碳环核苷类似物
碳环核苷类似物
1.本发明涉及5
‑
氮杂
‑2’‑
脱氧胞苷和5
‑
氮杂胞苷的新型水解稳定的碳环核苷类似物和它们作为低甲基化剂的用途。
[0002]5‑
氮杂
‑2’‑
脱氧胞苷(地西他滨,azadc)和5
‑
氮杂胞苷(维达莎,azac)为核苷类似物,其通过操纵表观遗传信息充当抗代谢物[1
‑
5]。dna中的表观遗传信息与在dna甲基转移酶(dnmt)和s
‑
腺苷甲硫氨酸(sam)作为甲基化辅因子的帮助下从2
’‑
脱氧胞苷(dc)形成5
‑
甲基
‑2’‑
脱氧胞苷(mdc)相关[6
‑
7,4]。启动子区域中dc到mdc的甲基化通常与基因的转录沉默有关[8
‑
9]。azadc是一种前药,其在细胞内转化为相应的活性三磷酸,并随后在细胞分裂期间并入基因组中。azac也是一种前药,其被2
’‑
脱氧并随后转化为相应的5
’‑
三磷酸以并入dna。azac也可在未事先2
’‑
脱氧的情况下转化为5
’‑
三磷酸并且并入到rna中。aza(d)c的作用模式涉及其亲电子的c6位置与dnmt活性位点硫醇亲核试剂的反应,如图1a所示[10
‑
11]。这产生了被sam辅因子甲基化、保持紧密的共价中间体。由于三嗪杂环第5位的n原子,通常会从dnmt酶中释放m(d)c的最终的β
‑
消除反应不再可能。结果是共价dna
‑
dnmt或rna
‑
dnmt交联的形成。
[0003]
由于施用aza(d)c,观察到了mdc水平的大幅下降(即低甲基化效应),这导致癌细胞中沉默的肿瘤抑制基因的重新激活[1],这允许重新分化癌细胞回到正常增殖的细胞或引起细胞死亡。aza(d)c目前用作治疗骨髓增生异常综合征(mds)[2]和急性髓性白血病(aml)[4]的药物。临床上,它分几个周期施用,每个周期包括一周的治疗和三周的暂停。
[0004]
与aza(d)c相关的一个问题是它在水溶液中按照图1b所示的路径水解。这种水解损害aza(d)c的活性,特别是在长的处理时间中。
[0005]
本发明人通过提供可去甲基化(并因此与s
‑
亲核试剂反应)同时阻止水解(与o
‑
亲核试剂反应)的aza(d)c类似物克服了这个问题。令人惊讶的是,他们发现用ch2基团取代脱氧核糖的氧对杂环的反应性有很大的远程影响。这些类似物,特别是碳环形式的aza(d)c仍然抑制通过dna甲基转移酶的甲基化,但为水解稳定的,如图1c所示。
[0006]
本发明的第一方面涉及式i的化合物:
[0007][0008]
其中r1为h,游离的或受保护的、修饰的或未修饰的磷酸类基团或羟基保护性基团,
[0009]
r2为h,游离的或受保护的、修饰的或未修饰的磷酸类基团或羟基保护性基团,
[0010]
r3为h、f、ch3、ch2f、chf2、cf3、oh或or6,其中r6为羟基保护性基团,
[0011]
r4和r5各自为h或形成氨基保护性基团,
[0012]
或其盐。
[0013]
在式i的化合物中,r1可选自h,游离的或受保护的、修饰的或未修饰的磷酸类基团,例如单磷酸酯/盐、二磷酸酯/盐或三磷酸酯/盐基团,或羟基保护性基团。
[0014]
术语“磷酸类基团”包括未修饰的磷酸酯/盐基团(例如未修饰的单磷酸酯/盐、未修饰的二磷酸酯/盐或未修饰的三磷酸酯/盐基团)或修饰的磷酸酯/盐基团(例如修饰的单磷酸酯/盐、修饰的二磷酸酯/盐或修饰的三磷酸酯/盐基团),其中一个或多个氧原子被碳、硫和/或氮原子取代。修饰的磷酸酯/盐基团的实例为膦酸酯/盐、氨基磷酸酯/盐或硫代磷酸酯/盐基团。术语“磷酸类基团”还包括游离的磷酸酯/盐基团和受保护的磷酸酯/盐基团,例如磷酸酯(包括单酯和二酯),和酰胺化物(包括单酰胺化物、二酰胺化物和酰胺酯)。在[28]和[29]中可找到对受保护的磷酸酯/盐和膦酸酯/盐基团的详细综述,其内容通过引用并入本文。因此,在药学应用中,r1可为受保护的磷酸酯/盐、二磷酸酯/盐或三磷酸酯/盐基团,以在细胞中裂解时释放磷酸酯/盐、二磷酸酯/盐或三磷酸酯/盐基团(前药概念)。
[0015]
羟基保护性基团是本领域已知的,特别是在核苷化学领域中,并且包括但不限于酰基(诸如乙酰基或苯甲酰基)、苄基、三苯甲基(诸如三苯甲基、甲氧基三苯甲基或二甲氧基三苯甲基)、甲硅烷基(诸如三甲基甲硅烷基、叔丁基二甲基甲硅烷基或三异丙基甲硅烷基),或烷基或取代的烷基(诸如甲基、乙氧基乙基或β
‑
甲氧基乙氧基甲基)。
[0016]
r2可选自h、如上所述的未修饰的或修饰的、游离的或受保护的磷酸类基团,例如单磷酸酯/盐、二磷酸酯/盐或三磷酸酯/盐基团,或如上所述的羟基保护性基团。因此,在药学应用中,r2可为受保护的磷酸酯/盐、二磷酸酯/盐或三磷酸酯/盐基团以在细胞中裂解时释放磷酸酯/盐、二磷酸酯/盐或三磷酸酯/盐基团(前药概念)。
[0017]
在某些实施方案中,r3可为h。在这种情况下,式i中的五元碳环可被认为是脱氧核糖,其中环的1’位的氧原子已被ch2基团取代。在另外的实施方案中,r3可为oh或or6,其中r6为如上所述的羟基保护性基团。在这些实施方案中,式i中的五元碳环可被认为是核糖,其中环的1’位的氧原子已被ch2基团取代。r3也可为ch3、ch2f、chf2、cf3或f。在这些实施方案中,式i中的五元碳环可被认为是核糖类似物,其中环的1’位的氧原子已被ch2基团取代。
[0018]
r4和r5各自可为h,因此提供伯氨基nh2或者它们可形成氨基保护性基团,例如其中一个或两个h原子被保护性基团取代。氨基保护性基团是本领域已知的,特别是在核苷化学领域中,并且包括但不限于酰基(诸如乙酰基或苯甲酰基)、苄基、苄氧羰基、甲苯磺酰基、9
‑
芴基甲氧基羰基或叔丁氧羰基。
[0019]
在某些实施方案中,r1为h。
[0020]
在某些实施方案中,r2为h或未修饰的或修饰的磷酸类基团,例如单磷酸酯/盐、二磷酸酯/盐或三磷酸酯/盐基团,包括游离的或受保护的单磷酸酯/盐、二磷酸酯/盐或三磷酸酯/盐基团。
[0021]
在某些实施方案中,r3为h或oh。
[0022]
在某些特定实施方案中,r1、r2、r3、r4和r5各自为h,或r1、r2、r4和r5各自为h并且r3为oh或or6。在特别具体的实施方案中,r1、r2、r3、r4和r5为h。该化合物为azadc(地西他滨)的碳环类似物,如上所述,其具有令人惊讶的高水解稳定性,同时对甲基转移酶,特别是dna甲
基转移酶保持抑制活性。在另外的特别具体的实施方案中,r1、r2、r4和r5为h并且r3为oh。该化合物为azac(维达莎)的碳环类似物。
[0023]
在另外的具体实施方案中,本发明还涉及azadc和azac的前药,特别涉及其中r1和/或r2不同于h的化合物。
[0024]
本发明还涉及式i的化合物的盐,特别是药学上可接受的盐。本发明的化合物能够通过氨基和/或磷酸酯/盐基团或与其相似的基团的存在形成酸和/或碱盐。酸加成的盐可利用无机酸和有机酸形成,并且包括但不限于乙酸盐、苯甲酸盐、氢溴酸盐、盐酸盐、柠檬酸盐、乳酸盐、磷酸盐、甲苯磺酸盐和三氟乙酸盐。碱加成的盐可在无机碱和有机碱中形成,包括但不限于铵盐、钠盐、钾盐、钙盐、镁盐或衍生自伯胺、仲胺和叔胺的盐。
[0025]
本发明的化合物通过降低表观遗传修饰5
‑
甲基
‑2’‑
脱氧胞苷(mdc)的水平而充当抗代谢物。它们可被并入增殖细胞的基因组和/或rna中,并抑制dna和/或rna甲基转移酶,导致dna中的mdc和rna中的mc减少。dna中mdc的缺失(启动子中的转录沉默子)会导致包括肿瘤抑制基因在内的基因重新激活,从而引发抗过度增殖作用。
[0026]
因此,本发明的另外的方面涉及如上所述的式i的化合物或其药学上可接受的盐用于药物、例如在兽医的或人类的药物中的用途。
[0027]
在某些实施方案中,本发明的化合物用于治疗过度增殖性疾病,包括恶性或非恶性过度增殖性疾病。术语“过度增殖性疾病”涉及由生物体内的细胞、组织和/或器官的功能失调性增加的增殖引起、与之相关和/或伴随的疾病。
[0028]
因此,本发明也涉及治疗过度增殖性疾病的方法,包括向有需要的受试者、特别是人类受试者施用治疗有效剂量的式i的化合物或其药学上可接受的盐。
[0029]
在具体的实施方案中,本发明的化合物用于治疗癌症。在某些实施方案中,本发明的化合物用于治疗白血病,例如急性髓系白血病(aml)或骨髓增生异常综合征(mds),包括先前治疗的或未治疗的原发性或继发性mds,诸如难治性贫血、难治性贫血伴环状铁粒幼细胞、难治性贫血伴原始细胞过多、难治性贫血伴转化中原始细胞过多和慢性粒单核细胞白血病以及中
‑
1、中
‑
2和高危mds。
[0030]
在另外的实施方案中,基于地西他滨的报道的结果[26],本发明的化合物可用于治疗肾功能不全。在某些实施方案中,基于地西他滨防止动脉粥样硬化病变形成并减少巨噬细胞产生炎性细胞因子的观察结果,本发明的化合物可用于治疗动脉粥样硬化[27]。
[0031]
式i的化合物或其药学上可接受的盐可作为包含活性剂和药学上可接受的载体的药学组合物施用给有需要的受试者。组合物可以任何合适的方式施用,例如口服、肠胃外、吸入和/或皮肤施用。载体可为任何合适的药物载体。式i的化合物以治疗有效剂量施用,该剂量可由熟练从业者基于受试者和待治疗的病症确定。
[0032]
在药学应用中,组合物可为固体剂型的形式(例如片剂、胶囊等)或液体剂型的形式(例如溶液、乳液、悬浮液等)。
[0033]
式i的化合物可在一个或几个治疗周期中施用,其中每个治疗周期可包括至少1、2、3、4、5、6、7或10天。
[0034]
式i的化合物可作为单一疗法或与另外的药物、特别是适合于治疗如上所述的过度增殖性疾病的药物组合施用。在某些实施方案中,式i的化合物可与不同于式i的抗癌药物共同作为联合疗法施用。
[0035]
此外,式i的化合物也可用于非医学应用,例如一般在基础研究、诊断和/或药物筛选领域。因此,在某些实施方案中,本发明涉及式i的化合物用于抑制包括dna和rna在内的核酸的甲基化、特别是用于抑制dna的甲基化、更特别是用于抑制基因组dna的甲基化的体外用途。因此,该化合物适用于细胞、例如真核细胞(诸如包括哺乳动物和人类细胞的动物细胞)中的基因组分析,特别是用于表观遗传分析。
[0036]
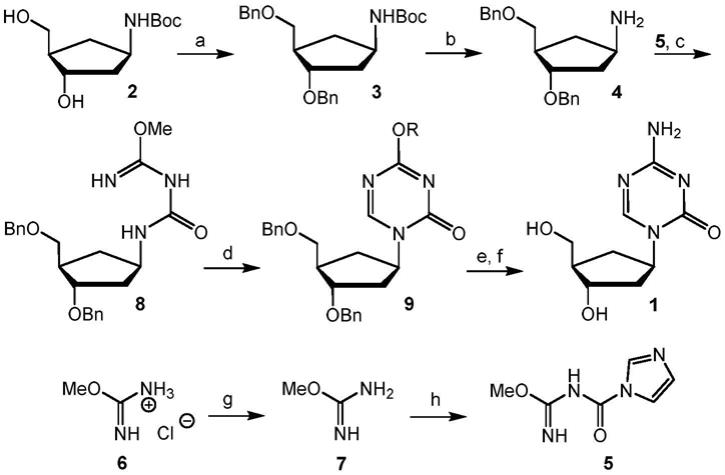
制备式i的化合物的一般方案描述于以下方案中。合成从叔丁氧羰基(boc)保护的氨基环戊烷衍生物2开始,该衍生物先前已用于合成dna损伤类似物[12
‑
15]。化合物2首先被苄基保护至3、boc去保护至4,并随后与碳咪唑5反应,后者由异甲基脲6分两步制备(在与氢氧化钾生成游离碱7和7与羰基二咪唑反应后)。这提供了氨基甲酰脲
‑
环戊烷核苷类似物8。随后用原甲酸三乙酯环化成三嗪碱9。9与nh3在甲醇中的反应和在二氯甲烷中的用bcl3对苄基的脱保护提供了作为游离核苷的最终的化合物cazadc 1。
[0037][0038]
化合物cazadc 1从热甲醇中重结晶得到无色针状晶体,这允许解析图2中描绘的晶体结构。有趣的是观察到cazadc 1在晶体中存在两种不同的环戊烷构象。这些构象中的一种构象是dna中2
’‑
脱氧核苷的典型构象,表明cazadc 1核苷可采用正确的dna类型构象,并有可能被磷酸化并整合到基因组中。
[0039]
下面描述了制备式i的化合物的另一个通用方案,显示了碳环版本的阿扎胞苷(维达莎)cazac,20的合成:
[0040][0041]
下面描述了制备式i的化合物的前药的一般方案,显示了cazac,20的5
’‑
磷酰胺化物前药24的合成:
[0042][0043]
另外,将通过以下附图和实施例更详细地解释本发明。
[0044]
附图图例
[0045]
图1:5
‑
氮杂
‑2’‑
脱氧胞苷(地西他滨,azadc)及其作用模式的描述。a)活性位点硫醇与azadc的c6位反应。b)与水分子和azadc的c6位(o反应性)反应密切相关的水解降解途径。这导致最终的碱基丢失和无碱基位点的形成。c)框内描述了碳环版本的cazadc 1。
[0046]
图2:碳环5
‑
氮杂
‑2’‑
脱氧胞苷(cazadc 1)的晶体结构,显示了观察到的c6
’‑
内构象(a)和c2
’‑
内构象(2t1)(b)中的分子。
[0047]
图3:基于hplc的稳定性测量,显示(a)5
‑
氮杂
‑2’‑
脱氧胞苷(azadc)溶液在不同ph值下严重水解分解,而(b)碳环化合物cazadc1在所有三个ph值下均稳定。(a)中的插图显示azadc在t1=10min和t2=20min之间的色谱图。azadc的信号用红色描述。
[0048]
图4:描述了通过uhplc
‑
ms2获得的碳环5
‑
氮杂
‑2’‑
脱氧胞苷处理的(cazadc 1)小鼠胚胎干细胞(mesc)的dna修饰的定量数据。对于每个条件,以技术三次重复测量三个生物学重复。对于每个技术重复,消化0.5μg的dna。条形图代表平均值,误差条代表标准偏差。lloq表示定量的下限。
[0049]
图5:地西他滨1(左)对比碳环化合物cazac 20(右)的中性溶液的hplc研究。将100mm的化合物溶液保存在纯水中,并在t=0h、1.5h、3h、4.5h、6h、24h、6d时进行测试。
实施例
[0050]
实施例1:cazadc(1)的合成程序
[0051]
叔丁基
‑
n
‑
[(1r,3s,4r)
‑3‑
羟基
‑4‑
(羟甲基)环戊基]氨基甲酸酯(2):
[0052][0053]
c
11
h
21
no4[0054]
231.29g/mol
[0055]
化合物2由叔丁基
‑
n
‑
((1s,2r,4r,5r)
‑4‑
((叔丁基二甲基甲硅烷氧基)甲基)
‑6‑
氧杂
‑
双环
‑
[3.1.0]
‑
己
‑2‑
基)
‑
氨基甲酸酯合成,如前所述[1]。
[0056]
叔丁基
‑
n
‑
[(1r,3s,4r)
‑3‑
苄氧基
‑4‑
(苄氧基甲基)环戊基]氨基甲酸酯(3):
[0057][0058]
c
25
h
23
no4[0059]
411.54g/mol
[0060]
将boc保护的2(3.107g,13.43mmol,1.0当量)溶解在干燥dmf(75ml)中。将该溶液冷却至0℃并分三份加入nah(在矿物油中60%的悬浮液,1.182g,29.55mmol,2.2当量)。在0℃15min后,滴加苄基溴(4.00ml,33.58mmol,2.5当量)。在0℃搅拌反应混合物1.5h并在室温搅拌额外的2h。用ch2cl2(30ml)稀释反应混合物,并用饱和碳酸氢钠(30ml)淬灭。用ch2cl2(500ml)萃取混合物。有机相用饱和碳酸氢钠(1
×
400ml,2
×
100ml)洗涤三次并在na2so4上干燥。真空除去溶剂。粗产物通过在硅胶上柱色谱用逐步梯度的ihex/etoac(9:1
→
7:1
→
4:1)纯化以得到产物3,为无色固体(3.39g,8.24mmol,61%)。
[0061]
r
f
=0.67(ihex/etoac=2:1)
[0062]
mp=74
‑
76℃
[0063]
hrms[esi+]:[c
25
h
33
no4na]
+
,[m+na
+
]
+
计算值:434.2292,发现值:434.2299。
[0064]1h
‑
nmr(599mhz,cdcl3):δ/ppm=7.39
‑
7.26(m,10h,ar
‑
h),4.76(s,1h,nh),4.53
‑
4.37(m,4h,c7‑
h and c8‑
h),4.18
‑
4.08(m,1h,c1‑
h),3.96
‑
3.87(m,1h,c3‑
h),3.53
‑
3.41(m,2h,c5‑
h),2.36
‑
2.25(m,2h,c6‑
h
a
,c4‑
h),2.11
‑
2.05(m,1h,c2‑
h
a
),1.77
‑
1.68(m,1h,c2‑
h
b
),1.43(s,9h,c2’
‑
h),1.29
‑
1.20(m,1h,c6‑
h
b
)。
[0065]
13
c
‑
nmr(151mhz,cdcl3):δ/ppm=155.5(c1’
),138.60,138.23,128.36,128.28,127.59,127.56,127.42(c
‑
ar),81.0(c3),79.2(c2’
),73.3(c8),71.9(c5),71.3(c7),50.4(c1),44.9(c4),39.6(c2),35.0(c6),28.6(c2’
)。
[0066]
ftir(atr):2973(w),2929(w),2858(w),1692(m),1496(m),1453(m),1390(w),1364(m),1274(m),1247(m),1166(s),1091(s),1067(s),1027(m),1012(m)。
[0067]
(1r,3s,4r)
‑1‑
氨基
‑3‑
苄氧基
‑4‑
(苄氧基甲基)环戊烷(4):
[0068][0069]
c
20
h
25
no4[0070]
311,42g/mol
[0071]
在室温搅拌boc
‑
保护的胺3(3.351g,8.14mmol,1.0当量)和三氟乙酸(12ml,30%(v/v))在干燥ch2cl2(28ml)中的溶液1h。真空除去溶剂后,将残余物重新悬浮在饱和na2co3(100ml)溶液中并搅拌10min。用etoac(3
×
150ml)萃取混合物,并在na2so4上干燥合并的有机相。真空除去溶剂,得到粘性棕色油状的游离胺4,由于其对o2不稳定,无需进一步纯化即使用。
[0072]
纯化的4的分析数据:
[0073]
hrms[esi
+
]:[c
23
h
29
n4o4na]
+
,[m+na
+
]
+
计算值:312.1958,发现值:312.1955。
[0074]1h
‑
nmr(400mhz,cdcl3):δ/ppm=7.38
‑
7.20(m,10h,c
‑
ar),4.57
‑
4.36(m,4h,c7,
c8),3.95
‑
3.85(m,1h,c1),3.62
‑
3.50(m,1h,c3),3.50
‑
3.39(m,2h,c5),2.41
‑
2.28(m,1h,c4),2.28
‑
2.13(m,1h,c6‑
h
a
),2.13
‑
1.96(m,3h,nh2,c2‑
h
a
),1.65
‑
1.50(m,1h,c2‑
h
b
),1.19
‑
1,.08(m,1h,c6‑
h
a
),
[0075]
13
c
‑
nmr(101mhz,cdcl3):δ/ppm=138.8,138.4,128.5,128.5,127.79,127.75,127.7,127.6(c
‑
ar),81.82(c3),73.23(c7),72.58(c5),71.10(c8),51.40(c1),45.63(c4),42.07(c2),38.08(c6)。
[0076]2‑
甲基
‑1‑
(1
‑
咪唑基羰基)异脲(5):
[0077][0078]
c6h8n4o4[0079]
168.16g/mol
[0080]
在
‑
15℃、不断搅拌下分批向o
‑
甲基异脲盐酸盐6(5.00g,45.65mmol,1.0当量)在et2o:h2o(39:1)中的溶液中加入koh粉末(51.22g,912.96mmol,20.0当量)。30min后,过滤混合物,并用冰冷的et2o(3
×
50ml)洗涤残余物。真空浓缩合并的有机相至20ml。冷却至
‑
20℃导致沉淀。在n2气流下真空过滤得到o
‑
甲基异脲7,为无色蜡(1.759g,23.74mmol,52%)。在室温搅拌7(1.24g,16.74mmol,1.0当量)和羰基二咪唑(2.90g,17.91mmol,1.07当量)在干燥thf(35ml)中的溶液3h。1h后,观察到无色沉淀。真空浓缩反应混合物至3ml并过滤。用冰冷的thf(2
×
3ml)洗涤残余物并冻干合并的有机相,得到无色固体5(1.946g,11.57mmol,69%)。
[0081]1h
‑
nmr(400mhz,cdcl3):δ/ppm=8.60(br s,1h,n
‑
h),8.35(s,1h,ar
‑
h),7.58(s,1h,ar
‑
h),7.04(s,1h,ar
‑
h),6.04(br s,1h,n
‑
h),3.96(s,3h,c2‑
h)。
[0082]
13
c
‑
nmr(101mhz,cdcl3):δ/ppm=165.7(c1),157.3(c3),137.5(ar),130.0(ar),117.2(ar),55.3(c2)。
[0083]1‑
[(1’r,3’s,4’r)
‑3’‑
苄氧基
‑4’‑
(苄氧甲基)环戊基]甲基异缩二脲(8):
[0084][0085]
c
23
h
29
n4o4[0086]
411.50g/mol
[0087]
将粗产物4溶解在干燥乙腈(75ml)中并加入羰基咪唑5(1.506g,8.96mmol,1.1当量)。回流混合物2h。真空除去溶剂。通过在硅胶上柱色谱用逐步梯度的ihex/etoac(2:1
→
1:1)纯化粗产物以得到黄色油状甲基异缩二脲8(2.613g,6.35mmol,78%)。
[0088]
r
f
=0.51(dcm/meoh=19:1)
[0089]
hrms[esi
+
]:[c
23
h
29
n4o4na]
+
,[m+na
+
]
+
计算值:434.2050,发现值:434.2049。
[0090]1h
‑
nmr(599mhz,cdcl3):δ/ppm=7.35
‑
7.16(m,10h,ar
‑
h),5.42(d,3j1’
,n1
=7.8hz,1h,n1‑
h),4.50
‑
4.34(m,4h,c7’
‑
h
a,b
,c8’
‑
h
a,b
),4.33
‑
4.16(m,1h,c1’
‑
h),3.93
‑
3.81(m,1h,c3’
‑
h),3.62(s,3h,c6‑
h),3.43
‑
3.31(m,2h,c5’
‑
h),2.34
‑
2.18(m,2h,c6’
‑
h
a
,c4’
‑
h),2.06(ddd,2j2’
a,2’b
=13,0hz,3j2’
a,1’/3’=6,9hz,3j2’
a,1’/3’=4,6hz,1h,c2’
‑
h
a
),1.73(ddd,2j2’
a,2b’=13.0hz,3j2’
b,1’/3’=7.0hz,3j2’
b,1’/4’=7.0hz,1h,c2’
‑
h
b
),1.28
‑
1.11(m,1h,c6’
‑
h
b
)。
[0091]
13
c
‑
nmr(151mhz,cdcl3):δ/ppm=163.8(c2),162.3(c4),138.9(c
ar
),138.5(c
ar
),128.51(c
ar
),128.41(c
ar
),127.76(c
ar
),127.74(c
ar
),127.70(c
ar
),127.56(c
ar
),81.1(c3’
),73.3(c7’
),71.9(c5’
),71.2(c8’
),53.8(c6),49.5(c1’
),45.0(c4’
),39.5(c2’
),35.0(c6’
)。
[0092]4‑
乙氧基
‑1‑
[(1’r,3’s,4’r)
‑3’‑
苄氧基
‑4’‑
(苄氧基甲基)环戊基]
‑
1h
‑
[1,3,5]
‑
三嗪
‑2‑
酮(9a)和4
‑
甲氧基
‑1‑
[(1’r,3’s,4’r)
‑3’‑
苄氧基
‑4’‑
(苄氧基甲基)环戊基]
‑
1h
‑
[1,3,5]
‑
三嗪
‑2‑
酮(9b):
[0093][0094]
将三氟乙酸(75μl)加入8(2.613g,6.35mmol,1.0eq)在原甲酸三乙酯(60ml)中的溶液中。回流混合物3h。真空除去溶剂。将残余物与meoh共蒸发一次,通过在硅胶上柱色谱用逐步梯度的ihex/etoac(2:1
→
1:1
→
1:2)纯化粗产物以得到9a(1.774g,4.07mmol,64%,无色固体)和9b(268.0mg,0.64mmol,10%,无色油)。
[0095]
9a:
[0096]
r
f
=0.71(ihex/etoac,1:3)
[0097]
hrms[esi
+
]:[c
25
h
30
n3o4h]
+
,[m+h
+
]
+
计算值:436.2232,发现值:436.2231。
[0098]1h
‑
nmr(400mhz,cdcl3):δ/ppm=8.23(s,1h,c6‑
h),7.38
‑
7.24(m,10h,ar
‑
h),5.07
‑
4.89(m,1h,c1’
‑
h),4.56
‑
4.39(m,6h,c7’
‑
h
a,b
,c8’
‑
h
a,b
,c7‑
h
a,b
),4.07
‑
3.08(m,1h,c3’
‑
h),3.61
‑
3.46(m,2h,c5’
‑
h
a,b
),2.52
‑
2.36(m,2h,c6’
‑
h
a
,c4’
‑
h),2.28(ddd,1h,2j2’
a,2’b
=13.3hz,3j=7.6hz,3j=2.7hz,c2’
‑
h
a
),2.11(ddd,1h,2j2’
a,2’b
=13.3hz,3j=10.0hz,3j=6.3hz,c2’
‑
h
b
),1.84
‑
1.68(m,1h,c6’
‑
h
b
),1.40(t,3h,3j
8,7
=7.1hz,c8‑
h),
[0099]
13
c
‑
nmr(101mhz,cdcl3):δ/ppm=169.3(c4),158.4(c6),155.0(c2),138.2(c
ar
),138.1(c
ar
),128.6(c
ar
),128.6(c
ar
),127.93(c
ar
),127.85(c
ar
),127.82(c
ar
),80.4(c3’
),
73.4(c7’
/8’),71.26(c7’
/8’),71.25(c5’
),65.1(c7),56.8(c1’
),44.9(c4’
),36.9(c2’
),32.6(c6’
),14.2(c8)。
[0100]
9b:
[0101]
r
f
=0.63(ihex/etoac,1:3)
[0102]
hrms[esi
+
]:[c
24
h
28
n3o4]
+
,[m+h
+
]
+
计算值:422.2074,测量值:422.2075。
[0103]1h
‑
nmr(599mhz,cdcl3):δ/ppm==8.23(s,1h,c6‑
h),7.38
‑
7.25(m,10h,ar
‑
h),5.04
‑
4.93(m,1h,c1’
‑
h),4.57
‑
4.43(m,6h,c7’
‑
h
a,b
,c8’
‑
h
a,b
),4.09
‑
3.98(m,4h,c3’
‑
h,c7‑
h),3.60
‑
3.50(m,2h,c5’
‑
h
a,b
),2.50
‑
2.39(m,2h,c6’
‑
h
a
,c4’
‑
h),2.28(ddd,1h,2j2’
a,2’b
=13.1hz,3j=7.4hz,3j=2.7hz,c2’
‑
h
a
),2.11(ddd,1h,2j2’
a,2’b
=13.1hz,3j=10.1hz,3j=6.3hz,c2’
‑
h
b
),1.80
‑
1.68(m,1h,c6’
‑
h
b
)。
[0104]
13
c
‑
nmr(101mhz,cdcl3):δ/ppm=169.9(c4),158.5(c6),155.0(c2),138.3(c
ar
),138.0(c
ar
),128.63(c
ar
),128.57(c
ar
),127.93(c
ar
),127.84(c
ar
),127.82(c
ar
),80.4(c3’
),73.5(c7’
/8’),71.3(c7’
/8’),56.9(c1’
),56.0(c7),44.9(c4’
),36.9(c2’
),32.6(c6’
)。
[0105]4‑
氨基
‑1‑
[(1’r,3’s,4’r)
‑3’‑
苄氧基
‑4’‑
(苄氧甲基)环戊基]
‑
1h
‑
[1,3,5]三嗪
‑2‑
酮(10):
[0106][0107]
c
23
h
26
n4o3[0108]
406.47g/mol
[0109]
将乙氧基三嗪9a(1.630g,3.74mmol,1.0当量)溶于甲醇的nh3(7n,在meoh中,60ml)中并在室温搅拌3h。用h2o(340ml)稀释混合物,产生无色沉淀,其用etoac(3
×
200ml)萃取。合并有机相,干燥并真空除去溶剂,得到苄基保护的cazadc 10(1.478g,3.63mmol,97%),为无色泡沫。从甲氧基三嗪9b开始并遵循相同的程序,也成功合成了10。
[0110]
r
f
=0.50(ch2cl2/meoh,9:1)。
[0111]
hrms[esi
+
]:[c
23
h
27
n4o3]
+
,[m+h
+
]
+
计算值:407.2078,发现值:407.2081。
[0112]1h
‑
nmr(599mhz,cdcl3):δ/ppm=8.01(s,1h,c6‑
h),7.35
‑
7.23(m,10h,ar
‑
h),5.78(s,2h,nh),4.92
‑
4.76(m,4h,c7’
‑
h和c8’
‑
h),4.59
‑
4.46(m,1h,c1’‑
h),4.07
‑
3.95(m,1h,c3’
‑
h),3.55
‑
3.45(m,2h,c5’
‑
h),2.44
‑
2.28(m,2h,c6’
‑
h
a
,c4’
‑
h),2.24
‑
2.16(m,1h,c2’
‑
h
a
),2.14
‑
2.05(m,1h,c2’
‑
h
b
),1.78
‑
1.67(m,1h,c6’
‑
h
b
)。
[0113]
13
c
‑
nmr(151mhz,cdcl3):δ/ppm=165.7(c4),157.1(c6),154.2(c2),138.3,138.3,128.6,128.5,127.84,127.78(c
‑
ar),80.4(c3’
),73.4(c8’
),71.4(c5’
),71.2(c8’
),56.7(c1’
),44.9(c4’
),36.8(c2’
),32.5(c6’
)。
[0114]4‑
氨基
‑1‑
[(1’r,3’s,4’r)
‑3’‑
羟基
‑4’‑
(羟甲基)环戊基]
‑
1h
‑
[1,3,5]三嗪
‑2‑
酮(cazadc,1):
[0115][0116]
c9h
14
n4o3[0117]
226.24g/mol
[0118]
将苄基保护的10(1.478g,3.63mmol,1.0当量)在ch2cl2(100ml)中的溶液冷却至
‑
78℃并逐滴加入bcl3(1m,在dcm中,12.7ml,12.7mmol,3.5当量)。在
‑
78℃搅拌混合物1h并在室温搅拌额外的2h。通过在恒定搅拌下加入meoh(85ml)淬灭反应。真空除去溶剂。通过在硅胶上柱色谱用逐步梯度的ch2cl2/meoh(9:1
→
7:3)纯化粗产物以得到cazadc(1,0.740g,3.27mmol,90%)。从热meoh中重结晶得到57%的cazadc(1,465.3mg,2.06mmol),为无色针状单晶。
[0119]
r
f
=0.1(ch2cl2/meoh,9:1)。
[0120]
mp.:198
‑
200℃。
[0121]
hrms[esi
+
]:[c9h
15
n4o3]
+
,[m+h
+
]
+
计算值:227.1138,发现值:227.1139。
[0122]1h
‑
nmr(400mhz,d2o):δ/ppm=8.29(s,1h,c6‑
h),4.90
‑
4.62(m,1h,c1’
‑
h,和d2o的信号交叠),4.29
‑
4.16(m,1h,c3’
‑
h),3.73(dd,2j5’
a,5’b
=11.2hz,3j5’
a,4’=5.7hz,1h,c5’
a
‑
h),3.62(dd,2j5’
a,5’b
=11.2hz,3j5’
b,4’=6.8hz,1h,c5’
b
‑
h),2.41
‑
2.29(m,1h,c6’
a
‑
h),2.28
‑
2.18(m,1h,c2’
a
‑
h),2.17
‑
2.02(m,2h,c2’
b
‑
h,c4’
b
‑
h),1.77
‑
1.55(m,1h,c6’
b
‑
h)。
[0123]
13
c
‑
nmr(101mhz,cdcl3):δ/ppm=165.5(c4),158.5(c6),156.4(c2),72.2(c3’
),62.8(c5’
),56.2(c1’
),48.11(c4’
),38.0(c2’
),32.0(c6’
)。
[0124]
ftir(atr):1653(m),1540(s),1437(s),1278
[0125]
(s),1036(s)。
[0126]
实施例2:cazadc的x射线晶体结构分析
[0127]
通过x射线晶体学分析cazadc单晶。表1显示了晶体中两种不同环戊烷构象的cazadc 1的解析结构。一种构象异构体采用c6
’‑
内向(p=88.2
°
,最大=47.8
°
)构象(图2a),而第二作为c2
’‑
内向
‑
c3
’‑
外向(南,p=150.8
°
,最大=45.4
°
)构象异构体存在(图2b)。后一种构象对于dna中的2
’‑
脱氧核苷是典型的。
[0128]
使用mercury 3.5.1程序(剑桥晶体学数据中心)生成了更多图。数据存放在ccdc 1910952下。
[0129]
表1:cazadc的晶体学数据:
[0130][0131]
实施例3:对水解稳定性的测试
[0132]
接下来,研究了cazadc 1的水解稳定性,并与药物azadc进行了直接比较。
[0133]
两种核苷都以100mm的浓度溶解在kh2po4水溶液缓冲液(100mm,ph=7.4、5.5和8.5)中。立即通过hplc分析溶液(t=0h),随后每1.5h或在指定时间点进行hplc分析。使用ec 250/4nucleosil 120
‑
3c18(macherey
‑
nagel)色谱柱作为固定相。流动相由水(缓冲液a)和乙腈(缓冲液b)组成,流速为0.5ml/min,如下所示:0
→
25min,0
→
5%缓冲液b;25min
→
28min,5%
→
80%;28min
→
38min,80%;38min
→
43min,80%
→
0%;43min
→
45min,0%。
[0134]
由于azadc的一个治疗周期超过4周,决定在与半个周期(14天)相关的时间点测量稳定性。将溶液保持在室温后测量nmr谱图。
[0135]
由于肿瘤细胞通常提供酸性微环境[16],微酸性ph值下的稳定性特别有用。从图3中显示的数据可明显看出,药物azadc在这14天内强烈降解。重要的是,在ph=5.5和ph=8.5时,几乎无法再检测到完整的azadc。在生理ph值(7.4),14天后azadc仍然存在,但降解
程度显著。
[0136]
与这些结果相反,惊讶地观察到cazadc 1在所有测试的ph值下都没有降解,包括ph=5.5。该结果令人惊讶地发现,简单的o
→
ch2交换会引起强烈的远程解除效应,这似乎改变了三嗪环的性质,从而停止了与水的反应。
[0137]
实施例4
‑
生物学功能测试
[0138]
用于cazadc处理的mesc细胞培养
[0139]
如前所述[25],在血清和lif存在下培养不依赖饲养层的野生型j1(129s4/svjae菌株)[24]细胞。它们通常保持在2i/l培养基中的糊化板上。对于引发实验,2i培养物在适用时在补充有fbs和lif的dmem中传代,如上所述但缺乏抑制剂。对于药物处理,通过从培养基中去除2i使细胞进入引发状态。细胞在6孔板(vwr)中在补充有fbs和lif的dmem中孵育2天。分裂后,将2x105个细胞转移到6孔板培养皿中,并补充1μm(在0.01%dmso中)或5μm cazadc(在0.05%dmso中)并处理72h。去除培养基并用dpbs洗涤细胞后,将它们直接用rlt
+
缓冲液裂解,如先前出版物[18]中所述。
[0140]
gdna分离、总酶消化和uhplc
‑
ms2[0141]
如前所述[18],分离gdna。将gdna直接进行总酶消化并使用uhplc
‑
ms2进行分析,如先前的出版物[19]中所述。外部校准曲线是通过连续稀释纯cazadc[20pmol、10pmol、5pmol、2.5pmol、1.25pmol、0.625pmol、0.3125pmol]并在每次测量之前以技术三次重复测量来生成的。线性回归由originpro 2016g完成。
[0142]
结果
[0143]
研究了实施例3中描述的解除效应是否会影响生物学功能。为此,使用在血清/lif中引发的小鼠胚胎干细胞(mesc)作为模型系统,因为mdc水平从初始状态增加到引发状态[17]。将两种不同浓度(1μm和5μm)的cazadc 1添加到已引发48h的mesc,并允许细胞在cazadc1存在的情况下在引发条件下进一步增殖72h。72h后,收获细胞,分离dna并使用我们描述的方案[18]将dna消化到核苷水平。
[0144]
使用同位素稀释uhplc
‑
ms2最终精确定量mdc的水平。为此,同位素标记的核苷标准品被掺入以进行精确定量[19,18]。除了mdc之外,还量化了5羟甲基
‑2’‑
脱氧胞苷(hmdc)(其通过tet酶的作用由mdc形成[20
‑
21])的水平。mesc中hmdc的绝对水平比mdc水平低十倍以上[20,22]。结果是即使在mdc大幅减少之后,也应该有足够的mdc来保持hmdc水平不变。因此,hmdc水平是否受到影响以及受到多大影响的问题可以告诉表观遗传重编程是如何组织的。
[0145]
并行于mdc和hmdc的量化,还量化了cazadc 1本身被并入mesc基因组的扩展。只有在用nabh4处理dna后才能在处理过的细胞的基因组中检测azadc。nabh4的应用减少了c(5)=c(6)双键,从而稳定了化合物,使其量化成为可能[23,19]。令人高兴的是,我们注意到cazadc 1的稳定性允许在没有这种预处理的情况下对其进行量化。还注意到,即使在存在大量cazadc 1的情况下,所应用的酶消化方案也允许消化基因组dna(gdna)。总之,使用外部校准曲线通过uhplc
‑
ms2对cazadc 1进行量化可以与典范和表观遗传碱基的量化并行。
[0146]
在1μm cazadc 1浓度下,检测到cazadc 1水平为每dn 5x10
‑4cazadc(图4a)。这相当于整合到基因组中的近300万个cazadc核苷酸。在5μm cazadc 1的较高浓度下,水平增加了3倍,达到每dn1.7x10
‑
3 cazadc,因此每个基因组的整合cazadc超过800万个。相比于
azadc的并入,当应用1μm时,每dn达到1.2x10
‑
3 azadc[19],cazadc 1的水平达到该水平的大约三分之一。数据清楚地表明azadc的碳环版本(cazadc 1)被并入,并且它在5μm浓度下最终达到了基因组中的可比水平。
[0147]
重要的是,在培养基中在1m cazadc下将mesc暴露72h后,检测到mdc值降低了近30%(图4b)。在培养基中5m的浓度下,mdc水平甚至下降到原始值的50%左右。也观察到azadc减少到50%。然而,在这里,更快地达到50%减少(24h),并且已经在较低的azadc浓度(1μm)情况下[19]。
[0148]
数据显示,碳环版本的cazadc 1只需要更多的时间来以相同的量影响mdc水平。我们认为这种效应是由cazadc 1向三磷酸酯的潜在较慢转化引起的。然而,鉴于临床应用的治疗时间较长,cazadc 1的较慢动力学不一定是缺点。
[0149]
非常有趣的是,发现在1μm实验中hmdc水平已经降低到约50%。在5μm时,甚至无法使用0.5μg的基因组dna检测到高于背景水平的hmdc。结果显示hmdc水平下降甚至比mdc水平还要快,尽管hmdc在基因组中的丰度要低十倍以上。这个结果很有趣。这表明hmdc可能主要在细胞分裂期间的mdc维持过程中产生。在这里看到化合物cazadc 1是一种完美的工具分子,现在允许进一步了解在dc到mdc的甲基化和mdc到hmdc的氧化之间的相互作用。在具有新化合物cazadc 1的情况下,可以将基因组的去甲基化与相应的细胞效应清晰地关联起来,而不会损害dna损伤效应。最后,cazadc 1可能不仅是一种有价值的工具化合物,而且可能是下一代表观遗传药物。
[0150]
总结
[0151]
我们表明,用ch2单元取代环内的o原子稳定了药物,从而停止其与水的亲核反应。新的核苷cazadc 1被细胞中的磷酸化酶接受,相应的cazadc
‑
三磷酸酯被有效地并入到基因组中。cazadc 1以数百万个核苷酸并入基因组中,它导致mdc水平相对于对照水平降低至70%。
[0152]
实施例5:cazac(20)的合成程序
[0153]
叔丁基
‑
(1r,4s,5r,6s)
‑
5,6
‑
二羟基
‑3‑
氧代
‑2‑
氮杂双环[2.2.1]庚烷
‑2‑
羧酸酯12:
[0154][0155]
将boc保护的文斯(vince)内酰胺11[30](11.66g,55.7mmol,1.0当量)溶解在214ml thf、92ml叔丁醇和31ml h2o中,加入22.59g n
‑
甲基吗啉
‑
n
‑
氧化物(167.2mmol,3当量)和1.03g k2oso4二水合物(2.79mmol,5mol%)并将混合物在室温搅拌16h。随后,加入200ml水和35.1g na2so3(278.5mmol,5.0当量)并在室温搅拌混合物额外的1h。随后,用etoac(3
×
300ml)萃取反应混合物。合并的有机层用饱和nacl水溶液(300ml)洗涤并在na2so4上干燥。在真空中蒸发溶剂后,通过在硅胶上柱色谱(ch2cl2:meoh 40:1
→
ch2cl2:meoh30:1)纯化残余物以产生2.91g的外型双羟基化产物12(12.0mmol,22%),为黄色泡沫。
[0156]1h
‑
nmr(400mhz,cdcl3):δ/ppm=4.35
‑
4.32(m,1h,1
‑
h),4.23(d,j=6.0hz,1h,5
‑
h),4.08(d,j=6.1hz,1h,6
‑
h),3.89
‑
3.80(br s,
‑
oh),3.78
‑
3.68(br s,
‑
oh)2.82
‑
2.76
(m,1h,4
‑
h),2.09(dt,j=10.9,1.5hz,1h,7
‑
h),1.97(m,1h,7
‑
h),1.51(s,9h,3
′‑
h)。
[0157]
13
c
‑
nmr(101mhz,cdcl3):δ/ppm=173.30(3
‑
c),149.20(1
′‑
c),83.82(2
′‑
c),70.57(6
‑
c),68.14(5
‑
c),62.29(1
‑
c),53.74(4
‑
c),31.99(7
‑
c),28.16(3
′‑
c)。
[0158]
ir(atr):ν(cm
‑1)=3415(w),2979(w),1777(m),1714(m),1457
[0159]
(w),1394(w),1368(m),1350(m),1326(m),1297(m),1256(m),1144(s),1081(s),1043(w),1031(w),1016(m),947(s),902(w),838(w),776(m),733(s),702(m)。
[0160]
r
f
(ch2cl2:meoh 20:1):0.24。
[0161]
叔丁基
‑
(1r,2s,3r,4r)
‑
2,3
‑
二羟基
‑4‑
(羟甲基)
‑
环戊基)
‑
氨基甲酸酯13:
[0162][0163]
将双羟基化内酰胺12(2.90g,11.9mmol,1.0当量)在40ml干燥meoh中的溶液冷却至0℃,并在15min内分5份添加nabh4(2.25g,59.61mmol,5.0当量)。将混合物在0℃进一步搅拌45min,升温至室温并在室温搅拌额外的1h。随后,加入100ml乙酸(10%v/v,在meoh中)和硅胶并将混合物真空浓缩至干。通过硅胶柱色谱纯化(干负载,ch2cl2:meoh 5:1
→
ch2cl2:meoh 4:1)并与干燥甲苯(2
×
150ml)共蒸发得到产物13,为其乙酸盐(2.96g,9.6mmol,81%),为略带棕色的油。
[0164]1h
‑
nmr(400mhz,dmso
‑
d6):δ/ppm=6.70(d,j=7.9hz,1h,nh),3.65
‑
3.49(m,2h,1
‑
h,3
‑
h),3.52(dd,j=6.3,5.2hz,1h,2
‑
h),3.37(dd,j=10.5,5.8hz,1h,5
‑
h),3.30(dd,j=10.5,6.2hz,1h,5
‑
h),1.98(dt,j=13.1,8.4hz,1h,6
‑
h),1.86
‑
1.77(m,4h,4
‑
h,oac),1.37(s,9h,3
′‑
h),1.04
‑
0.90(m,1h,6
‑
h)。
[0165]
13
c
‑
nmr(101mhz,dmso
‑
d6):δ/ppm=173.28(oac
‑
c1),155.23(c
‑1′
),77.37(2
′‑
c),75.93(2
‑
c),72.07(1
‑
c),63.02(5
‑
c),55.07(3
‑
c),44.94(4
‑
c),30.61(6
‑
c),28.30(3
′‑
c),22.49(oac
‑
c2)。
[0166]
r
f
(ch2cl2:meoh 8:1):0.43。
[0167]
叔丁基
‑
((1r,2s,3r,4r)
‑
2,3
‑
双(苄氧基)
‑4‑
((苄氧基)甲基)
‑
环戊基)
‑
氨基甲酸酯14:
[0168]
[0169]
将环戊烷13(2.93g,9.5mmol,1.0当量)在67ml干燥dmf中的溶液冷却至0℃并分3份添加nah(1.57g,39.3mmol,4.1当量,在矿物油中的60%溶液)。在0℃搅拌混合物15min后,缓慢加入5.31ml bnbr(44.6mmol,4.7当量)并将反应混合物在0℃下搅拌2.5h并在室温下搅拌3h。然后,添加220mg tbai(0.6mmol,6mol%)和另外3.18ml bnbr(26.8mmol,2.8当量),并将混合物加热至80℃3.5h。随后,将混合物冷却至室温,倒入300ml饱和nahco3水溶液中,并用ch2cl2(3
×
300ml)萃取。合并的有机层用饱和nahco3水溶液(300ml)、饱和nacl水溶液(300ml)洗涤,经无水na2so4干燥并在50℃真空浓缩。通过柱色谱(ihex:etoac 10:1
→
ihex:etoac8:1
→
ihex:etoac 7:1
→
ihex:etoac 6:1
→
ihex:etoac 4:1)纯化残余物以得到三苄基保护的产物14(2.47g,4.8mmol,51%)无色固体蜡。
[0170]1h
‑
nmr(400mhz,cdcl3):δ/ppm=7.45
‑
7.09(m,15h,3
×
och2ph),4.95(s,1h,nh),4.76
‑
4.25(m,6h,3
×
och2ph),4.15
‑
3.90(m,1h,1
‑
h),3.82(dd,j=6.7,4.4hz,1h.3
‑
h),3.79
‑
3.63(m,1h,2
‑
h),3.57
‑
3.34(m,2h,5
‑
h),2.51
‑
2.24(m,2h,4
‑
h,6
‑
h),1.50
‑
1.34(m,9h,3
′‑
h),1.31
‑
1.19(m,1h,6
‑
h)。
[0171]
13
c
‑
nmr(101mhz,cdcl3):δ/ppm=155.09(1
′‑
c),138.48(ar
‑
c),138.43(ar
‑
c),138.13(ar
‑
c),128.44(ar
‑
c),128.30(ar
‑
c),128.24(ar
‑
c),128.03(ar
‑
c),127.88(ar
‑
c),127.82(ar
‑
c),127.70(ar
‑
c),127.54(ar
‑
c),127.51(ar
‑
c),81.27(2
‑
c),79.29(3
‑
c),79.13(2
′‑
c),73.30(och2‑
ph),71.52(och2‑
ph),71.02(och2‑
ph),70.41(5
‑
c),52.85(1
‑
c),41.49(1
‑
c),30.34(6
‑
c),28.46(3
′‑
c)。
[0172]
hrms(esi
+
):对于c
32
h
40
no
5+
[m+h]
+
,计算值:518.2901;发现值:518.2911。
[0173]
ir(atr):ν(cm
‑1)=3331(br w),3029(w),2974(w),2867(w),1705(s),1495(m),1453(m),1390(w),1364(s),1245(m),1165(s),1092(s),1071(s),1027(m),732(s),695(s)。
[0174]
r
f
(ihex:etoac 4:1):0.69。
[0175]
(1r,2s,3r,4r)
‑
2,3
‑
双(苄氧基)
‑4‑
(苄氧基)甲基)
‑
环戊烷
‑1‑
胺15:
[0176][0177]
在ar气氛下,将1.02g boc保护的环戊烷14(1.97mmol,1.0当量)溶解在7.0ml干燥ch2cl2中,添加3.0ml tfa并将混合物在室温下搅拌1h。将红色溶液浓缩至干,加入20.0ml饱和nahco3水溶液并将混合物在室温下搅拌10min。然后,添加60ml etoac,分离各相,水层进一步用etoac(2
×
60ml)萃取。合并的有机层经na2so4干燥并真空浓缩至干,得到脱保护的胺15,为黄色油状物(882mg,1.97mmol,定量),由于其对氧化的不稳定性,无需进一步纯化即可用于下一步骤。
[0178]1h
‑
nmr(400mhz,cdcl3):δ/ppm=7.39
‑
7.26(m,15h,3
×
och2ph),4.63
‑
4.33(m,6h,3
×
och2ph),3.81(dd,j=5.0,3.1hz,1h,3
‑
h),3.49(dt,j=9.1,7.8hz,1h,1
‑
h),3.41(dd,j=9.2,5.1hz,1h,5
‑
h),3.34
‑
3.26(m,2h,2
‑
h,5
‑
h),2.47
‑
2.34(m,1h,4
‑
h),2.24
‑
2.13(m,1h,6
‑
h),1.06
‑
0.94(m,1h,6
‑
h)。
[0179]
13
c
‑
nmr(101mhz,cdcl3):δ/ppm=138.66(ar
‑
c),138.52(ar
‑
c),138.48(ar
‑
c),128.72(ar
‑
c),128.52(ar
‑
c),128.47(ar
‑
c),128.38(ar
‑
c),128.31(ar
‑
c),128.00(ar
‑
c),127.74(ar
‑
c),127.67(ar
‑
c),127.13(ar
‑
c),86.10(2
‑
c),78.11(3
‑
c),73.21(och2‑
ph),72.27(5
‑
c),71.86(och2‑
ph),70.89(och2‑
ph),54.32(1
‑
c),41.49(4
‑
c),32.19(6
‑
c)。
[0180]
hrms(esi
+
):对于c
27
h
32
no
3+
[m+h]
+
,计算值:418.2377;发现值:418.2378。
[0181]1‑
[(1’r,2’s,3’r,4’r)
‑2’
,3
’‑
双(苄氧基)
‑4’‑
((苄氧基)甲基)
‑
环戊基]
‑
甲基异缩二脲16
[0182][0183]
在ar气氛下,将882mg环戊基胺15(1.87mmol,1.97mmol,1.0当量)溶解在18.20ml干燥mecn中,添加431mg 2
‑
甲基
‑1‑
(1
‑
咪唑基羰基)
‑
异脲
[1]
(2.56mmol,1.3当量)。将混合物在92℃下回流2h,随后冷却至室温并蒸发至干。残余物通过在硅胶上柱色谱纯化(ihex:etoac 3:1
→
ihex:etoac 5:2
→
ihex:etoac 2:1
→
ihex:etoac 1:1)得到无色油状的甲基异缩二脲16(886mg,1.17毫摩尔,87%)。
[0184]1h
‑
nmr(400mhz,cdcl3):δ/ppm=7.43
‑
7.22(m,15h,3
×
och2ph),5.60(s,1h,1
‑
h),4.80
‑
4.61(m,2h,och2ph),4.55
‑
4.41(m,3h,och2ph),4.32(d,j=11.8hz,1h,och2ph),4.24(td,j=7.6,3.4hz,1h,1
′‑
h),3.87(dd,j=6.9,4.5hz,1h,3
′‑
h),3.82(dd,j=4.5,3.1hz,1h,2
′‑
h),3.61(s,2h,3
‑
h,5
‑
h),3.57
‑
3.45(m,2h,5
′‑
h),2.54
‑
2.39(m,2h,4
′‑
h,6
′‑
h),2.17(s,3h,6
‑
h),1.39
‑
1.31(m,1h,6
′‑
h)。
[0185]
13
c
‑
nmr(101mhz,cdcl3):δ/ppm=207.11(4
‑
c),162.27(2
‑
c),138.74(ar
‑
c),138.71(ar
‑
c),138.45(ar
‑
c),128.51(ar
‑
c),128.34(ar
‑
c),128.28(ar
‑
c),127.86(ar
‑
c),127.70(ar
‑
c),127.59(ar
‑
c),127.56(ar
‑
c),81.65(2
′‑
c),79.79(3
′‑
c),73.35(och2‑
ph),71.64(och2‑
ph),71.20(och2‑
ph),70.60(5
′‑
c),52.56(1
′‑
c),41.68(4
′‑
c),31.07(6
‑
c),30.57(6
′‑
c)。
[0186]
hrms(esi
+
):对于c
30
h
36
n3o
5+
[m+h]
+
,计算值:518.2650;发现值:518.2652。
[0187]
ir(atr):ν(cm
‑1)=3384(br w),3028(w),2857(w),1694(w),1635(s),1495(s),1453(m),1364(m),1300(w),1197(w),1096(s),1027(m)799(w),736(m),697(s)。
[0188]
r
f
(ihex:etoac 2:1):0.37。
[0189]4‑
甲氧基
‑1‑
[(1’r,2’s,3’r,4’r)
‑2’
,3
’‑
双(苄氧基)
‑4’‑
((苄氧基)甲基)
‑
环戊基]
‑
1h
‑
[1,3,5]
‑
三嗪
‑2‑
酮(17)和4
‑
乙氧基
‑1‑
[(1’r,2’s,3’r,4’r)
‑2’
,3
’‑
双(苄氧基)
‑4’‑
((苄氧基)甲基)
‑
环戊基]
‑
1h
‑
[1,3,5]
‑
三嗪
‑2‑
酮(18):
[0190][0191]
将甲基异缩二脲16(837mg,1.62mmol,1.0当量)溶解在15.30ml原甲酸三乙酯中,并添加20μl tfa。将混合物加热至90℃持续3h,随后冷却至室温并在50℃真空浓缩。残余物与干燥甲醇共蒸发,并通过在硅胶上柱色谱(ihex:etoac 3:1
→
ihex:etoac 5:2
→
ihex:etoac 2:1
→
ihex:etoac 1:1
→
ihex:etoac 1:2)纯化,得到373mg乙氧基三嗪18(0.69mmol,43%)无色蜡和389mg甲氧基三嗪17(0.73mmol,46%)无色固体。
[0192]
甲氧基三嗪17:
[0193]1h
‑
nmr(400mhz,cdcl3):δ/ppm=7.96(s,1h,6
‑
h),7.38
‑
7.17(m,15h,3
×
och2ph),4.70
‑
4.40(m,6h,1
′‑
h,2.5
×
och2ph),4.36
‑
4.24(m,2h,2
′‑
h,0.5
×
och2ph),3.98(s,3h,7
‑
h),3.93(dd,j=4.9,3.1hz,1h,3
′‑
h),3.54
‑
3.40(m,2h,5
′‑
h),2.51(ddt,j=9.8,4.4,2.3hz,1h,4
′‑
h),2.35(dt,j=13.3,9.4hz,1h,6
′‑
h),1.82(ddd,j=13.4,9.0,6.7hz,1h,6
′‑
h)。
[0194]
13
c
‑
nmr(101mhz,cdcl3):δ/ppm=169.87(4
‑
c),159.59(6
‑
c),154.67(2
‑
c),138.16(ar
‑
c),138.14(ar
‑
c),137.63(ar
‑
c),128.63(ar
‑
c),128.60(ar
‑
c),128.51(ar
‑
c),128.24(ar
‑
c),128.20(ar
‑
c),128.17(ar
‑
c),127.90(ar
‑
c),127.86(ar
‑
c),78.78(2
′‑
c),77.27(3
′‑
c),73.40(och2‑
ph),72.17(och2‑
ph),71.41(och2‑
ph),71.20(5
′‑
c),63.34(1
′‑
c),55.95(7
‑
c),41.14(4
′‑
c),26.75(6
′‑
c)。
[0195]
hrms(esi
+
):对于c
31
h
34
n3o
5+
[m+h]
+
,计算值:528.2493;发现值:528.2498。
[0196]
ir(atr):ν(cm
‑1)=3029(w),2867(w),1698(s),1613(s),1515(m),1464(m),1453(m),1398(m),1337(m),1265(w),1211(w),1096(m),1070(m),1027(m),913(w),802(m),733(s),696(s)。
[0197]
r
f
(ihex:etoac 2:1):0.27。
[0198]
乙氧基三嗪18:
[0199]1h
‑
nmr(400mhz,cdcl3):δ/ppm=7.97(s,1h,6
‑
h),7.39
‑
7.15(m,15h,3
×
och2ph),4.66
‑
4.21(m,10h,1
′‑
h,2
′‑
h,7
‑
h,3
×
och2ph),3.96
‑
3.87(m,1h,3
′‑
h),,3.54
‑
3.36(m,2h,5
′‑
h),2.56
‑
2.41(m,1h,4
′‑
h),2.33(ddd,j=14.5,9.5,4.7hz,1h,6
′‑
h),1.88
‑
1.74(m,1h,6
′‑
h),1.25(q,j=6.9hz,3h,8
‑
h)。
[0200]
13
c
‑
nmr(101mhz,cdcl3):δ/ppm=169.31(4
‑
c),159.53(6
‑
c),154.74(2
‑
c),138.18(ar
‑
c),138.16(ar
‑
c),137.64(ar
‑
c),128.79(ar
‑
c),128.64(ar
‑
c),128.61(ar
‑
c),128.51(ar
‑
c),128.25(ar
‑
c),128.20(ar
‑
c),128.19(ar
‑
c),127.90(ar
‑
c),127.87(ar
‑
c),79.11(2
′‑
c),77.36(3
′‑
c),73.46(och2‑
ph),72.28(och2‑
ph),71.55(och2‑
ph),71.20(5
′‑
c),65.06(7
‑
c),63.33(1
′‑
c),41.15(4
′‑
c),26.81(6
′‑
c),14.24(8
‑
c)。
[0201]
hrms(esi
+
):对于c
32
h
36
n3o
5+
[m+h]
+
,计算值:542.2650;发现值:542.2656。
[0202]
ir(atr):ν(cm
‑1)=3064(w),3030(w),2924(w),2962(w),1694(s),1615(s),1514(m),1495(m),1452(s),1361(m),1326(m),1270(m),1206(w),1096(s),1070(s),1027(m),803(w),734 8s),697(s)。
[0203]
r
f
(ihex:etoac 2:1):0.30。
[0204]4‑
氨基
‑1‑
[(1’r,2’s,3’r,4’r)
‑2’
,3
’‑
双(苄氧基)
‑4’‑
((苄氧基)甲基)
‑
环戊基]
‑
1h
‑
[1,3,5]
‑
三嗪
‑2‑
酮19:
[0205][0206]
将乙氧基三嗪18(358mg,0.66mmol)和甲氧基三嗪17(363mg,0.69mmol)的混合物溶于26.8ml 7n nh3的甲醇溶液中,并在室温下搅拌3h。随后,将125ml h2o加入到反应混合物中,然后用etoac(3
×
200ml)萃取。合并的有机层经na2so4干燥并真空蒸发至干,得到593mg呈无色泡沫状的胺化产物19(1.16mmol,86%),其无需进一步纯化即可用于下一步骤。
[0207]1h
‑
nmr(400mhz,cdcl3):δ/ppm=7.80(s,1h,6
‑
h),7.26(m,15h,3
×
och2ph),5.70(s,1h,nh),4.63
‑
4.42(m,6h,1
′‑
h,2.5
×
och2ph),4.39
‑
4.28(m,2h,2
′‑
h,0.5
×
och2ph),3.91(dd,j=5.0,3.5hz,1h,3
′‑
h),3.53
‑
3.42(m,2h,5
′‑
h),2.59
‑
2.35(m,1h,4
′‑
h),2.26(dt,j=13.3,9.3hz,1h,6
′‑
h),1.86(ddd,j=13.3,9.2,7.1hz,1h,6
′‑
h)。
[0208]
13
c
‑
nmr(101mhz,cdcl3):δ/ppm=165.84(4
‑
c),158.30(6
‑
c),154.11(2
‑
c),138.27(ar
‑
c),138.24(ar
‑
c),137.83(ar
‑
c),128.57(ar
‑
c),128.56(ar
‑
c),128.47(ar
‑
c),128.21(ar
‑
c),128.12(ar
‑
c),128.05(ar
‑
c),127.84(ar
‑
c),127.82(ar
‑
c),127.78(ar
‑
c),78.99(2
′‑
c),77.39(3
′‑
c),73.30(och2‑
ph),72.18(och2‑
ph),71.39(och2‑
ph),71.3(5
′‑
c)7,63.53(1
′‑
c),41.21(4
′‑
c),26.79(6
′‑
c)。
[0209]
hrms(esi
+
):对于c
30
h
33
n4o
4+
[m+h]
+
,计算值:513.2496;发现值:513.2496。
[0210]
ir(atr):ν(cm
‑1)=3331(br w),3182(br w),3060(w),3028(w),2925(w),2857(w),1633(s),1506(m),1495 8m),1470(m),1452 8s),1361(w),1262(w),1092(s),1070(s),1027(m),912(w),799(m),734(s),696(s)。
[0211]
r
f
(ch2cl2:meoh 15:1):0.42。
[0212]4‑
氨基
‑1‑
[(1’r,2’s,3’r,4’r)
‑2’
,3
’‑
双羟基
‑4’‑
(羟甲基)
‑
环戊基]
‑
1h
‑
[1,3,5]
‑
三嗪
‑2‑
酮20:
[0213]
[0214]
在ar气氛下,将苄基保护的5
‑
氮杂
‑
carbac 19(576mg,1.12mmol,1.0当量)的溶液溶解在30.8ml干燥ch2cl2中并冷却至
‑
78℃。然后,逐滴加入5.88ml bcl3(1m,在ch2cl2中,5.88mmol,5.25当量),将混合物在
‑
78℃下搅拌1h,在室温下搅拌2h。随后,加入40ml干燥meoh,将混合物在室温下再搅拌20min并真空浓缩至干。残余物通过在硅胶上柱色谱(干负载,ch2cl2:meoh 8:1
→
ch2cl2:meoh 5:1
→
ch2cl2:meoh 4:1
→
ch2cl2:meoh 7:3)纯化,得到228mg脱保护化合物20(0.94mmol,84%),为无色固体。
[0215]
对于细胞生物学应用,34.5mg化合物通过反相hplc(0%至3%mecn的h2o溶液,45min,流速5ml/min)进一步纯化,得到25.3mg经hplc纯化的20(73%)。
[0216]1h
‑
nmr(400mhz,d2o):δ/ppm=8.32(s,1h,6
‑
h),4.57
‑
4.33(m,2h,1
′‑
h,2
′‑
h),4.04(dd,j=5.3,4.4hz,1h,3
′‑
h),3.76
‑
3.57(m,2h,5
′‑
h),2.29(dt,j=13.0,8.2hz,1h,6
′‑
h),2.23
‑
2.15(m,1h,4
′‑
h),1.76
‑
1.59(m,1h,6
′‑
h)。
[0217]
13
c
‑
nmr(101mhz,d2o):δ/ppm=165.45(4
‑
c),159.03(6
‑
c),156.15(2
‑
c),73.12(2
′‑
c),71.77(3
′‑
c),63.55(1
′‑
c),62.90(5
′‑
c),44.42(4
′‑
c),27.17(6
′‑
c)。
[0218]
hrms(esi
+
):针对c9h
15
n4o
4+
[m+h]
+
,计算值:243.1088;发现值:243.1086。
[0219]
hrms(esi
‑
):针对c9h
13
n4o4‑
[m
‑
h]
‑
,计算值:241.0942;发现值:241.0941。
[0220]
r
f
(ch2cl2:meoh 7:3):0.29。
[0221]
在hplc稳定性测试中,表明碳环化合物cazac 20比阿扎胞苷(地西他滨)稳定得多(图5)。
[0222]
实施例6
[0223]
cazac前药24的合成程序
[0224]
为了改善向细胞的递送,还制备了cazac 20的5
′‑
氨基磷酸酯前药24。
[0225]4‑
氨基
‑1‑
((3as,4r,6r,6ar)
‑6‑
(羟甲基)
‑
2,2
‑
二甲基四氢
‑
4h
‑
环戊二烯并
‑
[d][1,3]二氧戊环
‑4‑
基)
‑
1,3,5
‑
三嗪
‑
2(1h)
‑
酮21
[0226][0227]
在加热干燥的烧瓶中,将5
‑
氮杂
‑
carbac 20(64mg,264μmol,1.0当量)悬浮在6.60ml干燥丙酮中。添加对甲苯磺酸(5mg,催化量)和2,2
‑
二甲氧基丙烷(49μl,396μmol,1.5当量),并将混合物在室温下搅拌18h。向所得溶液中加入150μl net3(1.08mmol)并将混合物在室温下再搅拌10min。随后,将混合物浓缩至干,所得残余物通过在硅胶上柱色谱(ch2cl2:meoh 10:1,含有1%net3)纯化,得到81mg异亚丙基
‑
乙缩醛保护的化合物21(182μmol,69%,与1.6当量net3的加合物),为无色固体。
[0228]1h
‑
nmr(400mhz,d3cod):δ/ppm=8.29(s,1h,6
‑
h),4.94(dd,j=7.0,4.9hz,1h,2
′‑
h),4.62
‑
4.39(m,2h,1
′‑
h,3
′‑
h),3.73
‑
3.51(m,2h,5
′‑
h),3.21(q,j=7.3hz,10h,n
(ch2ch3)3),2.32
‑
2.07(m,3h,4
′‑
h,6
′‑
h),1.50(s,3h,1
″‑
h),1.32(t,j=7.3hz,15h,n(ch2ch3)3),1.29(s,3h,1
″‑
h)。
[0229]
13
c
‑
nmr(101mhz,d3cod):δ/ppm=167.87(4
‑
c),159.88(6
‑
c),156.66(2
‑
c),114.31(2
″‑
c),83.95(2
′‑
c),82.98(3
′‑
c),66.26(1
′‑
c),64.17(5
′‑
c),47.88(n(ch2ch3)3),47.81(4
′‑
c),33.72(6
′‑
c),27.97(1
″‑
c),25.53(1
″‑
c),9.23(n(ch2ch3)3)。
[0230]
hrms(esi
+
):针对c
12
h
19
n4o
4+
[m+h]
+
,计算值:283.1401;发现值:283.1397。
[0231]
ir(atr):ν(cm
‑1)=3346(br w(,2983(w),2658(w),1634(s),1472(s),1210(m),1160(m),1065(m),868(w),800(m),682(w)。
[0232]
r
f
(ch2cl2:meoh 6:1):0.36。
[0233]
异丙基((((3ar,4r,6r,6as)
‑6‑
(4
‑
氨基
‑2‑
氧代
‑
1,3,5
‑
三嗪
‑
1(2h)
‑
基)
‑
2,2
‑
二甲基四氢
‑
4h
‑
环戊二烯并[d][1,3]二氧戊环
‑4‑
基)甲氧基)(苯氧基)磷酰基)
‑
l
‑
丙氨酸酯22:
[0234][0235]
将受保护的5
‑
氮杂
‑
carbac 21(69mg,155μmol,1.0当量,作为1.6当量net3‑
加合物)悬浮在2.40ml干燥thf中并冷却至
‑
78℃。然后加入855μl tbumgcl溶液(1.0m,在thf中,855μmol,5.5当量),将混合物直接加热至0℃并在该温度下搅拌30min。随后,将混合物再次冷却至
‑
78℃并滴加538μl氯化磷氧芳基酯[31]23的溶液(1.0m,在thf中,538μmol,3.5当量)。使反应混合物缓慢升温至室温并搅拌17h。通过加入h2o(20ml)淬灭所得澄清黄色溶液,并用etoac(3
×
25ml)萃取水层。合并的有机相用饱和nacl水溶液(25ml)洗涤,经干燥na2so4干燥并真空浓缩至干。残余物通过在硅胶上柱色谱(ch2cl2:meoh 60:1
→
ch2cl2:meoh 50:1
→
ch2cl2:meoh 40:1)纯化,得到83mg产物12(78μmol,51%,与5.0当量net3的加合物),作为p上的两种非对映异构体的混合物,为无色固体。
[0236]1h
‑
nmr(800mhz,cdcl3):δ/ppm=8.05(2
×
s,1h),7.32
‑
7.27(m,2h,oph),7.23
‑
7.17(m,2h,oph),7.15
‑
7.07(m,1h,oph),5.04
‑
4.87(m,2h,2
′‑
h,4
″′‑
h),4.59
‑
4.54(m,1h,3
′‑
h),4.31
‑
4.13(m,3h,1
′‑
h,5
′‑
h),4.02
‑
3.93(m,1h,2
″′‑
h),3.78(m,1h,nh),3.09(q,j=7.4hz,30h,n(ch2ch3)3),2.53
‑
2.17(m,3h,4
′‑
h,6
′‑
h),1.49
‑
1.47(m,3h,1
″‑
h),1.39(t,j=7.3hz,45h,n(ch2ch3)3),1.37
‑
1.34(m,3h,3
″‑
h),1.26
‑
1.24(m,3h,1
″‑
h),1.21
‑
.18(m,6h,5
″′‑
h)。
[0237]
13
c
‑
nmr(201mhz,cdcl3):δ/ppm=173.10(1
″′‑
c),165.39(4
‑
c),158.57(6
‑
c),153.13(o
‑
c
ar
),150.83(2
‑
c),129.73(ch
ar
),124.99(ch
ar
),120.45(ch
ar
),113.57(2
″‑
c),82.42(2
′‑
c),81.17(3
′‑
c),69.28(4
″′‑
c),67.50(1
′‑
c),65.93(5
′‑
c),50.50(2
″′‑
c),45.91(n(ch2ch3)3),44.93(4
′‑
c),32.34(6
′‑
c),29.79(5
″′‑
c),27.73(1
″‑
c),25.36(1
″‑
c),21.72(5
″′‑
c),21.13(3
″′‑
c),8.75(n(ch2ch3)3)。
[0238]
31
p
‑
nmr(162mhz,cdcl3):δ/ppm=2.46,2.36。
[0239]
hrms(esi
+
):对于c
24
h
35
n5o8p
+
[m+h]
+
,计算值:552.2218;发现值:552.2211。
[0240]
hrms(esi
‑
):对于c
24
h
33
n5o8p
‑
[m
‑
h]
‑
,计算值:550.2072;发现值:550.2074。
[0241]
ir(atr):ν(cm
‑1)=3379(m),2980(s),2946(m),2606(s),2498(s),1640(s),1476(s),1398(m),1383(m),1212(s),1158(m),1107(w),1070(m),1037(s),929(m),802(w),730(m)。
[0242]
r
f
(ch2cl2:meoh 20:1):0.25。
[0243]
异丙基((((1r,2r,3s,4r)
‑4‑
(4
‑
氨基
‑2‑
氧代
‑
1,3,5
‑
三嗪
‑
1(2h)
‑
基)
‑
2,3
‑
二羟基环戊基)甲氧基)(苯氧基)磷酰基)
‑
l
‑
丙氨酸酯24:
[0244][0245]
向异亚丙基乙缩醛保护的化合物22(38mg,69μmol,1.0当量)中加入1.0ml 80%tfa的ddh2o溶液,并将混合物在30℃下搅拌20min。然后,将混合物倒入5.0ml ddh2o中并冻干。将所得残留物溶解在500μl mecn、500μl dmso和4.0ml ddh2o(含0.1%tfa)的混合物中,过滤混浊溶液并通过反相hplc(5%mecn的ddh2o(含0.1%tfa)溶液至60%mecn的ddh2o(含0.1%tfa)溶液,45min,流速5ml/min)纯化,得到10.4mg目标化合物24(20μmol,29%),为无色固体。
[0246]1h
‑
nmr(400mhz,d3ccn):δ/ppm=8.11(s,1h),7.50(s,1h,nh2),7.41
‑
7.29(m,2h,oph),7.25
‑
7.15(m,3h,oph),6.61(s,1h,nh2),4.98
‑
4.88(m,1h,4
″‑
h),4.39
‑
4.23(m,3h,1
′‑
h,2
′‑
h,nh),4.20
‑
4.07(m,2h,5
′‑
h),3.97(dt,j=5.2,3.8hz,1h,3
′‑
h),3.93
‑
3.82(m,1h,2
″‑
h),2.33
‑
2.09(m,2h,4
′‑
h,6
′‑
h),1.83
‑
1.67(m,1h,6
′‑
h),1.30
‑
1.26(m,3h,3
″‑
h),1.19(dt,j=6.2,3.4hz,6h,5
″‑
h)。
[0247]
13
c
‑
nmr(101mhz,d3ccn):δ/ppm=174.00(1
″‑
c),165.92(4
‑
c),159.92(6
‑
c),153.84(2
‑
c),151.97(o
‑
c
ar
),130.62(ch
ar
),125.75(ch
ar
),121.36(ch
ar
),121.31(ch
ar
),74.12(2
′‑
c),72.50(3
′‑
c),69.64(4
″‑
c),68.53(5
′‑
c),64.96(1
′‑
c),51.44(2
″‑
c),44.44(4
′‑
c),28.08(6
′‑
c),21.89(5
″‑
c),21.81(5
″‑
c),20.80(3
″‑
c)。
[0248]
31
p
‑
nmr(162mhz,d3ccn):δ/ppm=3.08,2.92。
[0249]
hrms(esi
+
):对于c
21
h
31
n5o8p
+
[m+h]
+
,计算值:512.1905;发现值:512.1901。
[0250]
hrms(esi
‑
):对于c
21
h
29
n5o8p
‑
[m
‑
h]
‑
,计算值:510.1759;发现值:510.1757。
[0251]
参考文献列表:
[0252]
[1]m.daskalakis,t.t.nguyen,c.nguyen,p.guldberg,g.kohler,p.wijermans,p.a.jones,m.lubbert,blood 2002,100,2957
‑
2964.
[0253]
[2]h.kantarjian,j.p.issa,c.s.rosenfeld,j.m.bennett,m.albitar,
j.dipersio,v.klimek,j.slack,c.de castro,f.ravandi,r.helmer,3rd,l.shen,s.d.nimer,r.leavitt,a.raza,h.saba,cancer 2006,106,1794
‑
1803.
[0254]
[3]c.stresemann,f.lyko,int.j.cancer 2008,123,8
‑
13.
[0255]
[4]y.koh,y.a.kim,k.kim,j.
‑
a.sim,s.
‑
s.yoon,s.m.park,y.h.yun,blood 2016,128,2381
‑
2381.
[0256]
[5]m.nieto,p.demolis,e.b
é
hanzin,a.moreau,i.hudson,b.flores,h.ste熔点lewski,t.salmonson,c.gisselbrecht,d.bowen,f.pignatti,oncologist 2016,21,692
‑
700.
[0257]
[6]s.s.smith,b.e.kaplan,l.c.sowers,e.m.newman,proc.natl.acad.sci.1992,89,4744
‑
4748.
[0258]
[7]g.g.wilson,r.j.roberts,s.kumar,j.posfai,m.sha,s.klimasauskas,
×
.cheng,nucleic acids res.1994,22,1
‑
10.
[0259]
[8]a.bird,genes dev.2002,16,6
‑
21.
[0260]
[9]r.j.klose,a.p.bird,trends biochem.sci.2006,31,89
‑
97.
[0261]
[10]r.j
ü
ttermann,e.li,r.jaenisch,proc.natl.acad.sci.1994,91,11797
‑
11801.
[0262]
[11]s.gabbara,a.s.bhagwat,biochem.j.1995,307,87
‑
92.
[0263]
[12]m.ober,h.m
ü
ller,c.pieck,j.gierlich,t.carell,j.am.chem.soc.2005,127,18143
‑
18149.
[0264]
[13]h.m
ü
ller,t.carell,eur.j.org.chem.2007,2007,1438
‑
1445.
[0265]
[14]f.b
ü
sch,j.c.pieck,m.ober,j.gierlich,g.w.hsu,l.s.beese,t.carell,chem.eur.j.2008,14,2125
‑
2132.
[0266]
[15]t.h.gehrke,u.lischke,k.l.gasteiger,s.schneider,s.arnold,h.c.m
ü
ller,d.s.stephenson,h.zipse,t.carell,nat.chem.bio.2013,9,455.
[0267]
[16]y.kato,s.ozawa,c.miyamoto,y.maehata,a.suzuki,t.maeda,y.baba,cancer cell int.2013,13,89.
[0268]
[17]s.takahashi,s.kobayashi,i.hiratani,cell.mol.life sci.2018,75,1191
‑
1203.
[0269]
[18]f.r.traube,s.schiffers,k.iwan,s.kellner,f.spada,m.m
ü
ller,t.carell,nat.protoc.2019,14,283
‑
312.
[0270]
[19]s.schiffers,t.m.wildenhof,k.iwan,m.stadlmeier,m.m
ü
ller,t.carell,helv.chim.acta 2019,102,e1800229.
[0271]
[20]m.tahiliani,k.p.koh,y.shen,w.a.pastor,h.bandukwala,y.brudno,s.agarwal,l.m.iyer,d.r.liu,l.aravind,a.rao,science 2009,324,930
‑
935.
[0272]
[21]s.ito,l.shen,q.dai,s.c.wu,l.b.collins,j.a.swenberg,c.he,y.zhang,science 2011,333,1300
‑
1303.
[0273]
[22]m.m
ü
nzel,d.globisch,t.carell,angew.chem.int.ed.engl.2011,50,6460
‑
6468.
[0274]
[23]a.unnikrishnan,a.n.q.vo,r.pickford,m.j.raftery,a.c.nunez,a.verma,
l.b.hesson,j.e.pimanda,leukemia 2018,32,900
‑
910.
[0275]
[24]e.li,t.h.bestor,r.jaenisch,cell 1992,69,915
‑
926.
[0276]
[25]t.pfaffeneder,b.hackner,m.truss,m.m
ü
nzel,m.m
ü
ller,c.a.deiml,c.hagemeier,t.carell,angew.chem.int.ed.engl.2011,50,7008
‑
7012.
[0277]
[26]ravandi,f.;kantarjian,j.e.;issa,s.;jabbour,s.;santos,g.;mccue,d.;garcia
‑
manero,f.p.s.;pierce,e.;o’brien,j.p.;cort
é
s,j.e.;ravandi,f.(2010)."feasibility of therapy with hypomethylating agents in patients with renal insufficiency".clinical ly熔点homa,myeloma&leukemia.10(3):205
–
210.
[0278]
[27]dunn,j;thabet,s;jo,h(july 2015)."flow
‑
dependent epigenetic dna methylation in endothelial gene expression and atherosclerosis".arteriosclerosis,thrombosis,and vascular biology.35(7):1562
–
9.
[0279]
[28]hecker,s.j.;erion,m.d.(2008),"prodrugs of phosphates and phosphonates".j.med.chem 51:2328
‑
2345.
[0280]
[29]wiemer,j.;wiemer d.f.(2015),"prodrugs of phosphonates and phosphates:crossing the membrane barrier".top.curr.chem.360:115
‑
160.
[0281]
[30]wildenhof,t.m.,schiffers,s.,traube,f.r.,mayer,p.,carell,t.,angew.chem.int.ed.2019,58,12984
‑
12987.
[0282]
[31]davey,s.m.,malde,r.,mykura,r.c.,baker,a.t.,taher,t.e.,le duff,c.s.,willco
×
,b.e.,mehellou,y.,j.med.chem.2018,61,2111
‑
2117.
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1