(化合物37b)。
[0051]
在其他实施例中,所述对称合成的报告子系链(ssrt)包含含有选自以下的聚合物的易位减速区:(i)[((六乙二醇)(1,3-o-双(磷酸二酯-2-(4-(me-o-peg5)-1-(et-o-ac)-1,2,3-三唑)-丙烷(化合物35c))3(六乙二醇)2]、(ii)[((六乙二醇)(1,3-o-双(磷酸二酯-2-(4-(me-o-peg5)-1-(et-o-ac)-1,2,3-三唑)-丙烷(化合物35c))4(六乙二醇)2]、(iii)[((六乙二醇)(1,3-o-双(磷酸二酯-2s-o-(4-(me-o-peg7)-1-(et-obz)-1,2,3-三唑)-丙烷(化合物35d))4(六乙二醇)2]和(iv)[((六乙二醇)(1,3-o-双(磷酸二酯-2-(4-(me-o-peg5)-1-(et-2,2,2-三-(me-o-ac))-1,2,3-三唑)-丙烷(化合物37b))4(六乙二醇)2]。
[0052]
在再其他实施例中,对称合成的报告子系链(ssrt)的第一末端和第二末端包括键合部分,并且在某些实施例中,键合部分是叠氮基(-n3)基团。
[0053]
在另一方面,本发明提供了一种对靶核酸进行测序的方法,其包括以下步骤:一种用于对靶核酸进行测序的方法,其包括:a)提供通过模板定向合成产生的子链,所述子链包含多个xntp亚基,所述多个xntp亚基偶联在对应于所述靶核酸的全部或一部分的连续核苷酸序列的序列中,其中所述子链的各个xntp亚基包含报告子构建体、核碱基残基和可选择性切割的键,并且其中所述报告子构建体在所述可选择性切割的键的切割时允许延长所述子链的所述亚基;b)切割所述可选择性切割的键以产生长度长于所述子链的多个所述亚基的xpandomer,所述xpandomer包含用于解析对应于所述靶核酸的全部或一部分的所述连续核苷酸序列的序列中的遗传信息的所述报告子构建体;和c)检测所述xpandomer的所述报告子构建体。
[0054]
在一些方面,用于解析遗传信息的所述报告子构建体包括报告子代码和易位控制
元件,其中所述易位控制元件借由空间位阻提供易位控制,并在所述xpandomer穿过经受基线电压的纳米孔时暂停易位,其中所述易位控制元件在所述纳米孔的孔口内接合所述报告子代码,其中所述报告子代码由所述纳米孔感测。
[0055]
在一些实施例中,所述xpandomer借由脉冲电压的施加恢复通过所述纳米孔的易位,其中所述脉冲电压足以允许所述易位控制元件的易位,同时使所述xpandomer的下一个报告子构建体自由地与所述纳米孔接合。
[0056]
在其他实施例中,与所述纳米孔接合的所述报告子构建体的所述易位控制元件借由空间位阻,在所述脉冲电压的每个脉冲时易位。
[0057]
在一些实施例中,在所述脉冲电压的脉冲之间的时间段期间,由所述纳米孔感测目标构建体。
[0058]
在某些实施例中,基线电压为约55mv至约75mv且脉冲电压为约550mv至约700mv。
[0059]
在一些实施例中,脉冲电压具有约5μs至约10μs的持续时间和约0.5ms至1.5ms的周期性。
[0060]
在其他实施例中,纳米孔经受交流电流(ac)。
[0061]
在进一步的实施例中,xntp亚基中的一者或多者包括2
′
氟阿拉伯糖基差向异构体。
[0062]
在另一方面,本公开提供一种用于控制聚合物通过纳米孔的易位速率的缓冲剂,其包含至少一种选自由以下项组成的组的盐:nh4cl、mgcl2、licl、kcl、cscl、nacl和cacl2。
[0063]
在一些实施例中,缓冲剂还包含至少一种选自由3-甲基-2-噁唑烷酮(moa)、dmf、acn、dmso和nmp组成的组的溶剂,其中所述溶剂以约1%vol/vol至约35%vol/vol的范围存在。
[0064]
在其他实施例中,缓冲剂还包含至少一种选自由己酸钠(nahex)、edta、氧化还原试剂、peg、甘油、聚蔗糖等组成的组的添加剂。
[0065]
在另一方面,本公开提供一种用于控制聚合物通过纳米孔检测器的易位速率的缓冲剂系统,其包括顺式缓冲剂和反式缓冲剂,其中所述顺式缓冲剂具有第一盐浓度,并且所述反式缓冲剂具有第二盐浓度,其中所述第一盐浓度低于所述第二盐浓度。
附图说明
[0066]
图1a、图1b、图1c和图1d是说明概括性xntp的主要特征及其在展开测序(sbx)中功能的简明示意图。
[0067]
图2是说明xntp的一个实施例的更多细节的示意图。
[0068]
图3是说明xpandomer穿过生物纳米孔的一个实施例的示意图。
[0069]
图4是说明xpandomer穿过生物纳米孔的另一个实施例的示意图。
[0070]
图5a-图5d是说明报告子代码的替代实施例的示意图。
[0071]
图6是说明ssrt报告子构建体的固态合成的一个实施例的示意图。
[0072]
图7说明ssrt报告子构建体的替代结构实施例。
[0073]
图8a和图8b是说明ssrt和dntp-2c环化形成xntp的一个实施例的示意图。
[0074]
图9a和图9b是说明xpandomer通过纳米孔的易位控制的一个实施例的示意图。
[0075]
图10是说明生物素衍生物的一个实施例的示意图。
[0076]
图11是说明可切割延伸寡核苷酸的一个实施例的示意图。
[0077]
图12是说明经受棘轮穿过纳米孔的聚合物的一个实施例的示意图。
[0078]
图13a和图13b是描述报告子代码特性的代表性迹线。
[0079]
图14a和图14b是纳米孔衍生序列的比对读段群的直方图显示。
[0080]
图15是显示简单dna模板序列的代表性迹线。
[0081]
图16是显示cagt重复dna模板序列的代表性迹线。
[0082]
图17是显示复合dna模板序列的代表性迹线。
[0083]
图18是显示复合dna 222mer模板序列的代表性迹线。
[0084]
图19是纳米孔衍生序列的比对读段群的直方图显示。
[0085]
图20a是说明经受棘轮穿过纳米孔的xpandomer的一个实施例的示意图。
[0086]
图20b是经受棘轮的易位xpandomer的电流测量的实例。
具体实施方式
[0087]
通过参考以下对本发明的优选实施例和本文包括的实例的详细描述,可以更容易地理解本发明。除非另有说明,否则本文中使用的所有技术和科学术语所具有的含义与本发明所属领域普通技术人员通常理解的含义相同。
[0088]
贯穿本说明书的对“一个实施例”、“某一实施例”及其变体的引用,意指结合该实施例描述的特定特征、结构或特性包括在至少一个实施例中。因此,在本说明书中的不同位置出现的短语“在一个实施例中”或“在某一实施例中”不一定都是指同一个实施例。此外,具体特征、结构或特性可在一个或多个实施例中以任何合适的方式组合。
[0089]
如在本说明书和所附权利要求中所用,单数形式“一个”、“一种”、“该”和“所述”包括复数指代物,即一个或多个,除非上下文另外明确规定。还应注意的是,连接术语“和”和“或”通常以最广泛的含义使用以包括“和/或”,除非内容和上下文根据具体情况明确规定了包容性或排他性。因此,替代方案(例如,“或”)的使用应被理解为表示替代方案中的任一个、两者或其任何组合。此外,“和”和“或”的组成在本文中被引用为“和/或”时旨在涵盖包括所有相关项或想法的实施例,以及包括少于所有相关项或想法的一个或多个其他替代实施例。
[0090]
除非上下文另有要求,在整个说明书和随后的权利要求书中,词语“包括”及其同义词和变体,例如“具有”和“包括”,以及其变体,例如“包含”以开放的、包容性的意义来解释,例如,“包括但不限于”。术语“基本上由......组成”将权利要求的范围限制为特定的材料或步骤,或者对所要求保护的发明的基本和新特征没有实质性影响的那些。
[0091]
缩写,“例如”源自拉丁语exempli gratia,在此用于表示非限制性示例。因此,缩写“例如”与术语“举个例子”同义。还应理解,本文和所附权利要求中使用的单数形式“一个”、“一种”和“所述”包括复数形式,除非上下文另有明确规定,术语“x和/或y”表示“x”或“y”或“x”和“y”,名词后面的字母“s”表示该名词的复数和单数形式。此外,在根据马库什基团描述本发明的特征或方面的情况下,意图并且本领域技术人员将认识到,本发明包括并因此也在马库什基因的任何个体成员和任何成员亚基团方面进行描述,并且申请人保留修改申请或权利要求的权利,以明确提及马库什基团的任何个体成员或任何成员亚基团。
[0092]
本文件中使用的任何标题仅用于加快读者的审阅,不应被解释为以任何方式限制
本发明或权利要求。因此,本文提供的本公开的标题和摘要仅为方便起见,并不解释实施例的范围或含义。
[0093]
在本文提供数值范围的情况下,应当理解,介于该范围的上限和下限之间的每个中间值(以下限单位的十分之一为基准,除非上下文另有明确规定)和所述范围内的任何其它所述的或中间值均包括在本发明内。这些较小范围的上限和下限可独立地包括在该较小范围内,并且也为本发明所涵盖,以所指定范围内任何明确排除的限制为准。若所指定范围包括一个或两个限值,则排除那些所包括限制中的任意一个或两个的范围也包括在本发明中。
[0094]
例如,除非另有说明,本文提供的任何浓度范围、百分比范围、比例范围或整数范围应理解为包括所述范围内的任何整数的值以及在适当时其分数的值(例如一个整数的十分之一和百分之一)。此外,除非另有说明,否则本文引用的与任何物理特征(例如聚合物亚单元、尺寸或厚度)相关的任何数字范围应理解为包括所述范围内的任何整数。如本文所用,除非另有说明,否则术语“约”是指指示范围、值或结构的
±
20%。
[0095]
扩展测序
[0096]
由stratos genomics开发的“通过扩增测序”(sbx)方案(参见,例如kokoris等人,美国专利号7,939,259,“high throughput nucleic acid sequencing by expansion”)是基于称为“xntp”的高度修饰、非天然核苷酸类似物的聚合。一般而言,sbx使用生化聚合将dna模板的序列转录到称为“xpandomer”的可测量聚合物上。转录的序列沿xpandomer主链在高信噪比报告子中编码,所述报告子相隔约10nm,专为高信噪比、分化良好的反应而设计。这些差异在xpandomer相对于天然dna的序列读段效率和准确性方面提供了显著的性能增强。sbx过程的概述在图1a、图1b、图1c和图1d中描述。
[0097]
xntp是与模板依赖性酶促聚合相容的可展开的、5’三磷酸修饰的非天然核苷酸类似物。高度简化的xntp在图1a中示出,其强调了这些非天然底物的独特特征:xntp 100具有两个不同的功能区;即:可选择性切割的氨基磷酸酯键110,将5’α-磷酸115连接到核碱基105;以及连接在核苷三氨基磷酸酯内的某些位置处的对称合成的报告子系链(ssrt)120,该位置允许通过氨基磷酸酯键的切割来控制展开。ssrt包括连接基125a和125b,它们被可选择性切割的氨基磷酸酯键分开。每个连接基都连接到报告子代码130的一个末端。xntp 100以“受约束的构型”说明,这是xntp底物和模板依赖性聚合的子链产物的特征。聚合xntp的受限构型是扩展构型的前体,如xpandomer产品中所见。从受限构型到扩展构型的转变发生在子链的主要主链内的氨基磷酸酯的p-n键断裂时。
[0098]
xpandomer聚合物的合成总结在图1b和图1c中。在组装期间,单体xntp底物145(xatp、xctp、xgtp和xttp)通过使用单链模板140作为向导的模板导向聚合过程在新生子链150的可延伸末端聚合。通常,该过程从引物开始并沿5
′
到3
′
方向进行。通常,使用dna聚合酶或其他聚合酶来形成子链,并且选择条件以便获得模板链的互补拷贝。在合成子链之后,偶联的ssrt形成进一步形成子链的受约束的xpandomer。子链中的ssrt具有xntp底物的“受约束的构型”。ssrt的受约束的构型是展开的构型的前体,如xpandomer产物中所见。
[0099]
如图1c所示,从受约束的构型160到展开的构型165的转变是由于子链的主要主链内可选择性切割的氨基磷酸酯键(为简单起见用无阴影椭圆表示)的切割引起的。在该实施例中,ssrt包含一个或多个对它们所连接的核碱基具有特异性的报告子或报告子代码,
130a、130c、130g或130t,从而编码模板的序列信息。以这种方式,ssrt提供了一种手段来展开xpandomer的长度并降低母链序列信息的线性密度。
[0100]
图1d说明xpandomer 165通过纳米孔180从顺式储器175易位到反式储器185。通过纳米孔后,线性化xpandomer的每个报告子代码(在此图示中,标记为“g”、“c”和“t”)产生独特且可重复的电子信号(由叠加迹线190表示),对它所连接的核碱基具有特异性。
[0101]
图2更详细地描绘了xntp的一个实施例的概括结构。xntp 200包含具有连接臂部分220a和220b的核苷三氨基磷酸酯210,这两个连接臂部分被可选择性切割的氨基磷酸酯键230分开。ssrt在键合基团250a和250b处与核苷三磷酸酯相连,其中第一ssrt末端连接到杂环260(此处由胞嘧啶表示,尽管杂环可以是四种标准核碱基a、c、g、或t中的任一个),并且第二ssrt末端连接到核碱基主链的α磷酸酯270。本领域技术人员将理解,可以使用本领域已知的许多合适的偶联化学以形成最终的xntp底物产物,例如,可以通过三唑键合基团的形成来完成ssrt缀合。
[0102]
在该实施例中,ssrt 275包括若干功能元件或“特征”,诸如聚合酶增强区280a和280b、报告子代码285a和285b,以及翻译控制元件(tce)290a和290b。在其他实施例中,ssrt包括单个tce。这些特征中的每一个都在xpandomer通过纳米孔易位以产生一系列独特且可重复的电子信号期间执行独特的功能。ssrt 275设计用于通过空间位阻和/或电排斥的组合控制tce引起的xpandomer易位速率,如本文进一步讨论的。不同的报告子代码的大小可以阻止离子以不同的可测量水平流过纳米孔。使用通常用于寡核苷酸合成的亚磷酰胺化学可以有效地合成特定的ssrt聚合序列。可以通过从市售和/或专有库中选择特定亚磷酰胺的序列来设计报告子代码和其他特征。此类文库包括但不限于长度为1个至12个或更多乙二醇单元的聚乙二醇合长度为1个至12个或更多碳单元的脂肪族聚合物。在某些实施例中,ssrt在靠近核苷酸三磷酰胺酸二酯的ssrt末端包括称为“聚合酶增强区”的特征。聚合酶增强区可以包括带正电荷的多胺间隔区(例如,伯胺、仲胺、叔胺或季胺)或三胺间隔区(三个仲胺,每个由三个碳分开),其促进通过核酸聚合酶的xntp结构掺入。在某些实施例中,聚合酶增强区包括两个精胺重复单元,其中精胺部分由具有以下结构的亚磷酰胺单体提供(本领域技术人员将认识到,三氟乙酰胺保护基团在ssrt合成结束时被去除以暴露精胺上的胺基团):
[0103][0104]
如本公开通篇所用,术语“报告子构建体”是指包括报告子代码、对称化学分支和易位控制元件的ssrt的元件。在某些实施例中,报告子构建体是聚合物,其包括从第一末端到第二末端串联的第一报告子代码、带有易位控制元件的对称化学分支和第二报告子代码。术语“带有”是指对称分支和易位控制元件之间的共价连接,其产生易位控制元件相对于所述两个报告子代码的有利取向。如本文进一步讨论并参考图6-图8,对称化学分支可以由字母“y”表示,其中两个报告子代码接合到y的臂,而易位控制元件接合到y的茎。因此,两个报告子代码通过分支在线接合,而分支在相对于线性、在线、ssrt的垂直取向上带有易位控制元件。
读取。这种灵活性实现了本文进一步讨论的“棘轮”测序方法和其他方法,诸如基于电压施加的交流模式的“牙线”。
[0111]
图3显示了在易位α-溶血素纳米孔的过程中切割的xpandomer的一个实施例。该生物纳米孔嵌入脂质双层膜中,该膜将两个电解质储库分开并电隔离。典型的电解质具有缓冲至7.0的ph值的1摩尔kcl。当在双层上施加小电压(通常为100mv)时,纳米孔会限制离子电流的流动,并且是电路中的主要电阻。xpandomer报告子代码设计用于提供特定的离子电流阻塞水平,并且可以通过测量离子电流水平序列随着报告子代码序列易位纳米孔来读取序列信息。
[0112]
α-溶血素纳米孔通常是定向的,因此通过进入前庭侧并离开茎侧发生易位。如图3所示,纳米孔被定向以首先从茎侧捕获xpandomer。在某些情况下,与首先进入前庭时相比,此取向可能会导致更少的阻塞伪影。然而,根据本发明,α-溶血素纳米孔可以在任一方向上取向。随着xpandomer移位,报告子进入茎,直到其易位控制元件在茎入口处停止。报告子被保持在茎中,直到tce能够进入并通过茎,然后易位进行到下一个报告子。在该实施例中,通过易位控制部分与tce的解离实现tce进入茎中。有利地,本发明人已经发现由一类新型悬垂peg亚磷酰胺构造的tce基于固有的物理化学和空间特性提供显着改善的易位控制,从而避免依赖于反式作用的易位控制部分的结合和解离。
[0113]
用于ssrt特征设计的新型化合物
[0114]
通常用于自动寡核苷酸合成的亚磷酰胺化学为合成聚合ssrt提供了一种有效且方便的手段。然而,ssrt特征设计的最终潜力受到商业图书馆中可用的亚磷酰胺单体(ppa)的限制。商业ppa主要基于核苷核心结构,并且因此不提供设计更广泛的功能阵列所需的物理化学特性范围,所述特征提高纳米孔读取的效率和准确性。为了解决本领域中的这个缺点,本发明人设计并合成了大量新的ppa单体化合物。重要的是,这些化合物不是基于本领域众所周知的核苷核心结构,并且如上所述,限制了特征设计。
[0115]
如本文所用,缩写“ppa”是指为o-(2-氰乙基)-(n,n-二异丙基)-亚磷酰胺的亚磷酰胺。本领域技术人员容易理解,术语“亚磷酰胺”是指单体前体的结构;在ppa在线聚合成ssrt之后,单体被转化为磷酸二酯连接的寡聚产物。
[0116]
用于制造磷酸二酯主链聚合物的其他方法可用于合成ssrt。因此,与这些化学物质一起使用的单体也可以产生具有非核苷元素的ssrt。其他组装方法可能涉及使用在溶液相或固体支持物上完成的自动或手动组装策略。h-膦酸酯合成和磷酸三酯合成是本领域已知的实例。此外,使用酶促合成的方法可适用于合成ssrt(例如,用于酶促寡核苷酸合成的那些)。在一些实施例中,ssrt的合成可以基于任何上述合成方法的组合。
[0117]
以下是用于指导ppa单体设计的某些原则的简要、非限制性总结。1)磷酸盐间距。设计的化合物保持c3(3个原子)间距,所述间距模拟天然核苷酸主链的间距。在某些实施例中,其他合适的间距包括2至20个原子间距。出乎意料的是,发现原子间距会影响纳米孔易位的速率,从而可以对易位控制进行微调。2)亲水性。根据特定功能的需要,设计化合物以优化ssrt特征的亲水性。一些单体设计基于peg,因为它能够增加水溶性,这是例如报告子代码的重要特性。本发明人能够通过调整peg聚合物的长度,以及通过用甲基醚终止peg聚合物或将1,2,3-三唑引入聚合物中来微调ppa单体的亲水性,这具有意想不到的进一步提高水溶性的作用。3)空间体积。设计并测试了线性、支化、环状和树枝状ppa结构的几种替代
配置,以评估空间体积对通过纳米孔的电流的影响。4)主链中的手性。纳米孔是手性环境。对映体化合物被设计成确定关键纳米孔信号特性是否受到任何影响。5)电荷。除了反磷酸盐电荷外,某些化合物还带有正电荷,例如叔胺或负电荷,例如羧酸。(6)芳族:由多种芳烃和杂芳族结构构成的化合物被掺入到主链中,以确定与纳米孔的相互作用是否产生了期望的信号特性。
[0118]
ppa单体化合物除了减弱纳米孔信号特性(诸如易位速率控制或电流电平控制)外,还会影响xpandomer的物理化学特性。xpandomer作为半合成聚合物,表现出与天然聚合物(例如dna)和合成聚合物两者相关的特性。在一些实施例中,某些ppa单体可以使不期望的xpandomer间相互作用或xpanodmer与sbx工作流程的某些工艺要素之间的相互作用减弱。例如,有可能减少xpandomer自聚集、高阶玻璃或明胶的形成、隔离、被动吸附到容器表面、纳米通道壁或包含膜和纳米孔的制作装置的表面或与其相互作用。
[0119]
已证明当掺入ssrt特征时提供出色功能的一类化合物在本文中称为“悬垂peg”。这些结构基于分子核心,所述分子核心能够以相对于核心的悬垂配置连接一种或多种含peg的聚合物。可以在悬垂的peg化合物的聚合物和梳子之间进行结构类比,其中单个化合物之间的磷酸二酯键形成梳子的底部,并且基于peg的聚合物形成牙齿。有利地,可以针对特定的ssrt特征(例如,聚合物牙齿的间距、长度和组成中的一者或多者)定制悬垂的peg“牙齿”的若干特性。下面的结构1a、2a、3a和4a说明悬垂peg核心结构的四个示例性实施例。
[0120][0121]
在某些非限制性实施例中,x或x
′
可以表示-ch2o-[ch2ch2o-]mo-其中m为1-10且y或y
′
可表示-h、-ch3、、
[0122]
表1a-c列出了用于例如ssrt特征设计的新型亚磷酰胺单体化合物的非限制性集合。参考相关实例来提及每种化合物的合成方案,并且包括每种化合物的具体前体。表征纯化合成化合物的分析数据也列于表1a中。这些化合物可用于合成任何合适的聚合特征,例如ssrt报告子代码、易位控制元件和易位减速特征,如本文进一步详细描述的。表1a还提供了化合物的名称,参考它们在掺入合成聚合物后采用的串联结构。
[0123]
[0124]
[0125]
[0126]
[0127]
[0128]
[0129]
[0130]
[0131]
[0132]
[0133]
[0134]
[0135]
[0136]
[0137]
[0138]
[0139]
[0140]
[0141]
[0142]
[0143]
[0144]
[0145]
[0146]
[0147]
[0148]
[0149]
[0150]
[0151]
[0152]
[0153]
[0154]
[0155]
[0156]
[0157]
[0158]
[0159]
[0160]
[0161]
[0162]
[0163]
[0164]
[0165]
[0166]
[0167]
[0168]
[0169]
[0170][0171]
表1b
[0172]
示例性新型亚磷酰胺(ppa)单体-组b
[0173]
[0174]
[0175][0176]
表1c
[0177]
示例性新型亚磷酰胺(ppa)单体-组c
[0178]
[0179]
[0180]
[0181]
[0182][0183]
基于新型亚磷酰胺单体的易位控制元件(tce)。
[0184]
如本文所讨论的,ssrt的tce特征被设计为阻止xpandomer易位,以便将报告子代码定位在纳米孔孔口内进行测量。本发明的新的亚磷酰胺单体化合物的可用性使得能够设计下一代tce结构,所述结构通过空间位阻、电排斥和与纳米孔的优先相互作用中的一种或多种来控制易位速率。当定位在孔隙孔口处时,tce对离子电流的驱动力的阻力以及克服停滞和恢复易位所必需的随后施加的电压(即电压脉冲)的增加,可以通过调制tce的各种性质(以及在一些实施例中,报告子代码和ssrt的其他元素)例如体积、长度和/或电荷密度来定制。重要的是,由于易位速率受tce固有特性的控制,因此易位控制摆脱了依赖现有技术策略的负担,现有技术策略采用例如基于与可溶性寡核苷酸可逆相互作用的核苷酸杂交策略。
[0185]
在某些实施例中,tce是使用亚磷酰胺方法通过固相合成产生的聚合物,其具有以支化结构(即,“分支”)终止的合适单体结构单元。支化亚磷酰胺是本领域已知的,并且包括从例如glen research和chemgenes商购获得对称和不对称分支。在一个实施例中,tce分支
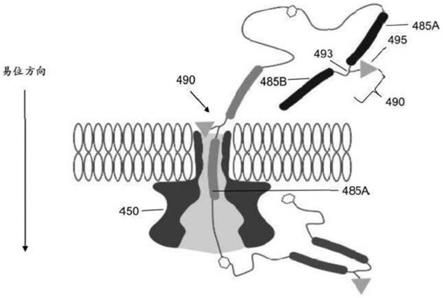
是对称支化ced亚磷酰胺,其中分支的每个臂与报告子代码连接。示例性对称化学分支包括1,2,3-o-三-(磷酸二酯)-丙烷、1,3-双-(5-o-磷酸二酯-戊基酰胺基)-2-o-磷酸二酯-丙烷和1,4,7-o-三-(磷酸二酯)-庚烷。
[0186]
图4以简化形式说明了tce如何通过纳米孔阻止xpandomer易位。此处,xpandomer的每个ssrt包括报告子代码485a和485b,所述报告子代码在tce的分支结构493的臂末端连接到tce 490。在该实施例中,tce 490包括结构495,其具有相对于报告子代码的物理体积更大的物理体积。当tce 490遇到孔隙孔口时,xpandomer易位通过纳米孔桶450被阻止。在某些实施例中,tce的体积和报告子代码的电荷密度(即,阻滞位点的局部电场)都有助于易位阻滞。在暂停期间,报告子代码495a被保持在纳米孔桶中,并以特征性和可检测的方式阻止电流通过所述孔隙。为了克服阻滞,向系统施加电压脉冲,迫使tce进入并通过孔隙。易位然后恢复,直到下一个tce遇到孔隙孔口。
[0187]
为了定制易位控制,可以调整tce的几个结构特性(以及在某些实施例中,ssrt的其他特征)。例如,可以修改tce的长度、体积和电荷密度中的一者或多者,以及纳米孔桶内带电荷元素的空间定位。在一些实施例中,通过将一种或多种悬垂peg亚磷酰胺掺入到聚合结构中来增加tce的体积。例如,tce可以掺入2至30、2至20、3至15或4至14个悬垂peg亚磷酰胺化合物。在其他实施例中,tce可以包括表1a-c中列出的任何合适数量亚磷酰胺化合物和其组合。例如,tce可以以任何顺序包括1到10、2到8、或2或3种不同的亚磷酰胺化合物;在某些实施例中,亚磷酰胺化合物中的至少一者是悬垂peg亚磷酰胺。在某些实施例中,整个tce的长度可包括2至30、2至20、3至15或4至14种亚磷酰胺化合物。在一些实施例中,tce的式可以由(ppa1)
n1
(ppa2)
n2
表示,其中ppa1和/或ppa2表示悬垂peg亚磷酰胺化合物并且n1=1至12和n2=0到10。发明人已经发现,基于该式的tce显著减少了测序错误(例如,插入或缺失事件)并实现了顺序ssrt之间的单脉冲转换。在某些实施例中,tce包括由具有以下序列的亚磷酰胺化合物合成的聚合物:[(1-o-dmt-3-o-ppa-2s-o-mpeg4-丙烷(化合物12b))]
n1
[(1-o-dmt-3-o-ppa-2-(4-me-o-peg3)-1-(et-o-ac)-1,2,3-三唑)-丙烷(化合物35b)]
n2
,其中n1是0至6且n2是6至10。在其他实施例中,tce包括一种或多种可通过uv辐射检测的亚磷酰胺发色团,例如苯并呋喃或含三唑的ppa。
[0188]
在其他实施例中,tce可以包括具有多于两个臂的分支结构。在一种实施例中,亚磷酰胺分支可具有末端支化结构。在该实施例中,分支具有四个臂,其中两个臂连接到报告子代码,并且其中两个臂有助于易位控制。在其他实施例中。可以定制分支以优化诸如尺寸、极性和稳定性等特征。在一个实施例中,分支包括异氰酸酯三聚物。
[0189]
在进一步的实施例中,本发明的tce可以包括两个分支(即,“双分支”tce),其中分支被多个未支化的亚磷酰胺隔开。分支可以是对称的或不对称的结构。不对称结构可以是单一的对映体或外消旋体。此外,外消旋体和/或两种对映体的组合可用于tce中的不同位置。在该实施例中,每个分支促进不同的易位暂停事件,其中第一个可以被称为“代码暂停”,它在纳米孔中保持报告子代码,而其中的第二个可以被称为“时钟”暂停”,它会产生独特的信号,表明检测系统已经“读取”了前面的报告子代码。
[0190]
表2列出了几个示例性tce序列。需要强调的是,本发明并不限于这些特定的实施例,因为技术人员将理解,基于本公开,可以设计广泛的不同tce文库以适应广泛的实验要求。在某些实施例中,以下列出的任何tce序列可以在具有一种或多种间隔化合物,包括c3、
苯并呋喃或peg3的分支的远侧末端终止。表2中的关键将其形式的化合物识别为亚磷酰胺单体。对本领域普通技术人员而言将显而易见的是,描述词“亚磷酰胺”仅适用于单体形式的化合物;适用于多聚体“在线”形式的化合物的描述词列于表1a中。
[0191]
表2
[0192]
示例性易位控制元件(tce)
[0193]
[0194]
[0195]
[0196]
[0197]
[0198][0199]
降低xpandomer易位过程中插入和缺失事件发生率的方法和构建体
[0200]
在其他方面,本发明提供通过xpandomer修饰结合设计到ssrt中的离散易位减速特征(在本文中也称为“d细胞”)的易位控制手段。如本文所公开的,xpandomer在合成后经受几个处理步骤,包括胺修饰步骤。在胺修饰过程中,xpandomer用琥珀酸酐处理,琥珀酸酐与ssrt聚合酶增强区的精胺成分上的仲胺基团(以及在某些情况下,酸切割步骤引入的伯胺基团)反应。胺基的琥珀酰化导致引入带负电荷的半琥珀酸酯基团。通过增加琥珀酰化程度,精胺基增强剂上的负电荷程度同样增加。每个精胺亚磷酰胺成分具有(+3)的净电荷;在标准修饰反应中,每个胺部分的电荷从(+1)变为(-1)。本发明人已经发现了产生不同程度的精胺琥珀酰化的条件,使得精胺成分的净电荷可以从(+3)到(-5)之间变化。在某些条件
下,可能需要增加增强子区的负电荷,以便在施加电压脉冲时增加xpandomer易位的速率(本文称为增强子电迁移率)。值得注意的是,已经发现这种电迁移率可以降低序列读取期间插入错误的百分比,并提高整体测序通量。
[0201]
因此,在某些实施例中,xpandomer处理包括胺修饰步骤。胺(例如精胺)修饰可以通过改变琥珀酰化反应条件,例如反应时间、温度、ph和/或反应中使用的琥珀酸酐的浓度中的一者或多者来实现。在其他实施例中,xpandomer处理进一步包括在胺修饰步骤之后的hepes洗涤步骤中的一者或多者以实现更完全的胺琥珀酰化。
[0202]
在另一方面,本公开提供了一个或多个易位减速特征或区(术语“特征”和“区”在本文中可互换使用),它们是永久带电的,例如叔胺或季胺和/或大体积化合物。可以在聚合酶增强子内或附近的位置处将易位减速特征引入到ssrt中。选择减速特征以便在xpandomer修饰(例如,琥珀酰化)反应期间不会改变。已发现将一个或多个减速特征包含到ssrt中的合适位置会减少缺失错误的百分比,这是由于增强子过度修饰导致易位率增加而引起的。不受理论束缚,据推测,大部分减速特征产生“摩擦”型力,其在遇到纳米孔孔口时降低xpandomer易位速率。通常,减速特征被引入到ssrt中聚合酶增强子和报告子代码之间的位置(即,与增强子相邻)。
[0203]
本发明的易位减速特征可以掺入表1a-1c中列出的亚磷酰胺化合物或市售亚磷酰胺的任何合适数量和组合。在一些实施例中,减速特征包括1至4个不同单体单元的组合。在其他实施例中,减速特征可以包括1或2个不同的单体单元。减速特征的整个长度可以是1至15个单体单元,或在其他实施例中,4至12或6至10个单体单元。表3列出了替代易位减速特征的非限制性实例。表3中的关键将其形式的化合物识别为亚磷酰胺单体。对本领域普通技术人员而言将显而易见的是,描述词“亚磷酰胺”仅适用于单体形式的化合物;适用于多聚体“在线”形式的化合物的描述词列于表1a中。
[0204]
表3
[0205]
示例性易位减速特征
[0206]
[0207][0208][0209]
报告子代码
[0210]
每个ssrt使用tce将报告子代码定位在具有高离子电流电阻的纳米孔区域内。在α溶血素中,该区域是茎。在该区域中,不同的报告子的大小被设计成阻止不同可测量水平的离子流。可以通过从表1a-1c和/或市售文库中设置的亚磷酰胺单体化合物集合中选择特定亚磷酰胺序列来设计报告子。合适的单体化合物也公开于申请人的美国专利号10,457,979中,其通过引用全文并入本文,包括peg3、peg6和c2。
[0211]
每种成分单体化合物根据其在纳米孔中的位置、其位移、其电荷、其与纳米孔的相互作用、其化学和热环境以及其他因素对净电阻做出贡献。
[0212]
报告子代码设计通过平衡测量特征来指导,所述测量特征包括:(i)归一化离子电流(i/io):其中i是离子电流,并且io是开放通道电流;(ii)离子电流噪声:包括多态响应、阻塞、随机尖峰等;和/或(iii)控制部分的释放时间或tce在茎入口处停滞的时间。
[0213]
图5a-5d说明了可如何设计不同的报告子代码以保持沿主链的相似电荷密度,同时提供由于每个独特单体成分占据的不同体积而导致的孔隙阻塞的特征水平。在这些插图中,纳米孔以横截面描绘,并标明了桶和前庭部分。报告子代码以简化形式描述,黑色圆圈表示由代码的亚磷酰胺成分引入的带电荷磷酸二酯部分。图5a说明了线性代码,在该实施例中识别为“零peg代码”,但本发明不旨在如此受限,例如,在某些实施例中,线性代码可以包括peg部分。图5b、图5c和5d说明了可如何从单个支化单体化合物的重复单元构建独特的代码,显示示例性基于peg的结构,例如,悬垂peg化合物。在这些实施例中,三个不同代码的支化部分在孔隙通道中占据不同的体积,并且从而产生可以彼此区分的独特信号。在这些实施例中,沿主链的电荷密度对于每个报告子代码是相同的。然而,如本文进一步讨论的,在其他实施例中,报告子代码可以设计为具有沿着主链的电荷密度梯度。
[0214]
报告离子电流阻塞及其双链体释放时间也通过测量条件来调节,所述测量条件为例如:(i)电压;(ii)电解质;(iii)温度;(iv)压力;和/或(v)ph,如本文进一步描述的。
[0215]
在一些实施例中,与报告子相关的tce也有助于离子流阻塞。
[0216]
对于一组给定的测量条件,报告子可以设计为定义测量动态范围的最小和最大i/io水平。其他报告子可以设计为在动态范围内具有不同的i/io水平。由于每个报告子在纳米孔中暂停,因此测量的i/io水平必须保持足够长时间的稳定并且具有足够低的噪声,以便可以唯一地区分报告子类型。通过选择低阻抗分子(报告子代码聚合物)的主链,将动态范围最大化,通常是那些具有小物理横截面和低线性质量密度的分子。
[0217]
表4列出了示例性报告子代码,但应理解,本发明考虑了掺入本文公开的亚磷酰胺化合物的任何合适组合和数量的报告子代码。表4中的关键将其形式的化合物识别为亚磷酰胺单体。对本领域普通技术人员而言将显而易见的是,描述词“亚磷酰胺”仅适用于单体形式的化合物;适用于多聚体“在线”形式的化合物的描述词列于表1a中。
[0218]
表4
[0219]
示例性报告子代码
[0220]
[0221]
[0222]
[0223]
[0224]
[0225]
[0226][0227]
ssrt和xntp的合成
[0228]
如本文所公开的,对称合成的报告子系链(ssrt)是使用标准自动化寡核苷酸合成方案合成的。图6以简化形式说明了ssrt合成。在步骤a中,将亚磷酰胺固定在固体支持物上;在步骤b中,使用亚磷酰胺偶联在支持物上聚合亚磷酰胺单体;在步骤c中,将对称分支添加到增长的ssrt结构中;在步骤d中,在对称分支的每个臂上聚合对称亚磷酰胺支链;在步骤e中,添加末端叠氮基,使ssrt能够经由点击反应与dntp-2c缀合;在步骤f中,以其最终形式从底物上释放ssrt。
[0229]
在一个实施例中,ssrt合成利用四步迭代过程,所述过程包括1)在固体支持物受控孔隙玻璃珠上合成ssrt聚合物(反映在步骤a-d的卡通片中)。在此步骤中,ssrt使用mermade
tm 12合成仪(可从bioautomation商购)以1μm的规模一次合成一个报告子构建体。首先制备mermade
tm
序列管理器,然后制备亚磷酰胺(例如,制备每种亚磷酰胺的0.067m溶液)。将每个亚磷酰胺的合适偶联时间编程到合成器中。ssrt合成循环基于传统的四步过程:脱三苯甲基化(使用溶剂,例如二氯甲烷中的3%dca),单体偶联(使用溶剂,例如乙腈中的0.25m ett),封端(使用溶剂例如,thf/二甲基吡啶/ac2o(cap a)和thf中的16%甲基咪唑(cap b))和氧化(使用溶剂,例如0.02m i2的thf/吡啶/h2o)。步骤2)通过手动转换对5
′
末端进行功能化,用叠氮取代br(反映在步骤e的卡通片中),即“叠氮修饰”。在该步骤中,合成柱用1ml dcm洗涤,并转移至2ml管中;制备叠氮化物转化溶液(dmf中的100mm碘化钠和100mm叠氮化钠)并在室温将1.6ml加入管中并孵育2小时;然后用1ml dmf冲洗支持物并转移到柱;用2ml dmf、然后是3ml acn和1ml dcm冲洗柱。步骤3)去除氰乙基保护基团。在本步骤中,在acn中制备10%的dea溶液,所述溶液可能包含0.1m硝基甲烷;在真空下,该溶液的稳定流穿过柱至少10
′
;然后用2ml acn然后是1ml dcm冲洗柱。步骤4)切割将ssrt从固体支持物上完全脱保护(反映在步骤f的卡通片中)。在此步骤中,将支持物转移到2ml管中,并且将500μl的30%nh4oh(可能包含100mm硝基甲烷)加入管中,并在55℃孵育30
′
;然后将管在
冰箱中冷却5’;将500μl 40%甲胺加入管中,并在65℃孵育1小时。然后将样品在冰箱中冷冻5分钟;然后通过排空柱并用15ml h2o洗涤来使样品脱盐;然后用100mm teaa从柱上洗脱ssrt并进行量化。
[0230]
图7说明了四种示例性ssrt产物,所述产物可用作形成xatp、xctp、xgtp和xttp的报告子构建体,其中每一种都被设计为在穿过纳米孔时产生独特的电子信号。关键说明了这些特定亚磷酰胺的化学结构,因为它们存在于最终的ssrt报告子构建体中。如本公开通篇所用,r表示叠氮六,q表示精胺,d表示peg6,x表示peg3,l表示c2间隔区,4表示悬垂peg 4,y表示对称化学分支,并且5表示苯并呋喃。在该实施例中,r提供叠氮化物缀合特征,qq聚合物提供增强子特征,y444444444444聚合物提供tce特征,并且ddllldx、ddd44lxxx、ddd4444lldx和xxl444444lllllll聚合物提供四种独特的报告子代码特征。
[0231]
图8a总结了示例性xntp经由ssrt和dntp-2c的环化,经由铜催化的点击反应来形成。如本公开通篇所用,“dntp-2c”是指可切割核苷三氨基磷酸酯类似物,其包括与核苷的杂环部分缀合的1,7-辛二炔基连接基和与三磷酰胺的α氨基磷酸酯部分缀合的5-己炔基连接基。这是包括以下步骤的三步过程:1)点击反应;在一个实施例中,将ssrt和dntp-2c添加到由0.2mm cuso4/0.6mm thpta/1.2mm naasc构成的点击反应溶液中并孵育30
′
;2)标准hplc纯化;以及3)使用本领域认可的方法进行脱盐。dntp-2c化合物的结构和合成的更多细节公开于申请人颁发的美国专利10,301,345中,该美国专利以引用方式全文并入本文。图8b说明详细化学结构的示例性xntp。
[0232]
在某些实施例中,xntp的核碱基可以是非天然类似物,例如7-脱氮杂腺嘌呤、7-脱氮鸟嘌呤等。
[0233]
额外的易位控制手段
[0234]
i.通过易位控制部分的可逆结合进行易位控制
[0235]
在图9a和图9b中以简化形式说明了易位控制的该实施例。在该实施例中,易位速率由可溶性易位结合部分与xpandomer的tce的结合和解离来控制。如图9a所示,第一可溶性易位结合部分与靠近xpandomer的第一报告子代码的第一tce元件的结合形成“代码易位控制复合体”。这种可逆的相互作用使纳米孔中的第一报告子代码停止、暂停或阻滞(为简单起见,这些术语在本文中可互换使用)并产生第一代码所独有的电流变化。这些代码信号用于识别xpandomer的每个单体单元。随后,如图9b所示,第一易位结合部分从第一tce解离,允许第一报告子代码通过,即退出,在它进入孔隙的相对侧上的纳米孔。然后,该易位事件通过第二易位结合部分与xpandomer中第一报告子代码远侧的第二tce的结合而停止。该第二易位控制复合体在本文中被称为“时钟易位控制复合体”。这种易位的中断会产生电流(即“时钟信号”)的变化,表明第一代码区通过纳米孔的完全易位。在某些实施例中,xpandomer的每个单元的时钟信号可以彼此相同或几乎无法区分。本领域技术人员将理解,代码信号本身足以确定xpandomer的序列信息。
[0236]
根据本发明,任何合适的可逆结合配偶体组均可用于易位控制。在一个实施例中,tce包括生物素衍生物,而易位控制部分由链霉亲和素提供。在该实施例中,生物素衍生物可以被工程设计成以比天然生物素低的亲和力结合链霉亲和素。合适的生物素衍生物的一个实例是脱硫生物素(dtb),如图10所示。在其他实施例中,生物素-sa tce系统可以通过例如使用形成较弱生物素-sa复合体的其他生物素类似物和/或使用形成较弱复合体的sa突
变体来控制。
[0237]
ii.通过调整运行条件来进行易位控制
[0238]
已经发现,对基于纳米孔的检测系统中使用的各种条件进行微调可以提高xpandomer易位控制和代码读取的准确性。因此,在其他方面,本公开提供了通过修改以下运行条件中的一者或多者来提高聚合物易位通过纳米孔的速率的手段:
[0239]
a.电压参数
[0240]
离子从本文所述的基于纳米孔的检测系统的顺式腔室到反式腔室的流动是由于跨膜施加电压电位的结果,所述电压电位可互换地称为“读取电压”或“基线电压”。在本发明的一个实施例中,通过改变基线电压来调节xpandomer易位速率。在一些实施例中,基线电压可以在从约40mv至约150mv的范围内。在其他实施例中,基线电压可以在从约90mv至约110mv的范围内。在再其他实施例中,基线电压可以在从约55mv至约75mv的范围内。在一些实施例中,可能需要更高的基线电压以更高的速率捕获报告子代码读段。
[0241]
如本文所讨论的,当靠近报告子代码的tce遇到孔隙的孔口时,xpandomer易位受到阻滞。报告子代码保持在孔隙中,直到施加足够强的电压脉冲以克服由保持在孔隙中的tce结构提供的电阻。因此,在另一个实施例中,通过改变脉冲电压的强度来调节xpandomer易位速率。在一些实施例中,脉冲电压在从约250mv至约2000mv的范围内。在其他实施例中,脉冲电压在从约550mv至约700mv的范围内。同样,电压脉冲的持续时间会影响xpandomer易位的速率。在一些实施例中,电压脉冲的持续时间在约1μs至约50μs的范围内。在其他实施例中,电压脉冲的持续时间在约5μs至约10μs的范围内。在另一个实施例中,可以优化脉冲电压的周期性。在一些实施例中,周期性在从约0.5ms至约20ms的范围内。在再其他实施例中,脉冲电压的周期为约0.5ms至约1.5ms。本领域技术人员将理解,最佳电压脉冲的强度、持续时间和周期性将取决于许多因素,例如tce的力。
[0242]
b.盐
[0243]
流过本文所述的基于纳米孔的检测系统的电流速率可受填充系统的顺式和反式腔室的缓冲剂的盐组成的影响。因此,在某些实施例中,xpandomer易位通过孔隙的速率可以通过盐组成调节。在这些实施例中,可以使用包含任何合适的一价或二价阳离子的盐。在一些实施例中,合适的盐包括但不限于nh4cl、mgcl2、licl、kcl、cscl、nacl和cacl2.在其他实施例中,合适的盐包括其中阴离子为乙酸根的盐。在需要较慢电流的条件下,具有较低离子迁移率的盐,例如licl,可能是有利的。在一些实施例中,反式腔室包含2m nh4cl和具有约0.2m的合适摩尔浓度的第二任选盐,并且顺式腔室包含具有范围在约0.4m至约1m的合适摩尔浓度的nh4cl和具有范围在约0.2m至约0.8m的合适摩尔浓度的第二任选盐。在其他实施例中,可能需要位于这些范围之外的其他摩尔浓度和/或盐的其他组合。
[0244]
c.离液剂
[0245]
在某些其他方面,本发明的基于纳米孔的检测系统的顺式腔室可以包括一种或多种离液剂以改善单个聚合物分析物例如线性化xpandomer的易位。可以使用任何合适的离液剂,例如尿素和/或盐酸胍(gucl)。在一些实施例中,顺式腔室的缓冲组合物包括范围在约200mm至约2m的gucl和/或尿素。
[0246]
d.渗透梯度
[0247]
在其他方面,本发明提供基于纳米孔的检测系统,其中跨膜建立渗透梯度以影响
xpandomer易位通过孔隙的速率。不受理论束缚,假设其中反式腔室中盐和/或其他添加剂的浓度相对于顺式腔室更高的梯度产生朝向纳米孔的水流,从而将xpandomer拉向孔隙。在这些条件下,可以在较低的运行电压下观察到事件频率(例如,代码读取)的速率增加。因此,在一些实施例中,运行条件包括跨膜建立约50%的渗透梯度;例如,在顺式腔室中约1m的盐(和/或其他添加剂)浓度和在反式腔室中约2m的盐(和/或其他添加剂)浓度。在进一步的实施例中,可以采用任何其他合适的渗透梯度。
[0248]
e.溶剂
[0249]
已经发现某些溶剂可以增强xpandomer的溶解性并提高通过纳米孔的易位速率。因此,在某些实施例中,本发明的样品缓冲剂包括一种或多种有机溶剂。合适的溶剂包括但不限于以约1%至约25%的范围使用的3-甲基-2-噁唑烷酮(moa)、dmf、acn、dmso和nmp。
[0250]
f.缓冲剂、添加剂和其他运行条件
[0251]
用于本发明的合适的缓冲剂包括但不限于具有约7.4的ph值的20mm-100mm hepes和具有范围在约25mm至约250mm的摩尔浓度且具有范围在约6至约10的ph值的双三丙烷缓冲剂。在其他方面,本发明的缓冲剂可以包含某些去污剂添加剂,例如己酸钠(nahex),以提高xpandomer易位的速率。在某些实施例中,本发明的样品缓冲剂包含约20mm nahex。其他合适的添加剂包括但不限于稳定剂,诸如edta和氧化还原试剂。任何缓冲剂的粘度也可以通过添加剂诸如peg、甘油、聚蔗糖等改变。
[0252]
在其他方面,xpandomer的易位率可以通过温度来调节。在一些实施例中,运行温度可以在从约4℃至约40℃的范围内。在其他实施例中,运行温度可以在约16℃至约22℃的范围内。
[0253]
可切割延伸寡核苷酸
[0254]
在另一方面,本公开提供用于xpandomer合成的可切割的延伸寡核苷酸(eo)。可切割的设计特征使eo能够在合成之后且在纳米孔分析之前从xpandomer中去除,即从xpandomer上切割下来。当不希望多核苷酸序列通过纳米孔易位时,该功能提供了优势。xpandomer的合成、处理和纳米孔序列分析如已在例如申请人的pct专利申请号pct/us18/67763中描述,其以引用方式全文并入本文。
[0255]
可切割延伸寡核苷酸的一个实施例以简化形式示于图11中。eo的3
′
端被修饰以包括可切割键,例如,酸可切割氨基磷酸酯键,此处由“p-nh”表示。通过可切割连接基连接到eo的核碱基为xpandomer合成提供了游离3
′
羟基,其方向由虚线箭头指示。相同核碱基的碱基部分被修饰以提供纳米孔易位所需的其他特征,例如在本文中也称为“悬垂前导序列”的前导基团。因此,在eo延伸形成xpandomer之后,酸处理会释放寡核苷酸引物,而前导基团和其他接合特征仍然与xpandomer相关联。有利地,发明人已经发现xpandomer合成不受在可切割延伸寡核苷酸上添加悬垂前导序列的影响。
[0256]
棘轮
[0257]
本领域中实施的基于纳米孔的检测系统的一个缺点是由于在连续施加直流电压期间发生的电解质耗尽导致电流随时间消耗。例如,在电解质电路基于亚铁氰化物-铁氰化物氧化还原对的情况下,纳米孔阵列中的每个孔具有有限的体积,并且因此含有有限数量的这些氧化还原离子种类。在直流电压下,一种物质会转化为另一种物质,并将导致电流下降。为了克服电流消耗的这个问题并随着时间保持更平衡的电流,本公开提供了用于使用
基于纳米孔的检测系统检测聚合物分析物的手段,该检测系统替代地依赖于电压施加的交流(ac)模式。这种模式在本文中被称为“棘轮”。棘轮的一般概述示于图12中。图12的顶部面板说明了电压施加的示例性模式;在本实施例中,施加了+70mv的“正向”读取电压,在中间,系统经受+500mv的短暂(5μs)脉冲电压。然后在“正向”读取电压之后是-70mv的“反向”读取电压,并且重复这个+70mv正向读取/500mv脉冲/+70mv正向读取/-70mv反向读取的循环,直到整个聚合分析物穿过纳米孔。棘轮协议的一个显著优势是在每个正向读取-反向读取循环中补充电解质分布。
[0258]
图12的底部面板说明了在棘轮过程中聚合物易位的方向性如何变化。在该实施例中,聚合物分析物的每个单元包括由易位控制元件例如tce 1215分开的两个相同的报告子代码(例如1210a和1210b)。如图12所示,施加+70mm正向运行电压导致聚合物通过孔隙从膜的顺式侧移动到反式侧,直到报告子代码1210a通过由tce 1215诱导的易位暂停被阻滞在孔隙中。由于阻滞的报告子代码1210a引起的通过孔隙的电流变化被读取为信号“l1+”。应用500mv脉冲迫使tce 1215和报告子代码1210b通过孔隙。在5μs脉冲之后,读取电压返回到+70mv,随后串联的下一个报告子代码1220a被其相应的tce 1225阻滞在孔隙中。由于阻滞的报告子代码1220a引起的流过孔隙的电流变化被读取为信号“l2+”。接下来,通过向系统施加-70mm的反向读取电压来使电压反向,导致聚合物易位方向的反转。在此期间,报告子代码1220a被推回通过孔隙到膜的顺侧。易位继续进行,直到tce 1215遇到孔隙,因此易位被阻滞,从而将报告子代码1210b定位在孔隙中以产生测量为电平“l1
‑”
的电流变化。接下来,电压恢复到+70mm正向运行电压,因此聚合物易位的方向反转回顺式到反式方向,直到由于tce 1225的itc效应而发生阻滞,将报告子代码1220a定位在孔隙中。由此产生的电流变化读作电平“l2+”。在此棘轮协议期间,表征聚合物中每个单元的报告子代码被读取三次(报告子代码“a”被读取两次,并且报告子代码“b”被读取一次)。这种冗余为序列读段提供了一种质量控制手段,并提高了所得序列数据的准确性。插入和缺失错误很容易被识别为与预期模式的偏差,例如l1+/l2+/l1-/l2+。
[0259]
尽管图12中描绘的棘轮图案示出了在正向读取电压的“中间”期间施加的脉冲电压,但本发明设想了其他脉冲模式。例如,在一些实施例中,可以在施加正向读取电压之前施加脉冲,而在其他实施例中,可以在正向读取电压结束时,恰好在施加反向读取电压之前施加脉冲。
[0260]
在其他实施例中,棘轮提供用于补偿或校正由于不同报告子代码的电阻中的脉冲和不对称引起的电流消耗中的一者或两者的手段。例如,在一些实施例中,可以增加反向读取电压以补偿由于在正向读取电压期间施加的脉冲造成的电流损失。当不同的报告子代码具有不同的固有电阻时,也可以调整反向读取电压的百分比增加以平衡电流。
[0261]
在棘轮方案的其他变体中,可以改变正向、脉冲和反向电压的顺序。例如,在一个实施例中,棘轮循环可以如下运行:(正向读取电压)、(正向读取电压)、(反向读取电压)。在另一个实施例中,棘轮循环可以如下运行:(正向读取)n(反向读取)n,其中“n”表示由纳米孔测量的聚合物中单体单元的总数。
[0262]
在一个相关的想法中,提出了“牙线”,其中可以在纳米孔中沿其全长读取dna,停止,然后在使电压极性反向时可以在另一个方向上读取(参见,例如,kasianowicz,john j.“nanopores:flossing with dna.”nature materials 3,no.6(2004):355-56.https://
doi.org/10.1038/nmat1143)。这是阵列中效率较低的方法,因为dna聚合物不会被同步捕获或停止,而棘轮是在这些时间尺度上连续向前的过程。
[0263]
基于sbx的诊断方法和试剂盒
[0264]
在另一方面,本发明公开了用于检测和诊断目标样品中的遗传改变/突变的方法和试剂盒,所述目标样品可以是实体组织或体液。遗传改变可以是种系或体细胞突变。本发明可用于与癌症、自身免疫疾病、器官移植排斥、遗传性胎儿异常、病原体和其他合适的病症相关的检测和诊断。
[0265]
实例
[0266]
材料和方法
[0267]
除非另有说明,否则以下材料(具有所示缩写)均得自美国的上述来源。2-苯基-1,3-二恶烷-5-醇,tbdps-cl(叔丁基二苯基氯硅烷)、dmap(4-二甲基氨基吡啶)、(r)-(+)-缩水甘油、(+)-2,3-o-异亚丙基-l-苏糖醇、异山梨醇、4,4
′‑
双(羟基甲基)-2,2
′‑
联吡啶、tbta(三[(1-苄基-1h-1,2,3-三唑-4-基)甲基]胺),来自tci america(porrland,or)。nah(氢化钠)、meoh(甲醇)、甲苯、thf(四氢呋喃)、tbaf(四丁基氟化铵)、dcm(二氯甲烷)、hcl(浓盐酸)、dmso(二甲基亚砜)、na抗坏血酸(抗坏血酸钠)、碳酸氢钠、硫酸铜、炔丙基丙二酸二甲酯、硼氢化锂和乙酸得自sigma-aldrich(st.louis,mo)。dmt-cl(4,4
′‑
二甲氧基三苯甲基氯)和ppa-cl(n,n-二异丙基氨基氰乙基磷酰胺)来自chemgenes corporation(wilmington,ma)。tea(三乙胺)、己烷、乙酸乙酯、edta(乙二胺四乙酸)、乙醚来自emd millipore(billerica,ma)。m-peg4-tos由m-peg4-oh(cat.no.bp-23742)制成。氟[3,2-c]吡啶-4(5h)-酮(combi-blocks,san diego,ca)。
[0268]
高效液相色谱(hplc)在agilent technologies,inc.(santa clara,ca)的prostar helix
tm hplc系统上进行,所述系统由两个泵(prostar 210溶剂输送模块)组成,所述泵具有10ml钛泵头、柱温箱(prostar 510 air oven),设置为292nm的紫外检测器(prostar 320紫外/可见检测器)。该系统由star色谱工作站软件(6.41版)控制。使用的柱是来自imtakt usa(porrland,or)的cadenza guard column system cd-c18(2.0mm x 5mm)。所用的缓冲剂是缓冲剂a(100mm三乙基醋酸铵,ph 7.0)和缓冲剂b(100mm三乙基醋酸铵,ph 7.0,含95%体积的乙腈)。自动固相亚磷酰胺合成在mermade
tm 12合成仪(bioautomation corp,plano,tx)上进行。mermade
tm
的合成溶液购自glen research(sterling,va)。
[0269]
实例1
[0270]
外消旋2-(3,6-二氧杂庚氧基)-1,3-丙二醇的dmt亚磷酰胺的合成悬垂代码2-甘油基peg-2亚磷酰胺[外消旋体]
[0271]
将2-苯基-1,3-二恶烷-5-醇(1,2.7g,15mmol)溶解在30ml无水thf中。添加氢化钠(1.08g,27mmol)以产生醇盐。当鼓泡停止时,将mpeg4-tos(4.94g,18mmol)溶解在10ml thf中并分批加入。将反应升温至40℃并在搅拌下孵育3小时,然后使其降至室温过夜。用1ml meoh淬灭过量的nahh,然后用水稀释并用dcm萃取。在减压下浓缩合并的有机层。将残余物重新悬浮在甲苯中,与剩余的盐分离,并通过快速色谱纯化得到产率为73%的2。
[0272]
将苄基啶保护的2b(3.05g,10.8mmol)溶解在10ml meoh中并添加hcl(0.2ml,2.3mmol)。将溶液孵育20分钟,然后用碳酸氢钠(200mg)中和并在减压下干燥。将残余物重
新悬浮在dcm中,并通过快速色谱纯化得到产率为72%的二醇3。
[0273]
将二醇3(1.52g,7.8mmol)溶解在20ml dcm和tea(2.17ml,15.6mmol)中。在90分钟内分批加入dmt-cl(1.85g,5.46mmol)在10ml dcm中的溶液以使单三苯甲基化最大化。添加meoh(1ml)并将反应在减压下干燥。将残余物重新悬浮在甲苯中并与盐分离,然后通过快速色谱纯化得到以产率为48%的单三苯甲基产物4以及回收的起始二醇。
[0274]
将单三苯甲基4(1.84g,3.7mmol)溶解在10ml dcm和tea(1.03ml 7.4mmol)中。添加ppa-cl(1.05g,4.4mmol)并将反应孵育15分钟。将反应在减压下干燥并重新悬浮在含有1%tea的甲苯中,然后通过快速色谱纯化。得到产率79%为亚磷酰胺5(2.03g,2.9mmol)并通过1h和31p nmr确认。
[0275][0276]
实例2
[0277]
对映体2-(3,6-二氧杂庚氧基)-1,3-丙二醇(12b)的dmt亚磷酰胺(悬垂代码2-甘油基peg-2亚磷酰胺[对映体]的合成
[0278]
将2,3-异亚丙基-sn-甘油6溶解在无水dcm和tea中。添加dmap和tbdps-cl。用dcm从水中萃取反应并通过快速色谱纯化以得到产物7。
[0279]
将甲硅烷基醚7溶解在meoh中并添加hcl。将溶液孵育20分钟,然后用碳酸氢钠中和并在减压下干燥。将残余物重新悬浮在dcm中,并通过快速色谱纯化得到二醇8。
[0280]
将二醇8溶解在dcm和tea中。分批加入dmt-cl的dcm溶液。添加meoh并将反应在减压下干燥。将残余物重新悬浮在甲苯中并与盐分离,然后通过快速色谱纯化得到以单三苯甲基产物9。
[0281]
将仲醇9溶解在无水thf中。添加氢化钠以产生醇盐。当鼓泡停止时,将mpeg4-tos
溶解在thf中并分批加入。将反应升温至40℃并在搅拌下孵育3小时,然后使其降至室温过夜。用1ml meoh淬灭过量的nah,然后用水稀释并用dcm萃取。在减压下干燥合并的有机层。将残余物重新悬浮在甲苯中,与剩余的盐分离,并通过快速色谱纯化得到10。
[0282]
将mpeg4醚10b重新悬浮在thf中并添加tbaf。在减压下浓缩反应并通过快速色谱法纯化以得到11。
[0283]
将dmt peg4醇11b溶解在dcm和tea中。添加ppa-cl并将反应孵育15分钟。将反应在减压下干燥并重新悬浮在含有1%tea的甲苯中,然后通过快速色谱纯化。分离亚磷酰胺12并通过1h和31p nmr确认。
[0284][0285]
实例3
[0286]
对映体1-(3,6-二氧杂庚氧基)-2,3-丙二醇(16)的dmt亚磷酰胺的合成悬垂代码1-甘油基peg-2亚磷酰胺
[0287]
将2,3-异亚丙基-sn-甘油6溶解在无水thf中。添加氢化钠以产生醇盐。当鼓泡停止时,将mpeg2-tos(broadpharm cat.no.bp-2-982)溶解在thf中并分批加入。将反应孵育24小时。用meoh淬灭过量的nah,然后用水稀释并用dcm萃取。在减压下浓缩合并的有机层。将残余物重新悬浮在甲苯中,与剩余的盐分离,并通过快速色谱纯化得到13。
[0288]
将peg2产物13溶解在meoh中并添加hcl。将溶液孵育20分钟,然后用碳酸氢钠中和并在减压下干燥。将残余物重新悬浮在dcm中,并通过快速色谱纯化得到二醇14。
[0289]
将二醇14溶解在dcm和tea中。分批加入dmt-c1的dcm溶液。添加meoh并将反应在减压下干燥。将残余物重新悬浮在甲苯中并与盐分离,然后通过快速色谱纯化得到以单三苯甲基产物15。
[0290]
将单dmt 15溶解在dcm和tea中。添加ppa-cl并将反应孵育15分钟。将反应在减压下干燥并重新悬浮在含有1%tea的甲苯中,然后通过快速色谱纯化。分离亚磷酰胺16并通过1h和31p nmr确认。
[0291][0292]
实例4
[0293]
1-(5h-呋喃并[3,2-c]吡啶-4-酮)-2,3-丙二醇(20)的dmt亚磷酰胺的合成悬垂代码1-甘油基杂环亚磷酰胺
[0294]
将(r)-(+)-缩水甘油17溶解在dcm和tea中。分批加入dmt-cl的dcm溶液。添加meoh并将反应在减压下干燥。将残余物重新悬浮在甲苯中并与盐分离,然后通过快速色谱纯化得到dmt醚18。
[0295]
将dmt醚18溶解在无水dmf中。添加氢化钠以产生醇盐。当鼓泡停止时,将氟[3,2-c]吡啶-4(5h)-酮溶解在thf中并分批加入。将反应带至100℃并在搅拌下孵育12小时。用meoh淬灭过量的nah,然后用水稀释并用dcm萃取。在减压下浓缩合并的有机层。将残余物重新悬浮在甲苯中,与剩余的盐分离,并通过快速色谱纯化得到19。
[0296]
将仲醇19溶解在dcm和tea中。添加ppa-cl并将反应孵育15分钟。将反应在减压下干燥并重新悬浮在含有1%tea的甲苯中,然后通过快速色谱纯化。分离亚磷酰胺20并通过1h和31p nmr确认。
[0297][0298]
实例5
[0299]
异山梨醇(23)的在线代码dmt亚磷酰胺的合成双仲醇代码主链
[0300]
将异山梨醇21溶解在dcm和tea中。分批加入dmt-cl的dcm溶液。添加meoh并将反应在减压下干燥。将残余物重新悬浮在甲苯中并与盐分离,然后通过快速色谱纯化得到单三苯甲基22。
[0301]
将单三苯甲基22溶解在dcm和tea中。添加ppa-c1并将反应孵育15分钟。将反应在减压下干燥并重新悬浮在含有1%tea的甲苯中,然后通过快速色谱纯化。分离亚磷酰胺23并通过1h和31p nmr确认。
[0302][0303]
实例6
[0304]
联吡啶(26)的在线代码dmt亚磷酰胺的合成双伯醇代码主链
[0305]
将4,4
′‑
双(羟甲基)-2,2
′‑
联吡啶24溶解在dcm和tea中。分批加入dmt-cl的dcm溶液。添加meoh并将反应在减压下干燥。将残余物重新悬浮在甲苯中并与盐分离,然后通过快速色谱纯化得到单三苯甲基25。
[0306]
将单三苯甲基25溶解在dcm和tea中。添加ppa-c1并将反应孵育15分钟。将反应在减压下干燥并重新悬浮在含有1%tea的甲苯中,然后通过快速色谱纯化。分离亚磷酰胺26并通过1h和31p nmr确认。
[0307][0308]
实例7
[0309]
2-烷基三唑的悬垂代码dmt亚磷酰胺的合成peg-21,3-丙二醇(31a)。悬垂三唑peg代码
[0310]
将炔丙基丙二酸二甲酯27滴加到硼氢化锂在乙醚中的冷悬浮液中。然后将反应温热至室温,并且孵育过夜。用甲醇、然后水和乙酸淬灭反应。用乙醚萃取溶液,在减压下浓缩合并的有机层。通过快速色谱法纯化粗制材料得到二醇28。
[0311]
将二醇28溶解在dcm和tea中。分批加入dmt-c1的dcm溶液。添加meoh并将反应在减压下干燥。将残余物重新悬浮在甲苯中并与盐分离,然后通过快速色谱纯化得到dmt醇29。
[0312]
将dmt醇29溶解在dmso中并添加叠氮化物(cat.no.bp-20988broadpharm)。单独地,将tbta溶解在dmso中并且合并抗坏血酸钠和硫酸铜。在搅拌下将tbta溶液分批添加至炔烃/叠氮化物溶液中。孵育45分钟后,用edta淬灭反应。将溶液用水稀释并用乙酸乙酯萃取,然后在减压下浓缩有机层并通过快速色谱法纯化以得到30。
[0313]
将1,2,3-三唑30a溶解在dcm和tea中。添加ppa-cl并将反应孵育15分钟。将反应在减压下干燥并重新悬浮在含有1%tea的甲苯中,然后通过快速色谱纯化。分离亚磷酰胺31并通过1h和31p nmr确认。
[0314][0315]
实例8
[0316]
悬垂peg-2、peg-4和peg-6亚磷酰胺[异构体纯](35a)的合成
[0317]
将1-o-tbdps-3-o-dmtr-丙烷-1,2,3-三醇9(来自实例2)溶解在无水thf中。添加氢化钠以产生醇盐。当鼓泡停止时,分批加入甲苯磺酸酯(经由cat.no.bp-21657的甲苯磺酰化制备,broadpharm)。将反应在搅拌下孵育48小时。用水淬灭过量的nah,然后将溶液转移到分液漏斗中并用乙酸乙酯萃取。将合并的有机层用盐水洗涤,经硫酸钠干燥,过滤并在减压下浓缩。将残余物重新悬浮在甲苯中,与剩余的盐分离,并通过快速色谱纯化得到32。
[0318]
将炔烃32a重新悬浮在thf中并添加tbaf。在减压下浓缩反应并通过快速色谱法纯化以得到33。
[0319]
向炔烃33a在dmso中的溶液中添加2-叠氮基乙酸乙酯。在单独的小瓶中,将抗坏血酸钠溶解在水中并添加dmso,然后添加1m cuso4,以制备催化剂混合物。在10分钟内将催化剂混合物滴加到炔烃/叠氮化物溶液中。完成后,用0.5m edta淬灭并搅拌15分钟。用水稀释并用乙酸乙酯萃取三次。用盐水洗涤合并的有机萃取物并经硫酸钠干燥。将残余物重新悬浮在甲苯中,并通过快速色谱纯化得到34a。
[0320]
将三唑34a溶解在dcm和tea中。添加ppa-cl并将反应孵育15分钟。将反应在减压下干燥并重新悬浮在含有1%tea的甲苯中,然后通过快速色谱纯化。分离亚磷酰胺35并通过1h和31p nmr确认。
[0321][0322]
实例9
[0323]
基于peg的报告子代码(37b)的合成
[0324]
向产物33在dmso中的溶液中添加2-(乙酰氧基甲基)-2-(叠氮甲基)丙烷-1,3-二
乙酸二酯(通过将2-(溴甲基)-2-(羟甲基)-1,3-丙二醇溶解在dmf中,然后加入nan3制备)。将反应在110℃孵育、浓缩并通过快速色谱法纯化。分离产物后,将残余物与乙酸酐反应并纯化)。在单独的小瓶中,将抗坏血酸钠溶解在水中并添加dmso,然后添加1m cuso4,以制备催化剂混合物。在10分钟内将催化剂混合物滴加到炔烃/叠氮化物溶液中。完成后,用0.5m edta淬灭并搅拌15分钟。用水稀释并用乙酸乙酯萃取三次。用盐水洗涤合并的有机萃取物并经硫酸钠干燥。将残余物重新悬浮在甲苯中,并通过快速色谱纯化得到36。
[0325]
将伯醇36b溶解在dcm和tea中。添加ppa-cl并将反应孵育15分钟。将反应在减压下干燥并重新悬浮在含有1%tea的甲苯中,然后通过快速色谱纯化。分离亚磷酰胺37并通过1h和31p nmr确认。
[0326][0327]
实例10
[0328]
基于peg的报告子代码(40)的合成
[0329]
将1-o-tbdps-3-o-dmtr-丙烷-1,2,3-三醇9(来自实例2)溶解在无水thf中。添加氢化钠以产生醇盐。当鼓泡停止时,将甲苯磺酸酯(通过cat.no.bp-21036,broadpharm的顺序甲苯磺酸化和甲硅烷基保护来制备)溶解在thf中并分批加入。将反应在搅拌下孵育过夜。用水淬灭过量的nah,并用dcm萃取。在减压下干燥合并的有机层。将残余物重新悬浮在甲苯中,与剩余的盐分离,并通过快速色谱纯化得到38。
[0330]
双甲硅烷基醚38重新悬浮在thf中并添加tbaf。在减压下浓缩反应并通过快速色谱法纯化。将纯化的材料重新悬浮在dcm中并添加tea。滴加bzcl。将反应在室温搅拌,直到完成。在减压下浓缩反应并通过快速色谱法纯化以得到醇39。
[0331]
将醇39溶解在dcm和tea中。添加ppa-cl并将反应孵育15分钟。将反应在减压下干燥并重新悬浮在含有1%tea的甲苯中,然后通过快速色谱纯化。分离亚磷酰胺40并通过1h和31p nmr确认。
[0332][0333]
实例11
[0334]
基于双peg的报告子代码(45a-d和47e-i)的合成
[0335]
o,o
′‑
亚苄基季戊四醇(41,cat.no.b2682,tci)溶解在无水thf中。添加氢化钠以产生醇盐。当鼓泡停止时,将甲苯磺酸盐(经由cat.no.bp-21397,broadpharm或cat.no.bp-21657,broadpharm的甲苯磺酰化制备)溶解在thf中并分批加入。将反应在搅拌下孵育过夜。用水淬灭过量的nah,并用dcm萃取。在减压下干燥合并的有机层。将残余物重新悬浮在甲苯中,与剩余的盐分离,并通过快速色谱纯化得到42a-h。
[0336]
将产物42a-h溶解在meoh中并添加hcl。将反应在室温孵育过夜,然后用碳酸氢钠中和。将其减压浓缩并通过快速色谱纯化以得到43a-h。
[0337]
将产物43a-h溶解在dcm和tea中。分批加入dmt-c1的dcm溶液。将反应在减压下干燥。将残余物重新悬浮在甲苯中并与盐分离,然后通过快速色谱纯化得到单三苯甲基44a-h。
[0338]
将产物44a-d溶解在dcm和tea中。添加ppa-c1并将反应孵育15分钟。将反应在减压下干燥并重新悬浮在含有1%tea的甲苯中,然后通过快速色谱纯化以得到亚磷酰胺45a-d。
[0339]
向产物44(e-h)在dmso中的溶液中加入2-叠氮基乙酸乙酯。在单独的小瓶中,将抗坏血酸钠溶解在水中并添加dmso,然后添加1m cuso4,以制备催化剂混合物。在10分钟内将催化剂混合物滴加到炔烃/叠氮化物溶液中。完成后,用0.5m edta淬灭并搅拌15分钟。用水稀释并用乙酸乙酯萃取三次。用盐水洗涤合并的有机萃取物并经硫酸钠干燥。将残余物重新悬浮在甲苯中,并通过快速色谱纯化得到46e-h。
[0340]
将产物46e-h溶解在dcm和tea中。添加ppa-cl并将反应孵育15分钟。将反应在减压下干燥并重新悬浮在含有1%tea的甲苯中,然后通过快速色谱纯化以得到亚磷酰胺47e-h。
[0341][0342]
实例12
[0343]
基于peg的报告子代码(52)的合成和测试
[0344]
将2,2-双(溴甲基)-1,3-丙二醇(48,cat.no.d1808,tci)溶解在dmf中并添加nan3。将反应在110℃孵育、浓缩并通过快速色谱法纯化以得到产物49。
[0345]
向49在dmso中的溶液中加入苯甲酸2-(2-(丙-2-炔-1-基氧基)乙氧基)乙酯(通过商业醇前体的苯甲酰化来制备)。在单独的小瓶中,将抗坏血酸钠溶解在水中并添加dmso,然后添加1m cuso4,以制备催化剂混合物。在10分钟内将催化剂混合物滴加到炔烃/叠氮化物溶液中。完成后,用0.5m edta淬灭并搅拌15分钟。用水稀释并用乙酸乙酯萃取三次。用盐水洗涤合并的有机萃取物并经硫酸钠干燥。将残余物重新悬浮在甲苯中,并通过快速色谱纯化得到50。
[0346]
将产物50溶解在dcm和tea中。分批加入dmt-cl的dcm溶液。将反应在减压下干燥。将残余物重新悬浮在甲苯中并与盐分离,然后通过快速色谱纯化得到51。
[0347]
将产物51溶解在dcm和tea中。添加ppa-cl并将反应孵育15分钟。将反应在减压下干燥并重新悬浮在含有1%tea的甲苯中,然后通过快速色谱纯化以得到亚磷酰胺52。
[0348][0349]
实例13
[0350]
基于peg的报告子代码(62)的合成和测试
[0351]
将2-(溴甲基)-2-(羟甲基)-1,3-丙二醇(53,cat.no.b4057,tci)溶解在dmf中并添加nan3。将反应在110℃孵育、浓缩并通过快速色谱法纯化以得到产物54。
[0352]
将产物54溶解在dcm和tea中。添加苯甲酰氯。将溶液在室温孵育过夜。用dcm从水中萃取反应并通过快速色谱纯化以得到产物55,双-bz,将其与任何单-或三-保护的物种分离并划分。
[0353]
将一部分产物55溶解在dcm和tea中。分批加入dmt-cl的dcm溶液。添加meoh并将反应在减压下干燥。将残余物重新悬浮在甲苯中并与盐分离,然后通过快速色谱纯化得到三苯甲基化56。
[0354]
将41溶解在无水thf中。添加氢化钠以产生醇盐。当鼓泡停止时,将炔丙基溴溶解在thf中并分批加入。用1ml meoh淬灭过量的nah,然后用水稀释并用dcm萃取。在减压下干燥合并的有机层。将残余物重新悬浮在甲苯中,与剩余的盐分离,并通过快速色谱纯化得到57。
[0355]
将产物57溶解在meoh中并添加hcl。将反应在室温孵育过夜,然后用碳酸氢钠中
和。将其在减压下浓缩并通过快速色谱法纯化以得到58。
[0356]
将产物58溶解在dcm和tea中。添加苯甲酰氯,并将反应孵育60分钟。用dcm从水中萃取反应并通过快速色谱纯化以得到产物59。
[0357]
将产物56和59溶解在9∶1 dmso:h2o中。添加tbta、抗坏血酸钠和硫酸铜的溶液并将反应孵育60分钟。用dcm从水中萃取反应并通过快速色谱纯化以得到产物60。
[0358]
将产物60和55溶解在9∶1 dmso:h2o中。添加tbta、抗坏血酸钠和硫酸铜的溶液并将反应孵育60分钟。用dcm从水中萃取反应并通过快速色谱纯化以得到产物61。
[0359]
将产物61溶解在dcm和tea中。添加ppa-cl并将反应孵育15分钟。将反应在减压下干燥并重新悬浮在含有1%tea的甲苯中,然后通过快速色谱纯化。分离亚磷酰胺62并通过1h和31p nmr确认。
[0360][0361]
实例14
[0362]
对映体(9s,10s)-2,5,8,11,14,17-六氧杂十八烷-9,10-二醇(悬垂代码c2双-peg-2亚磷酰胺[对映体])(67)dmt亚磷酰胺的合成
[0363]
将(+)-2,3-o-异亚丙基-l-苏糖醇63在无水dmf中的溶液缓慢加入nah在无水dmf中的混合物中(注意:h2气体剧烈放出)。当鼓泡停止时,将mpeg2-tos(broadpharm cat.no.bp-20983)溶解在dmf中并分批加入以在环境温度下搅拌过夜。将反应混合物倒在水中,用乙酸乙酯萃取并通过快速色谱纯化,得到64。
[0364]
将产物64溶解在meoh中并添加hcl。将溶液孵育20分钟,然后用碳酸氢钠中和并在减压下干燥。将残余物重新悬浮在乙酸乙酯中,并通过快速色谱纯化得到65。
[0365]
将产物65溶解在dcm和tea中。分批加入dmt-c1的dcm溶液。将反应在减压下干燥。将残余物重新悬浮在甲苯中并与盐分离,然后通过快速色谱纯化得到66。
[0366]
将产物66溶解在dcm和tea中。添加ppa-cl并将反应孵育15分钟。将反应在减压下干燥并重新悬浮在含有1%tea的甲苯中,然后通过快速色谱纯化。将亚磷酰胺67通过1h和31p nmr来确认。
[0367][0368]
实例15
[0369]
1-o-dmt-3-o-ppa-2,2-双(me-o-mpeg4)-丙烷(72)的合成
[0370]
向季戊四醇(68,cat.no.p0039,tci)在dmf中的溶液中添加对甲苯磺酸。将反应用三乙胺中和,浓缩并通过快速色谱纯化,得到69。
[0371]
将产物69添加到edc-hcl、dmap和乙酰丙酸在thf中的搅拌溶液中,并在环境温度下搅拌过夜。将溶液浓缩并通过快速色谱法纯化以得到70。
[0372]
将产物70溶解在dcm和tea中。分批加入dmt-cl的dcm溶液。将反应在减压下干燥。将残余物重新悬浮在甲苯中并与盐分离,然后通过快速色谱纯化得到71。
[0373]
将产物71溶解在dcm和tea中。添加ppa-cl并将反应孵育15分钟。将反应在减压下干燥并重新悬浮在含有1%tea的甲苯中,然后通过快速色谱纯化。将亚磷酰胺72通过1h和31p nmr来确认。
[0374][0375]
实例16
[0376]
基于peg的报告子代码的合成和测试
[0377]
在本实例中,报告子代码是用基于peg的亚磷酰胺合成的;值得注意的是,这些代码不包含核苷酸。表5中列出了四个示例性报告子代码。
[0378]
表5
[0379]
代码指示符序列l1dddlllll2dxxx3ll3xxlll3dxll44xxxlllll4dxxl4444llxxll
[0380]
关键:d=peg6:x=peg3;l=c2;3=点击peg-2;4=悬垂peg-4
[0381]
通过合成源自hiv-2基因组的序列的100mer xpandomer拷贝来评估每个代码的级别区分(即可区分的电子信号)和易位时间,所述序列掺入xntp,其中四个xntp中的每一个都包含来自表4所列组的唯一代码。xpandomer合成、处理和纳米孔序列分析如申请人的pct专利申请号pct/us18/67763所述来实施,所述专利以引用方式全文并入本文。作为对照,对掺入不同已知代码的相同hiv-2序列的xpandomer拷贝进行并行测序。图13a(对照-旧代码)和13b(测试-新的、基于peg的对照)示出了说明每个代码的级别区分和易位时间的代表性轨迹。
[0382]
为了评估xpandomer序列信息的准确性,通过显示来自sbx反应的序列读段群的直方图显示来分析序列数据。分析软件将每个序列读段与模板序列对准,并修整与正确模板序列不对准的读段末端的序列范围。100mer模板的sbx测序的代表性直方图呈现在图14a(对照)和图14b(新的,基于peg的代码)中。值得注意的是,掺入无核苷酸的基于peg的代码的xpandomer产生了该模板的高度准确的序列读段。
[0383]
实例17
[0384]
对基于peg的tce-2个代码级别进行易位控制
[0385]
在本实例中,通过使用sbx方案对由tg二核苷酸重复序列组成的简单60mer模板进行测序,评估了掺入悬垂peg亚磷酰胺的tce的易位控制。xatp和xctp底物均设计为掺入以下tce:y22222222222255,其中“y”表示对称亚磷酰胺分支;“2”表示悬垂peg2;且“5”表示苯并呋喃。xatp底物被设计为掺入以下报告子代码:ddddddllll,其中“d”表示peg6,且“l”表示c2。xctp底物被设计为掺入以下报告子代码:ddddxx44xxdl,其中“x”表示peg3,且“4”表示悬垂peg4。
[0386]
为了产生60mer模板的xpandomer拷贝,使用4pm的延伸寡核苷酸和250pm的每个xntp进行引物延伸反应。10μl延伸反应包括以下试剂:50mm triscl、ph 8.84、200mm nh4oac、20%peg8k、5%nms、0.75nmol聚磷酸盐pp-60.20、2μg ssb、0.5m尿素、5mm pem添加剂(合适的聚合酶增强分子公开在申请人的未决pct专利申请号pct/us18/67763中,所述专利通过引用全文并入本文)和1.2μg纯化的重组dna聚合酶(dpo4聚合酶的合适工程变体公开在申请人的pct专利申请号wo2017/087281、pct/us2018/030972和pct/us1864794中,所述专利申请通过引用全文并入本文)。在42℃进行延伸反应30分钟。
[0387]
接下来使用sbx方案对延伸反应的xpandomer产物进行测序。简而言之,将受约束的xpandomer产物切割以产生线性化的xpandomer。这通过首先淬灭延伸反应并用2m琥珀酸酐对xpandomer进行胺修饰来实现。然后通过用11.7m dcl在23℃处理样品30分钟来切割xpandomer的氨基磷酸酯键。通过乙醇沉淀纯化线性化的xpandomer,并重新悬浮在补充有34%acn和15%dmf的缓冲剂中。
[0388]
对于测序,将xpandomer添加到2.8m nh4cl、1.2m guancl、20mm nahex、10%dmf、2mm edta和20mm hepes ph 7.4的样品缓冲剂中。通过将α-溶血素插入缓冲剂中的dphpe/十六烷双层成员中来制备蛋白质纳米孔,所述缓冲剂含有2m nh4cl和100mm hepes,ph 7.4。该实验在顺式孔中使用了0.4m nh4cl、600mm guancl和100mm hepes,ph 7.4的缓冲剂,并且在检测系统的反式孔中使用了2m nh4cl和100mm hepes,ph 7.4。将xpandomer样品加热至70℃达2分钟,完全冷却,然后将2μl样品添加至顺式孔中。运行的电压参数如下:60mv/300mv/10μs/2ms(读段电压/脉冲电压/脉冲电压持续时间/脉冲频率)。数据经由labview采集软件获得。该运行的代表性迹线如图15所示。
[0389]
如图15中叠加在迹线上方的级别数所示,本实验中的检测系统正确读取了所有报告子代码,证明了100%的准确性。缺失或插入错误不存在验证了基于悬垂peg的tce作为能够严格调节xpandomer易位的结构的功效。值得注意的是,通过本实验中使用的基于悬垂peg的tce,在单个电压脉冲之后观察到两个代码级别之间的转换。利用单个电压脉冲在xpandomer的序列报告子代码之间转换的能力是本领域的重大进步,并且能够实现更高的测序通量。
[0390]
实例18
[0391]
通过扩增基于悬垂peg的tce-4个代码级别进行测序
[0392]
在本实例中,通过使用sbx方案对由序列重复序列catg组成的60mer模板进行测序,评估了对掺入悬垂peg亚磷酰胺的tce的易位控制。所有xntp底物均设计为掺入以下tce:y444444444444455,其中“y”表示对称亚磷酰胺分支;“4”表示悬垂peg4;且“5”表示苯并呋喃。xatp底物被设计为掺入以下报告子代码:ddddddlldx;xctp底物被设计为掺入以下
报告子代码:ddddddllll;xttp底物被设计为掺入以下报告子代码:dddddd44lxxx;并且xgtp底物被设计为掺入以下报告子代码:ddddxxl444444xllll,其中“d”表示peg6,“l”表示c2,“x”表示peg3,并且“4”表示悬垂peg4。
[0393]
为了产生60mer模板的xpandomer拷贝,使用4pm的延伸寡核苷酸和1000pm的每个xntp末端进行引物延伸反应。10μl延伸反应包括以下试剂:50mm triscl、ph 8.84、200mm nh4oac、20%peg8k、10%nmp、3nmol多磷酸盐pp-60.20、2μg ssb、1m尿素、10mm pem添加剂和1.8μg纯化重组dna聚合酶。延伸反应在37℃进行30分钟。
[0394]
接下来使用sbx方案对延伸反应的xpandomer产物进行测序。简而言之,将受约束的xpandomer产物切割以产生线性化的xpandomer。这通过首先淬灭延伸反应并用2m琥珀酸酐对xpandomer进行胺修饰来实现。然后通过用11.7m dcl在23℃处理样品30分钟来切割xpandomer的氨基磷酸酯键。通过乙醇沉淀纯化线性化的xpandomer,并重新悬浮在补充有34%acn和15%dmf的缓冲剂中。
[0395]
对于测序,将xpandomer添加到0.8m nh4cl、1.2m guancl和200mm hepes,ph 7.4的样品缓冲剂中。通过将α-溶血素插入缓冲剂中的dphpe/十六烷双层成员中来制备蛋白质纳米孔,所述缓冲剂含有2m nh4cl和100mm hepes,ph 7.4。该实验在顺式孔中使用了0.4m nh4cl、600mm guancl和100mm hepes,ph 7.4的缓冲剂,并且在反式孔中使用了2m nh4cl和100mm hepes,ph 7.4。将xpandomer样品加热至70℃达2分钟,完全冷却,然后将2μl样品添加至顺式孔中。运行的电压参数如下:70mv/650mv/6μs/1.5ms(读段电压/脉冲电压/脉冲电压持续时间/脉冲频率)。数据经由labview采集软件获得。该运行的代表性迹线如图16所示。
[0396]
图16中叠加在迹线上方的级别数对应于由xctp代码(l1)、xttp代码(l2)、xatp代码(l3)和xgtp代码(l4)生成的信号。在本实验中,检测系统正确读取了所有报告子代码,证明了100%的准确性。缺失和插入错误不存在强调了基于悬垂peg的tce在瞬时暂停孔通道中的报告子代码以在“读段电压”期间产生准确的信号读段并允许在“脉冲电压”期间恢复易位方面的功效。同样,使用本实验中使用的基于悬垂peg的tce,通过单个电压脉冲实现了顺序报告子代码之间的转换。值得注意的是,在使用以下电压参数的相同xpandomer样品的等分试样的不同运行中观察到相同水平的准确性:65/700/6/1.5;65/650/6/1.5;60/650/6/1.5和60/600/6/1.5。进一步观察到,代码读段的通量会受到各种电压参数的影响。
[0397]
实例19
[0398]
通过扩增具有基于悬垂peg的tce的复杂模板进行测序在本实例中,通过使用sbx方案对复杂100mer模板进行测序,评估了掺入悬垂peg亚磷酰胺的tce的易位控制。每个xntp底物都用以下tce合成:y22222222222255,其中“y”表示对称亚磷酰胺分支;“2”表示悬垂peg2;且“5”表示苯并呋喃。xntp底物是用以下报告子代码合成:(xc)ddddddllll,其中“d”表示peg6,“l”表示c2;(xt)dddddd44ldx,其中“x”表示peg3,并且“4”表示悬垂peg4;(xa)ddddxx44xxdl;和(xg)ddddxxl。
[0399]
为了产生100mer模板的xpandomer拷贝,使用1pmol的xatp和xctp以及1.5pmol的xgtp和xttp进行固态引物延伸反应(其中延伸寡核苷酸共价结合到芯片底物上的固态xpandomer合成描述于申请人的临时专利申请号62/826,805中,所述专利申请通过引用全文并入本文)。50μl延伸反应包括以下试剂:50mm triscl、ph 8.84、200mm nh4oac、20%
guancl/200mm hepes,ph 7.4)中。
[0408]
通过将α-溶血素插入2m nh4cl和100mm hepes,ph 7.4的缓冲剂中的dphpe/十六烷双层成员中来制备蛋白质纳米孔。对顺式孔灌注含有0.4m nh4cl、600mm guancl、100mm hepes;ph 7.4的缓冲剂,并且对反式孔灌注含有2m nh4cl、100mm hepes;ph值7.4的缓冲剂。将xpandomer样品加热至70℃2分钟,完全冷却并涡旋,然后将2μl等分试样加入顺式孔中。电压参数运行如下:60mv/650mv/6μs/1.0ms(读段电压/脉冲电压/脉冲电压持续时间/脉冲频率)。数据经由labview采集软件获得。该运行的代表性迹线如图18所示。
[0409]
如图18所示,在本实验中,检测系统正确读取了所有报告子代码的序列,证明了100%的准确性。缺失或插入错误的不存在再次验证了基于悬垂peg的tce作为能够高度可靠地读取聚合模板序列的结构的功效。同样,在这些条件下对均聚物的运行进行了准确测序,从而强调了具有基于悬垂peg的tce的sbx的准确性。
[0410]
实例21
[0411]
通过扩增具有基于悬垂peg的tce和d细胞的复杂222mer模板进行测序
[0412]
在本实例中,通过使用sbx方案对复杂222mer模板进行测序,评估了对基于悬垂peg的tce和d细胞特征的易位控制。合成每一xntp底物以包含以下tce:y(32)(32)(32)(32)(32)(32)(61)(61)(61)(61)(61)(61),其中“y”表示对称亚磷酰胺分支;“32”表示悬垂mpeg4(ppa032);且61”表示悬垂peg(ppa061)。每个xntp还包括以下d细胞特征:d(63)d(63)d(63)dd其中“d”表示peg6,且“63”表示悬垂peg(ppa063)。xntp底物是用以下报告子代码合成:(xc)ddlllx;(xt)lxxx;(xa)dd(32)(32)(32)lllllll;和(xg)xxl(32)(32)(32)(32)(32)(32)lllllll,其中“d”表示peg-6,“l”表示c2,“x”表示peg-3,且“32”表示悬垂mpeg-4(ppa032)。
[0413]
为了产生222mer模板的xpandomer拷贝,使用5000pmol的每种xntp、4pmol的模板和20pmol的e-oligo引物进行固态引物延伸反应(其中延伸寡核苷酸共价结合到芯片底物上的固态xpandomer合成描述于申请人的临时专利申请号62/826,805中,所述专利申请通过引用全文并入本文)。50μl延伸反应包括以下试剂:50mm triscl、ph 8.84、200mm nh4oac、50mm gucl 20%peg8k、10%nmp、15nmol聚磷酸盐pp-60.23、2.5μg kod ssb、0.1m尿素、15mm pem添加剂和13μg纯化的重组dna聚合酶(dpo4聚合酶的变体)。延伸反应在37℃进行60分钟。
[0414]
接下来使用sbx方案对xpandomer产物进行测序。简而言之,将受约束的xpandomer产物在缓冲剂b.064(1%tween-20/3%sds/5mm hepes,ph 8.0/100mm napo4/15%dmf)中洗涤并通过添加200μl缓冲剂c.001(7.5m dcl)并在23℃孵育30分钟进行切割以产生线性化的xpandomer。然后通过添加2000μl缓冲剂b.064并在室温孵育2’来中和样品。然后通过在缓冲剂b.065中添加500μmol琥珀酸酐并在23℃孵育5分钟,对xpandomer样品进行胺修饰。然后在缓冲剂d.102(50%acn)中洗涤样品,并且将xpandomer通过光切割从底物释放并在60μl洗脱缓冲剂中洗脱。
[0415]
通过将α-溶血素插入2m nh4cl和100mm hepes,ph 7.4的缓冲剂中的dphpe/十六烷双层成员中来制备蛋白质纳米孔。对顺式孔灌注含有0.4m nh4cl、600mm guancl、100mm hepes;ph 7.4和5%甘油的缓冲剂ag242,且对反式孔灌注含有0.4m nh4cl、600mm guancl、5%乙酸乙酯、10mm hepes;ph值7.4的缓冲剂ab080。将xpandomer样品加热至70℃2分钟,完
全冷却并涡旋,然后将2μl等分试样加入顺式孔中。电压参数运行如下:70mv/625mv/6μs/1.0ms(读段电压/脉冲电压/脉冲电压持续时间/脉冲频率)。数据经由labview采集软件获得。
[0416]
为了评估xpandomer序列信息的准确性,通过显示来自sbx反应的序列读段群的直方图显示来分析序列数据。分析软件将每个序列读段与模板序列对准,并修整与正确模板序列不对准的读段末端的序列范围。222mer模板的sbx测序的代表性直方图呈现在图19中。可以看出,sbx实验产生了222mer模板的高度准确读段。值得注意的是,该实验的通量非常出色。这些结果表明,掺入基于悬垂peg的tce和d细胞特征的ssrt可以高度准确和有效地控制xpandomer通过纳米孔的易位。
[0417]
实例22
[0418]
棘轮
[0419]
在此棘轮的实例中,单个溶血素纳米孔在脂质双层中制备,在反面有前庭,并在顺式储器中具有由0.4m nh4c1、600mm guancl、100mm hepes;ph 7.4构成的试剂混合体;且在反式储器顺式中具有由2m nh4c1、100mm hepes;ph 7.4构成的试剂混合体。通过axopatch 200b放大器测量位于每个储器中的ag/agcl电极之间通过的电流,并以100k样品/秒的速度进行数字化。为了驱动电流通过纳米孔,向反式储器施加占空比在+70mv和-50mv之间交替变化的50%方波,同时在从正电压向负电压(所有电压均以顺式储器电势为基准)转变之间施加+600mv的6μs脉冲。采用这种施加的脉冲序列,假设理想的易位没有缺失或插入,测量了掺入xpandomer中的每种xntp的两个报告子,一个用+70mv,而另一个用-50mv。对每个碱基进行两次测量提供冗余,从而为匹配结果提供更高的置信度,并且还有助于识别非均聚物序列中的缺失和插入。图20a说明+70mv/600mv脉冲/-50mv的循环如何影响xpandomer通过纳米孔易位并导致c代码的两次测量,然后是a代码的两次测量(以及g代码的第1(一)次测量)。模式代码显示了在这个非均聚物序列中,每个碱基的2个报告子测量值可如何用于识别插入和缺失。当xpandomer不前进到下一个报告子(在纳米孔中)或它跳过下一个报告子时,会导致插入和缺失。
[0420]
使用sbx合成和纯化方案,将从已知序列的合成dna模板产生的xpandomer样品引入顺式储器并进行测量。使xpandomer易位的电流测量的实例示于图20b中。所述图按比例缩放,因此+70mv测量值(14、20、27和35pa)的四个报告子电流电平和-50mv测量值(-12、-20、-25和-29pa)的4(四)个报告子电流电平如水平虚线所示。将数据与预期的dna模板序列比对,并由图表上的数字序列表示。四个数字中的每一个都表示一个基数。蓝色序列号表示与模板的确认碱基匹配。箭头表示错误。用数字1表示的箭头是非均聚物插入,因为碱基调用表明xpandomer没有前进,因此可以识别这些插入。标有数字2的箭头表示未识别的均聚物插入,因为碱基调用均处于同一水平。
[0421]
实例24
[0422]
氟阿拉伯糖基xntp差向异构体的合成
[0423]
本实例描述了每种xntp的2
′
氟(f)差向异构体(2
′
fana xntp)的合成。这些差向异构体基于氟化核苷,称为“氟代阿拉伯糖核酸”(fana)。预计2
′
f差向异构体将在酸处理过程中表现出增加的稳定性,这是产生线性化的xpandomer产物的合成途径中的关键步骤。以下是产生每种2
′
fana xntp的合成方案。
[0424]
a.
[0425]2′
fana xttp的制作方法
[0426][0427]
在第一步中,将氟尿嘧啶(化合物1,可从tci america获得)经由菌头反应与1-8辛二炔偶联(参见,例如bag,s.,jana,s.和kasula,m.(2018)。sonogashira cross-coupling:alkyne-modified nucleosides and theirapplications.in palladium-catalyzed modification of nucleosides,nucleosides,and oligonucleotides(pp.75-146).elsevier)。在第二步中,用约一当量的dmtrcl在吡啶中处理化合物2以产生化合物3。在第三步中,按照授予kokoris等人、标题为“phosphoramidate esters and use and synthesis thereof”的美国专利号10,301,345将化合物3转化为三磷酸酰胺,所述美国专利通过引用全文并入本文。
[0428]
b.
[0429]2′
fana xctp的制作方法
[0430][0431]
在第一步中,将fialcitabine(化合物5,可从trc canada获得)经由菌头反应(如上所述)与1-8辛二炔偶联,以产生化合物6。在第二步中,用约一当量的dmtrcl在吡啶中处理化合物6以产生化合物7。在第三步中,由乙酰基来保护化合物7的环外胺(参见,例如fan,y.,gaffney,b.,和jones,艮(2004).transient silylation of the guanosine o6 and the amino groups faciltates n-acylation.organic letters,6,15,2555-2557.),并且随后转化为酰胺三磷酸酯8,如授予kokoris等人的美国专利号10,301,345中所述。
[0432]
c.
[0433]2′
fana xgtp的制作方法
[0434]
商购自granlen
[0435][0436]
在第一步中,将7-脱氮-7-碘代鸟苷(可从granlen获得的化合物9;cas:444020-71-7)用1当量的1,3-二氯-1,1,3,3-四甲基二硅氧烷处理以提供化合物10(参见例如markiewicz,w.t.和wiewiorowski,m.(1978)a new type of silyl protecting groups in nucleoside chemistry.nuc.acids res.5,s185-ss190)。在第二步中,通过使用氟化剂dast将化合物10转化为化合物11(参见,例如pankiewicz,k.,kreminski,j.,ciszewski,l.,ren,w.,和watanabe,k.(1992).a synthesis of 9-(2-deoxy-2-fluoro-b-d-arabinofuranosyl)adenine and-hypoxanthine.an effect of c3
’‑
endo to c2
’‑
endo conformational shift on the reaction course of 2
’‑
hydroxyl group with dast.j.of organic chem.57,2,553-559.)在第三步中,如上所述用苯氧基乙酰基保护化合物11中的环外胺。在第四步中,所得化合物12通过上述菌头反应与1-8辛二炔偶联,得到化合物13。在第五步中,如上所述将硅氧烷基团脱保护将得到化合物14。在第六步中,用1当量dmtrcl在吡啶中处理化合物14产生化合物15。在第七步中,将化合物15转化为鸟苷酰胺三磷酸酯16,如授予kokoris等人的美国专利号10,301,345所述。
[0437]
同样的方案可用于合成以下三磷酸腺苷类似物:
[0438][0439]
来自起始化合物7-脱氮-7-碘代腺苷(可从granlen获得,cas:24386-93-4)。
[0440]
本说明书中提及和/或申请数据表中列出的所有美国专利、美国专利申请公开、美国专利申请、国外专利、国外专利以及非专利公开,包括但不限于2019年5月23日提交的美国临时专利申请号62/852,262、2019年7月22日提交的美国临时专利申请号62/877,183和
2019年8月12日提交的美国临时专利申请号62/885,746,均通过引用全文并入本文。为了描述和公开例如出版物中描述的材料和方法的目的,可以通过引用并入这些文件,其可以与当前描述的发明结合使用。提供以上和通篇讨论的出版物仅用于它们在本技术的申请日之前的公开。本文中的任何内容均不应被解释为承认发明人无权凭借在先发明早于任何引用出版物。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1