功能性RISC关联小RNA的纯化方法和试剂盒
本发明涉及用于纯化所有生物体、器官、组织、细胞或生物流体中功能性RISC关联小RNA的方法和试剂盒。
背景技术
在绝大多数真核生物中,RNA沉默是一种基本的基因调控机制,它还对转座子和病毒等入侵核酸起到必要的防御功能。在迄今为止研究的所有真核生物中,RNA沉默的核心组分是RNA诱导沉默复合物(RISC),它由Argonaute家族(AGO)蛋白与长度为17-33个核苷酸(nt)的小RNA(sRNA)关联组成。
内源性微小RNA(microRNA)
在健康生物体中,大多数sRNA是细胞来源的,在这种情况下它们在通过各种机制产生它们的特定位点被编码。这些机制中的一种产生了一个称为微小RNA(miRNA)的内源性RNA大家族,其涉及Dicer家族中高度保守的RNase-III蛋白。miRNA大小为21-24nt,由位于蛋白质编码基因之间或内含子中的特定核基因编码。MIRNA基因编码非编码初级转录物(pri-miRNA),其总是包含双链RNA(dsRNA)茎环结构。一种较短的pri-miRNA的衍生物称为miRNA前体或pre-miRNA,与dsRNA茎环结构相对应并在一精确位置被Dicer切割生成显性的成熟miRNA双链体。当并入到一个或多个AGO效应蛋白中时,双链体中的一条链被选作引导链,而称为过客链或miRNA*的互补链被降解。然后产生的miRNA-RISC扫描细胞的转录组,寻找表现出与miRNA部分序列或扩展序列互补的mRNA,然后通过各种方式执行这些mRNA的转录后RNA沉默。
这些包括通过许多(尽管不是全部)AGO蛋白质所含核酸酶核心的内切核苷酸裂解(称为切割),和/或通过各种可能机制进行的翻译抑制,几乎总是伴随着适度但一致的加速mRNA降解。
循环miRNA
在过去的10年,已认识到在若干种包括血液、淋巴、乳液、唾液和尿液在内的哺乳动物体液中发现数量不等(但通常非常少)的潜在的大批miRNA。由于尚未明确的原因,miRNA在人类血浆中特别且惊人地稳定。这些miRNA的确切来源仍然存在争议:它们可以见于例如从死细胞产生的凋亡小体中,和/或在来自通常源自例如免疫细胞的微囊泡或外泌体中活跃分泌或在肿瘤中高水平产生。体液携带的miRNA,尤其是血液携带的miRNA,无论其来源、特定状态或假定的生理作用如何,其身份和数量都可以潜在地在人类健康和兽医应用中作为几种疾病和病理病症的诊断和预后支持特征。
病毒编码的miRNA
被以庞大的DNA基因组为特征的DNA病毒(主要是疱疹病毒科,例如,单纯疱疹病毒-HSV1/2、EB病毒EBV、巨细胞病毒CMV、卡波西肉瘤相关疱疹病毒KHSV)感染的分化哺乳动物细胞经分析揭示了这类病毒编码自己的一套miRNA。这些miRNA是病毒基因组在细胞核中转录时通过篡夺宿主miRNA机制产生的。与可以对抗入侵者的病毒小干扰RNA(vsiRNA)不同,病毒源miRNA对其来源病毒起着重要的有益作用,包括逃避宿主免疫反应、调节病毒蛋白丰度或使病毒进入持续性而非裂解性的感染状态。在受感染细胞分泌的细胞外囊泡中,已经发现了几种生理浓度下的病毒源miRNA,因此,当它们被感染周围的细胞摄取时可能作为基因表达调节器发挥作用。与其宿主编码的相对物一样,其也在血液中、可能还在其他体液中被发现并且可用于诊断感染,尽管考虑到在体液中检测细胞miRNA已经遇到的困难而言疱疹病毒科通常为潜伏状态并不利于它们的检测。病毒源miRNA是治疗的潜在靶点,尤其是对于KSHV的情形而言——其是免疫受损患者(如感染艾滋病的患者)死亡的主要原因之一。目前尚不清楚miRNA的现有技术分析是否已经揭示了由这些DNA病毒产生的病毒编码的miRNA全部组群,因为如上所述,它们大多处于潜伏状态(因此它们在体内的转录水平很低,而在体外它们主要在选定的细胞类型中进行研究)。间接证据表明,可能存在另外由至少一些RNA病毒编码的miRNA。
小干扰RNA
其他Dicer依赖的sRNA也有外源来源。在植物、无脊椎动物和至少一些哺乳动物细胞中,长dsRNA是病毒复制的几乎不变的产物,其由细胞内Dicer样蛋白检测并转化为小干扰RNA(siRNA)。与离散miRNA不同,这些病毒衍生的siRNA或vsiRNA以通常定义出特定显性序列寄存器——一种称为“定相”(phasing)的Dicer加工读出的方式,通过沿着dsRNA连续切割作为群体产生。尽管其生物发生与miRNA不同,但siRNA与一种或几种AGO蛋白缔合,形成靶向病毒RNA本身的抗病毒RISC,它们通过切割和/或可能的翻译抑制将其破坏。这种抗病毒RNA沉默反应是独特的,因为它纯粹是天生的,也就是说,它可以潜在地适应每一种病毒。事实上,它完全是由病毒基因组的结构和核苷酸序列特征编程的。在植物和一些无脊椎动物中,这种反应也具有非细胞自主成分,并先于病毒使远离感染前沿的未感染细胞免疫。抗病毒RNAi也在一些哺乳动物细胞的体内和体外发挥作用,但在其他细胞中则不起作用,原因目前尚不清楚。原则上,考虑到多数病毒的高复制率,感染组织中大量的vsiRNA可用于诊断特定病毒疾病,甚至诊断指定病毒的特定毒株,因为现在可以通过将序列重叠的vsiRNA相连来重构全病毒基因组部分。或者,通过更具针对性的方法,可以将特定的vsiRNA用作疾病标志物。
异染色质siRNA和其他内源性siRNA
siRNA也不一定是病毒来源的,因为对裂变酵母、其他真菌、植物、线虫、果蝇(可能还有许多其他生物体)中的分析已经揭示了大量的内源性siRNA。这些所谓的内源性siRNA主要来源于转座元件(TE)和DNA重复序列,并促进这些位点的染色质凝聚和转录基因沉默,因此可能有助于基因组完整性的维持。其他类型的内源性siRNA也在植物、蠕虫和果蝇中积累,在此它们发挥着各种发育和基础基因调控作用。已在哺乳动物生殖系中主要是在卵母细胞中以及在胚胎干细胞中检测到了内源性RNA。它们是否在体内其他细胞类型、组织、器官或体液中积聚尚待确定。
PIWI关联sRNA(piRNA)和扫描RNA(scnRNA)
在果蝇以及后来在蠕虫和哺乳动物中发现了一种与AGO样蛋白(称为PIWI蛋白)关联的生殖系特异性sRNA,但在植物或真菌中没有发现。piRNA在转录(组蛋白甲基化)和转录后(通过PIWI蛋白质进行的切割)水平上靶向生殖系中的转座子。在果蝇中,这些sRNA在受精卵中也起着重要作用,受精卵受母系中储存的PIWI结合的piRNA的保护,以抵抗雄性基因组带来的转座子的潜在有害活动。piRNA途径的缺陷通常会导致哺乳动物的生殖系发育异常,而在果蝇中,则会导致杂交不育。
最后,在纤毛虫(例如草履虫、四膜虫)中检测到了piRNA的变体,其中25-29nt的扫描RNA(scnRNA)的复杂群体在减数分裂早期积累。这些scnRNA与微核中发现的所有类型的生殖系序列都具有同源性,并且在装载到PIWI样蛋白质中后,可促进子代体细胞大核中外源序列(通常是转座子和重复序列)的物理DNA切除。scnRNA是通过Dicer样蛋白从长dsRNA产生的,迄今为止仅在原生动物中检测到。
sRNA检测和分析的标准方法
从历史上看,检测所有生物体中已知siRNA的方法都基于Northern分析,其涉及在聚丙烯酰胺凝胶上从整个生物体或特定器官、组织或细胞类型中提取的总RNA中分离低分子量(LMW)RNA种类。然后将凝胶印迹到尼龙膜上,随后与同感兴趣的sRNA互补的DNA或RNA探针进行交联和杂交。这些探针通常用放射性同位素标记,但也有其他灵敏度较低的方法。单个物种的miRNA通常用互补的末端标记的寡核苷酸检测,但这些寡核苷酸通常不能将在包括植物和哺乳动物在内的各种物种中发现的通常只有一个或几个核苷酸不同的许多miRNA亚型和旁系同源物区分开。与离散miRNA物种不同,siRNA作为群体积累,以低丰度到极低的丰度单独积累。因此,为检测群体而不是来自转座子、病毒或内源性位点的大量siRNA的特定成员,使用长的随机引物DNA探针。这种方法的缺点是无法获得关于单个siRNA物种的信息。Northern分析siRNA群体和单个miRNA物种的另一个常见缺点是该技术的检测极限:低丰度的sRNA通常无法检测到,即便使用大量的总RNA、甚至是特别富集的LMW RNA(几十微克)也如此。
另一可选的用于检测已知sRNA物种的技术主要用于miRNA,其依赖于使用专门设计的寡核苷酸进行的定量反转录PCR(RT-qPCR)。与Northern分析相比,基于RT-qPCR的sRNA检测是高度定量的,非常敏感(只需要1-10ng总RNA),因此它可以检测来自细胞群甚或是单个细胞的sRNA。然而,同Northern分析中使用的探针一样,RT-qPCR探针并不总是能区分仅有一些核苷酸不同的特定的miRNA亚型和类似物。这种方法也几乎没有效率,而且确实很少用来量化来自其来源群体的单个siRNA。
作为RT-qPCR的替代方法,包含给定物种中所有或部分已知miRNA的微阵列也可用于量化单个miRNA物种。在这种情况下,总RNA或专门制备的LMW RNA部分在3'端用荧光染料(例如Cyanine3-pCp)标记,并与微阵列杂交。该方法的灵敏度与RT-qPCR相当,动态范围很广。
然而,Northern、RT-qPCR和基于微阵列的sRNA检测方法的一个主要共同缺点在于,它们都依赖于已验证的sRNA的先验知识。因此,这些方法无一允许对感兴趣的生物体、器官、组织或细胞类型的sRNA含量进行无偏见的探索,更不用说在生物/非生物胁迫、细胞代谢失调或疾病环境下了。然而,众所周知,这些环境会改变——有时是深刻地改变——所述生物体/器官/组织/细胞类型的sRNA图谱,并诱导通常在健康或非应激细胞中低于检测水平的特定sRNA的积累。
由于sRNA生物学在基础研究和应用研究中的影响,已经开发了多种方法和方案通过大规模并行测序(这里称为“深度测序”或“deep-seq”)接近在多种生物体、器官、组织和细胞类型中获得的sRNA。与前面提到的技术不同,deep-seq允许在基因组规模上访问sRNA群体,而无需事先了解其序列。学术、临床和公司研究当前负担得起的所有的sRNA deep-seq技术都是基于连接到sRNA的3'和5'适配体。这使得RNA可以反转录为cDNA,然后进行几个PCR扩增循环以生成所谓的“sRNA文库”。然后,对文库进行deep-seq,测序的深度取决于所使用的平台(例如454,SOLID,lllumina),生成测序文件,管理时从中提取全基因组sRNA信息。进一步的基于计算机的分析于是允许在任何给定样本中进行sRNA定性和定量分选,以及样本或队列之间的差异分析。因此,可以识别例如由特定发育、应激或病理状态等诱发的sRNA库的可再现变异,从中可以选择“精英”sRNA(哺乳动物中主要是miRNA)作为这些特定细胞状态、生理或病理条件的潜在生物标志物。
然而,几乎所有的从样品进行总sRNA测序的实施方式都需要通过各种方式从较长的RNA中预先分离大小,以避免有时较丰富的出现于<70nt部分的tRNA、rRNA或mRNA降解产物的克隆。这在一些生物模型中包括例如植物或果蝇中尤其重要,在这些模型中,例如经Trizol提取后直接克隆sRNA会产生充满污染物的文库,因此这样的大部分文库无法使用。同样的问题也适用于充满RNA污染物的生物液体(例如血浆),其中仅含有相对少量的真正的sRNA。从几种生物体的其他“正常”组织制备的不进行大小选择的Trizol提取的sRNA文库可能是可接受的,尽管其质量几乎总是次优的。尽管专业的实验室在进行初步、常规分析或担心样品丢失(见下文)时避免或绕过了该步骤,但大多数商业供应商使用的总sRNA测序的金标准要求在文库克隆和测序之前进行凝胶大小分离。尽管其他方法已经被商业化开发,但耗时费力的基于丙烯酰胺凝胶的分离仍然是最可靠的技术。
在这种基于凝胶的分离过程中,总RNA在高浓度聚丙烯酰胺凝胶上通过电泳分离,在其旁边有(通常是放射性标记的)RNA梯度条带用作大小参照物。这使得能够切取富含感兴趣的同源sRNA的凝胶部分(siRNA和miRNA通常为18-25nt;piRNA和scnRNA通常为27-32nt)。然后在文库的恰当制备之前,从凝胶中重新提取剪切的RNA。通过多个繁琐步骤对样本进行长时间处理容易使RNA降解,也容易使较长的、不相关的RNA最终成为污染物。此外,可变但通常重要的部分sRNA材料在这个程序中被丢失,导致总sRNA产量很低甚至极低。对大量起始生物材料的固有要求(通常在数微克总RNA的范围内)对易降解(例如活检)、数量有限(例如胚胎、卵巢)和/或sRNA含量有限(例如生物液体)的样品提出了相当大的挑战。由于复杂性、易降解性和产量低的特点,为了可靠性,sRNA大小筛选和随后的文库制备通常外包给专业公司。文库制备的外包因涉及人工而有很高的成本,讽刺的是,测序反应本身的成本往往超过deep-seq试剂不断下降的成本一个数量级。
无论其形式如何,制备sRNA文库之前尺寸筛选程序的另一主要限制是,较长RNA的降解产物和/或在感兴趣的大小范围内的高丰度RNA(如果蝇的2S rRNA)很难分离。这通常导致序列数据被高背景混淆,导致大量假阳性。此外,此类污染物可能占据大量的测序空间。在果蝇中,由共迁移2S rRNA造成的问题是,它需要在连接之前使用另一称为核糖体RNA去除(ribodepletion)的步骤,作为整个凝胶分离程序的一部分,从而进一步增加了样本RNA降解的风险。尽管有核糖体RNA去除,rRNA通常还是会占果蝇标准sRNA文库所有测序读段的20-40%。考虑到piRNA独特的2'-O-甲基化状态能保护它们免受高碘酸盐攻击,就这一点而言,关注于通常是来自解剖果蝇卵巢的piRNA的分析在一定程度上更为有利,因为经另一步骤(此时称为氧化)可以除去核糖体RNA去除后剩余的大部分rRNA。同样的步骤通常用于哺乳动物生殖系中的piRNA的deep-seq分析。然而,缺点在于,这一增加的步骤需要对从非常有限量的繁琐分离的组织(卵巢、睾丸)中提取的RNA进行更多的处理,从而更增加了降解或材料损失的风险。总之,由于起始材料量少、繁琐、有降解风险和污染严重,对来自解剖的果蝇卵巢的piRNA的deep-seq可被视为全基因组或定向sRNA分析中最具挑战性的环境之一的基准。
在所有生物体中,专用的AGO/PIWI蛋白质装载有特定的sRNA以执行特定的生物功能。因此,对于大多数应用,有必要识别RISC关联sRNA,而不是感兴趣样本中存在的一般sRNA池。到目前为止,解决这一必要性的最常见方法是开发适用于免疫沉淀(IP)的高度特异性抗AGO/PIWI抗体。IP之后,结合到感兴趣AGO的sRNA群体随后经苯酚提取、克隆,并按照总sRNA测序中使用的相同程序进行deep-seq。然而,一个主要本质差异是,AGO蛋白及其货物的纯化使sRNA能够大量富集,避免了污染RNA或分解产物。目前,能够对从AGO/PIWI IP中提取的RNA直接操作适配体连接、扩增和deep-seq(即没有在凝胶上进行尺寸筛选)。最近,基于称为GW182(或TNRC6)的哺乳动物蛋白对表现出人类AGO2的高亲和力,开发了AGO IP的替代方法,上述蛋白为翻译和mRNA降解水平的miRNA介导的靶调控所需。GW182包含几个重复的GW残基,形成一个称为“AGO-Hook”的结构域。AGO-hook也见于其他生物体中,它们可能在GW二元体密度(dyad density)和空间组织方面表现出高度多态性。在这些生物体中,AGO-hook有助于将专门的功能赋予一些(尽管不是全部)AGO。例如,在植物中,一种特定的AGO-hook蛋白使AGO4、AGO6和AGO9能够触及DNA,从而引导经来自转座子和重复序列的异染色质siRNA的染色质修饰。因此,无凝胶sRNA分离的一个实施方式需要使用融合到GST的短GW182衍生肽以高亲和力结合至少一些与sRNA复合的AGO蛋白质。这种方法被称为“经肽的AGO蛋白质亲和纯化”(AGO-APP)。
AGO-APP方法的主要限制是,只有一些AGO对GW重复显示出的亲和力足以通过该技术被拉下,这大大限制了其在RISC分离方面的广泛应用。例如,应用于植物,AGO-APP只能显著纯化拟南芥10种AGO蛋白中的2种。因此,AGO-APP的专精性(proficiency)是不可预测和可变的,这取决于影响AGO蛋白与AGO钩相互作用的AGO蛋白的内在特征。在各界中,一些AGO可能尚未进化到作为它们参与的通路的一部分与这些蛋白质相互作用。
基于IP的sRNA测序对于许多实验应用具有重要意义。然而,开发适合IP的高质量AGO/PIWI抗体可能需要数年的时间,并且这种抗体并不总能区分通常在单个生物体内发现的AGO/PIWI大家族的个体成员。在哺乳动物中,AGO IP对AGO1和AGO2的作用良好,但由于缺乏合适的内部或商业抗体,对AGO3和AGO4的作用极不理想。此外,AGO/PIWI抗体通常不会发生交叉反应,即使在相关物种中也是如此,这通常限制了IP耦合的深度测序用于模式生物。IP不仅繁琐、耗时且技术要求高,它们还内在地依赖于任何给定样本中都存在AGO的预设之见,而这种知识很少可得。AGO免疫原性(由此还有抗体效力/特异性)的差异或者仅仅是IP专精抗体的不可得,意味着该方法天然存在偏差,在不同AGO的IP之间和内部比较效果不良,并且通常不能很好地反映感兴趣样本中存在的AGO sRNA货物的完整组合。
IP和AGO-APP方法的另一主要限制是,它们仅适于使用少量样品进行的实验室工作。事实上,由于其多步骤性质和高技术性(珠耦合、多次洗涤、AGO-APP裂解缓冲液处理)以及涉及4℃下长时间孵育(AGO-APP过夜),可以想到,两者都不容易适应中/高通量环境,例如涉及来自大的患者队列的许多样本的临床探索所需的环境。即使对于涉及小样本数的靶向应用,田间农学家、兽医或临床医生也不愿使用AGO-IP或AGO-APP,这对于非专家用户来说仍然很麻烦,要求也很高。
待解决的客观技术问题
因此,需要一种快速、简单和可靠的方法来纯化功能性(即AGO负载/RISC关联的)sRNA,该方法不需要事先知晓样品的AGO含量,并由此可加以应用而无论感兴趣的生物体、组织、生物液体或细胞类型如何。这种方法应允许用户友好、高通量和高质量地纯化功能性sRNA,而不考虑RNA污染物或样品的RNA降解状态。最后,该方法应允许从众所周知的顽固性组织(例如淀粉植物、血清和血浆)和/或微量起始材料中稳健地分离sRNA。
技术实现要素:
通过一种用于纯化RISC关联sRNA的方法来解决问题,所述方法包括以下步骤:
a)提供来自含有RISC关联sRNA的生物样本的天然样品;
b)使用天然裂解缓冲液裂解样品;
c)选择性地从裂解物中去除非RISC关联核酸;和
d)收集包含RISC关联sRNA的RISC。
在另一个实施方式中,通过提供纯化RISC关联sRNA的试剂盒来解决该问题,该试剂盒包括:
-天然裂解缓冲液;
-洗脱缓冲液;和
-具有包含阴离子交换树脂的主体的柱。
附图说明
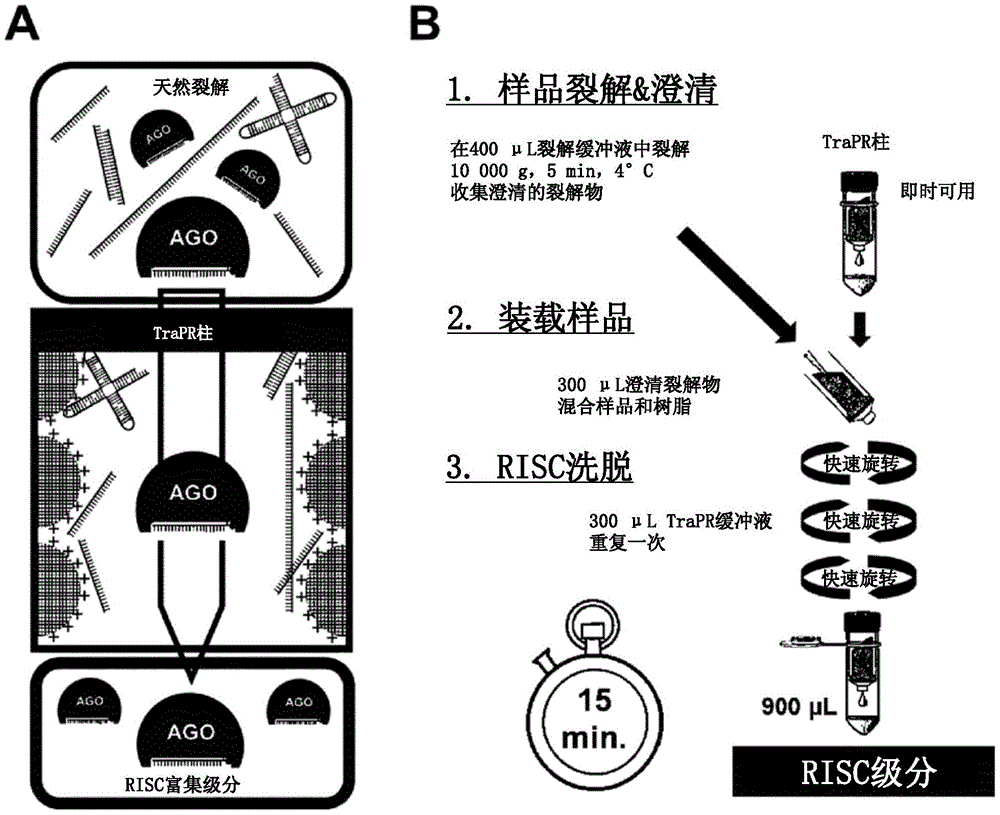
图1A显示根据本发明的方法的基本原理。将天然的澄清裂解物与带正电的基质混合,收集流出液,并使用逐渐增加的盐浓度进行洗脱。RISC及其关联sRNA在给定的盐浓度之前洗脱,而带负电的游离核酸仍然停留在柱上。
图1B显示根据本发明的方法的示意性概述。根据本发明的方法需要三个主要步骤:样品的天然裂解和澄清,将澄清的裂解物装载到柱上并与阴离子交换树脂混合,以及使用三次短离心洗脱RISC关联sRNA。该程序通常在15分钟内进行。
图2A显示在拟南芥(Arabidopsis thaliana)中表达的九种AGO蛋白质的系统发育的示意图,包括三个主要分支。
图2B显示拟南芥花序中2种主要AGO蛋白质(AGO1和AGO4)的蛋白质印迹(上)分析,其根据本发明方法提取,使用渐增的醋酸钾(KoAc)浓度进行分离,并使用针对内源蛋白质的抗体进行检测。提取各部分中存在的RNA,在17%变性聚丙烯酰胺凝胶上进行迁移,并在溴化乙锭染色后检测(下)。对于每个洗脱步骤,测量缓冲液的电导率(Cond,mS/cm2)。黑色箭头表示细胞AGO内容物被洗脱的级分,虚化箭头表示缓冲液中使长RNA保留在树脂上的最大盐浓度。
图2C显示根据本发明方法提取并用针对内源性蛋白质的抗体检测的拟南芥花序样品中主要AGO蛋白质的蛋白质印迹分析(右侧道)。通过对凝胶左侧上分离自单个拟南芥ago突变体与野生型植物(Col-0)的总裂解物进行比较分析,证实了抗体的特异性。(I:澄清裂解物,E:RISC级分,HS:AGO洗脱后树脂的高盐洗涤)。“Flag”是纯化后添加到每个级分中的刺突蛋白(protein spike),作为蛋白质提取步骤的对照。
图2D显示根据本发明方法从长角果(授粉后1-5天)提取并用抗Flag抗体检测的在AGO3内源启动子下表达的拟南芥Flag-AGO3的蛋白质印迹分析(I:澄清裂解物,E:RISC级分,HS:AGO洗脱后树脂的高盐洗涤)。
图2E显示根据本发明方法从2周龄幼苗中提取并用抗Flag抗体检测的在AGO7内源启动子下表达的拟南芥Flag-AGO7的蛋白质印迹分析(I:澄清裂解物,E:RISC级分,HS:AGO洗脱后树脂的高盐洗涤)。
图2F显示根据本发明方法提取的拟南芥花序RISC关联sRNA在17%变性聚丙烯酰胺凝胶上的RNA印迹分析。在凝胶迁移和转移到尼龙膜上之前,使用T4多核苷酸激酶(PNK)对从提取的级分中纯化的RNA进行放射性标记(I:澄清裂解物,E:RISC级分,HS:AGO洗脱后树脂的高盐洗涤)。被用作RNA大小梯状标尺(核苷酸)。
图2G显示根据本发明方法提取的拟南芥花序RISC关联sRNA在17%变性聚丙烯酰胺凝胶上的低分子量RNA分析。从提取物中纯化的RNA在凝胶上分离然后转移到尼龙膜上。使用放射性标记的寡核苷酸作为探针来揭示特定的已知拟南芥sRNA,如右侧所示(I:澄清裂解产物,E:RISC级分,HS:AGO洗脱后树脂的高盐洗涤)。Spike是一种合成的22nt RNA序列,其添加到纯化后的每个级分中,作为RNA提取步骤的对照。
图3显示从标注的各种生物体中提取的RISC在17%变性聚丙烯酰胺凝胶上的RNA印迹分析。(I:澄清裂解物,E:RISC级分,HS:AGO洗脱后树脂的高盐洗涤)。被用作RNA大小梯状标尺(核苷酸)。
图4A显示根据本发明方法的一个实施方式分离RISC关联sRNA的工作流程的示意图,其中使用商用硅酸盐柱(右)而不是标准沉淀(左)回收sRNA。标注了每个sRNA回收工序的大致持续时间。
图4B显示在17%变性聚丙烯酰胺凝胶上对根据本发明方法的一个实施方式提取的拟南芥花序RISC关联sRNA进行的比较低分子量RNA分析,其中sRNA使用商业硅酸盐柱而不是标准沉淀进行回收。如图2G所阐释,检测到特定的已知拟南芥sRNA物种。每个测试的商业试剂盒显示两个平行。Spike是一种合成的22nt RNA序列,其添加到纯化后的每个级分中,作为RNA提取步骤的对照。
图4C显示根据本发明方法提取RISC的拟南芥花序样品中miR159和miR171丰度的定量RT-PCR分析(n=3)。表达表示为相对于粗裂解物(I级分)的检测水平。snoRNA 85用作非RISC装载RNA的对照(I:澄清裂解物,E:RISC级分,HS:AGO洗脱后树脂的高盐洗涤)。
图4D显示来自小鼠肝脏样品的miR16和miR21的定量RT-PCR分析(n=3)。snoRNA 202用作非RISC装载RNA的对照(I:澄清裂解物,E:RISC级分,HS:AGO洗脱后树脂的高盐洗涤)。
图5A显示总RNA(上)、经聚丙烯酰胺凝胶选择的sRNA(中)或根据本发明方法提取的纯化RISC关联sRNA(下)在基于TRUseq的测序后的拟南芥花序RNA长度分布和基因组来源(每种情况n=3)。
图5B显示经聚丙烯酰胺凝胶选择、核糖体RNA去除的sRNA(上)或额外进行氧化(中)、或根据本发明方法提取的纯化RISC关联sRNA(下)在定制的测序方案后的果蝇卵巢RNA长度分布和基因组来源(每种情况n=2)。
图6如图5B所示,根据本发明的方法被应用于50(TraPR50)、25(TraPR25)、10(TraPR10)或2对卵巢(TraPR2)(每种情况n=2)。
图7A显示基于所有已知果蝇miRNA的特性和丰度,对图5B和图6中制备的单个定制测序文库进行的聚类分析。
图7B显示miRNA(左)和映射到转座元件的sRNA(右)基于其在图5B和图6制备的文库中的特性和丰度的相关性分析(n=2)。
图8A显示图5A所示的总RNA、凝胶提取的sRNA或根据本发明方法提取的RISC关联sRNA的5'核苷酸组成。显示21nt长(上)和24nt长(下)sRNA的核苷酸身份(每种情况n=3)。
图8B显示根据本发明方法从花序中提取并用抗AGO1抗体检测的AGO1内源启动子下表达的拟南芥Flag-AGO1的蛋白质印迹分析(上图,TraPR)(I:澄清裂解物,E:RISC级分,HS:AGO洗脱后树脂的高盐洗涤)。或者,Flag-AGO1(上图,IP Flag)是从澄清的裂解物(总)或根据本发明方法的含有RISC的级分中免疫沉淀的(Ub:未结合,IP:免疫沉淀)。中间的图是在17%变性聚丙烯酰胺凝胶上对图2G中检测到的拟南芥miR160进行的低分子量RNA分析。下图显示在17%变性聚丙烯酰胺凝胶上对图2F所示的拟南芥花序RISC关联sRNA进行的RNA印迹分析。被用作sRNA大小梯状标尺(核苷酸)。
图9A显示来自完整的或在室温下在30分钟期间用100U RNAse T1处理的小鼠肝脏的总RNA(总RNA)和根据本发明方法纯化的RISC关联sRNA(TraPR)在17%变性聚丙烯酰胺凝胶上的低分子量RNA分析(I:澄清裂解物,E:RISC级分,HS:AGO洗脱后树脂的高盐洗涤)。
图9B显示用来自完整(上)的和来自RNase T1处理过的(下)小鼠肝脏中的总RNA(左)或根据本发明方法纯化的RISC关联sRNA(TraPR,右)产生的小鼠sRNA文库中的总RNA在基于TRUseq的测序后的小鼠肝脏RNA长度分布和基因组来源(每种情况n=3)。
图9C显示基于图9B中制备的文库中miRNA的身份和丰度的相关分析(n=3)。
图10A显示通过Lexogen sRNA文库试剂盒使用来自小鼠血浆的总RNA或根据本发明方法(TraPR)纯化的RISC关联sRNA生成的文库中小鼠sRNA的比例和基因组来源(n=4)。
图10B与图10A相同,但包含了sRNA长度分布。
图10C显示来自总RNA(上)和根据本发明方法纯化的RISC关联sRNA(TraPR,下)生成的如图10A所释的单个文库中miRNA丰度的相关性。
图10D显示如图10A所释从总RNA或RISC关联sRNA制备的文库中miRNA的离散度。根据miRNA丰度(WiIcoxon秩和检验,***<4.5.10-5,**<5.10-4)以四分位数描述离散度。
图11A显示来自小鼠血浆样品的miR16和miR21的定量RT-PCR分析(n=3)。(I:澄清裂解物,E:RISC级分,HS:AGO洗脱后树脂的高盐洗涤)。
图11B显示通过Lexogen sRNA文库试剂盒(n=4)或TRUseq(n=3)使用来自小鼠血浆的总RNA或根据本发明方法(TraPR)纯化的RISC关联sRNA生成的单个文库中miRNA丰度的相关性。
具体实施方式
在一方面,本发明涉及用于纯化RISC关联sRNA的方法,包括以下步骤:
a)提供来自含有RISC关联sRNA的生物样本的天然样品;
b)使用天然裂解缓冲液裂解样品;
c)选择性地从裂解物中去除非RISC关联核酸;和
d)收集包含RISC关联sRNA的RISC。
根据本发明的方法称为TraPR,代表“Transkingdom rapid and affordable Purification of RISCs”(跨界且快速实惠的RISC纯化)。
在此,术语“sRNA”涉及长度为18至40个核苷酸的小RNA分子。该术语旨在涵盖微小RNA(miRNA)、Piwi相互作用RNA(piRNA)、小干扰RNA(siRNA)、扫描RNA(scnRNA)。特别地,该术语指任何与Argonaute(AGO)-家族蛋白功能相关联作为RISC的一部分、可参与基因调控的sRNA分子。术语“sRNA”、“sRNA分子”和“sRNAs”可互换使用。
本文中的术语“RISC关联sRNA”是指例如通过与AGO家族蛋白缔合或装载到其上而在功能上并入RISC的sRNA。该术语不涵盖不与RISC相互作用和/或在纯化时不装载到RISC上的游离RNA。RISC关联sRNA是最有趣的RNA类型,因为它们与AGO家族蛋白相互作用和装载到其上是它们传递mRNA调控的先决条件。因此,高度优选的是分离RISC装载的sRNA而不是感兴趣样本中存在的一般sRNA池。换句话说,本发明涉及一种仅分离纯化时装载在RISC上的那些sRNA(即RISC负载的sRNA)的方法。
术语“Argonaute(AGO)-家族蛋白质”在本文中指Argonaute蛋白质家族的成员,它们构成了在RNA或DNA水平上发挥作用的任何RISC的核心成分。AGO蛋白在真核生物中进化保守,可分为AGO和PIWI亚家族。所有AGO/PIWI蛋白包括三个关键结构域:PIWI、PAZ和Mid,并结合不同类别的sRNA,通过核苷酸序列互补性(碱基配对)将其引导至特定目标。AGO结合的sRNA可能是功能惰性的,或者可能促进mRNA切割、增强的mRNA降解和/或翻译抑制、或是染色质压实和/或转录改变、或是物理基因组修饰/编辑。虽然完整的RISC组分套件尚未完全阐明,但AGO蛋白已被证实是此类复合物不变的关键元素。因此,术语“AGO关联”、“AGO结合”、“AGO装载”、“RISC关联”、“RISC结合”和“RISC装载”在此可互换使用。
本文中的术语“关联”或“负载”是指sRNA分子与AGO家族蛋白质的非共价结合。
与基于IP或AGO-PAPP的方法一样,本发明的原理基于最有用的sRNA信息将包含在功能性RISC中的概念,即,这些sRNA可能通过引导AGO而参与基因调控。分离AGO结合的sRNA也同时提供了显著消除与sRNA无关的其他核酸的优势。然而,与IP或AGO-PAPP不同,根据本发明的方法并不是基于免疫富集或AGO-AGO-钩亲和力来分离RISC关联sRNA,而是通过利用装载sRNA货物的所有已知AGO/PIWI蛋白质所展示的保守生化特性,这主要包括在生理条件下介于9.3和9.8之间的等电点。
相比之下,在这种条件下,所有游离核酸都重度带负电荷。因此,能够从天然裂解物中选择性地去除非RISC关联核酸。例如,可将裂解物暴露于带正电的树脂,以使所有污染的游离核酸(RNA、DNA)保持与其结合,从而从裂解物中去除。清洗树脂后,可使用温和的盐浓度收集RISC关联的sRNA。因此,根据本发明的方法的分离原理基于保留而不是富集(参见图1A)。
本发明涉及从样品中纯化RISC关联sRNA,尤其是纯化RISC装载的sRNA。样品可来自生物样本,例如细胞、生物液体、组织活检或动物、真菌、原生动物或植物器官。在一个实施方式中,生物样本是动物、真菌、原生动物或植物的整个器官。同样,样品可源自动物、真菌、原生动物或植物细胞的细胞培养生物标本。在这方面,样品还可包含来自细胞培养样本的上清。样本也可由一个或几个完整的生物体组成。
在优选实施方式中,样品是来自哺乳动物的生物样本,尤其是来自人类受试者或人类患者的样本。样本可以是全血样品、血清样品、血浆样品、脑脊液样品、唾液样品、泪液样品、尿液样品、粪便样品、淋巴样品、乳液样品、精液样品、腹水或羊水样品。
在另一个实施方式中,从中获得标本的动物是任何兽医感兴趣的动物,包括但不限于动物园动物、宠物、牛、家禽和鱼。在另一实施方式中,该动物为线虫或酵母。
在另一个实施方式中,样本是来自任何植物的生物液体,例如木质部或韧皮部。
生物样本可以是新鲜的或冷冻的,因为冷冻不会改变根据本发明的方法的应用所需的生化特性。
用于获得根据本发明的天然样品的生物样本可以在用作本发明方法中的样品之前进行处理,以促进sRNA的纯化。例如,可使用标准缓冲液(用于细胞培养的PBS、用于线虫的M9缓冲液或用于动物活检的无菌生理水)清洗样本。一旦去除洗涤缓冲液,干燥团块可在液氮或干冰中急速冷冻,并可用作根据本发明的样品。
可使用合适的标准程序分离收集的细胞。技术人员知道如何选择合适的程序来分离样本中的不同细胞。合适的方法可以是(GE Healthcare,17-1440-02)、LymphoprepTM(STEMCELL Technologies,07801)或荧光激活细胞分选(FACS)。分选后,可通过离心将细胞团块化。一旦洗涤缓冲液从团块细胞去除,干燥团块可在液氮或干冰中急速冷冻,并可用作根据本发明的样品。
根据本发明,可收集生物流体并随后将其在液氮或干冰中急速冷冻。收集的样品可冷冻储存,优选在-80℃下。技术人员知道应尽量减少冻结/解冻周期,以保持材料质量。在优选实施方式中,在冷冻前制备等分试样,例如250万个细胞、10对果蝇卵巢、20mg植物/动物材料、50至100μL完整线虫或真菌颗粒、150μL生物流体。
在本发明的一个实施方式中,生物样本是通过体外、细胞内或体内RISC生产而产生的含有RISC的样品。
如本文所用,术语“裂解”是指使用洗涤剂使样品的细胞质膜、囊泡、细胞器和核膜去稳定化,以触及其蛋白质内容物。术语“天然裂解”是指在优化浓度下以低严格条件使用洗涤剂进行的裂解,以保持蛋白质-蛋白质相互作用(蛋白质复合物)、RNA-蛋白质相互作用(核糖核蛋白)及其酶活性。
本领域技术人员知道,在任何纯化方法中用于裂解和洗脱的不同缓冲液应彼此兼容。因此,在本发明的一些实施方式中使用的不同缓冲液是基于相同的基础缓冲液,彼此的不同之处仅在于加入特定的化合物或者调整其他性质,例如,期望目的(即,柱保存、裂解、洗脱)所需的保藏剂、洗涤剂或盐浓度。
这些实施方式使用的缓冲液经优化以(i)溶解RISC,同时保留sRNA和AGO蛋白质之间的非共价相互作用,(ii)有利于将所有其他核酸保留在带正电的基质上,和(iii)允许基于等电点差异分离AGO结合的sRNA。这些组合的生物化学特性是使用特定的盐浓度获得的——这是由发明人惊人地发现。在优选实施方式中,乙酸钾(CH3CO2K)用作盐。
本发明实施方式中使用的不同缓冲液中的特定盐浓度通过电导率测量进行监测。本文中的术语“电导率”指电解质溶液导电的能力。电导率测量是一种工业过程中常用的用于测量溶液的离子含量(盐浓度)的快速、廉价和可靠的方法。电导率的国际单位为西门子每米(S/m)。在这些实施方式中使用的缓冲液的CH3CO2K浓度通过监测缓冲剂电导率进行调整,直到达到特定值。
在优选实施方式中,基础缓冲液由以下组成:调整至pH=7.9的20mM HEPES-KOH,以允许在一定温度范围内具有更好的pH稳定性;10-20%(v/v)甘油,以保持AGO-sRNA相互作用;还原剂如1mM二硫苏糖醇(DTT),以防止氧化;和0.2mM EDTA,以在用1.5mM MgCl2添加过量Mg2+之前螯合二价阳离子。
在优选实施方式中,基础缓冲液补充有2mM NaN3作为保藏剂,以及最终100mM CH3CO2K以获得存储缓冲液(测量电导率介于7.5和8.5mS/cm2之间)。该缓冲液确保了树脂在柱内最佳储存。
在优选实施方式中,基础缓冲液补充有0.1%(v/v)TRITON-X100作为两性离子洗涤剂以溶解用于裂解的最终在100mM CH3CO2K的基础缓冲液中的RISC内容物(测量电导率介于7.5和8.5mS/cm2之间)。
在优选实施方式中,本发明中使用的缓冲液在0.22μm过滤并脱气。缓冲液可进一步通过电导率测量和/或基准sRNA分离来验证。缓冲液还可包含适合于蛋白质和RNA稳定化的额外的市售化合物。
根据本发明,样品可以是固体或液体。使用适于各生物模型的标准程序将固体样品以机械方式转化为粉末,然后在裂解缓冲液中均质化。液体样品与裂解缓冲液以1:1(v/v)的比例混合。裂解破坏细胞壁、膜和核膜,导致随后的RISC溶解。本领域技术人员充分了解各种裂解剂的性质以及如何选择适当量的裂解剂。此外,本领域技术人员知道如何使用给定的盐及在给定的pH下测量裂解剂对给定生物化学相互作用、给定树脂的影响。
由于本发明的方法旨在获得天然状态RISC,因此裂解缓冲液可以不含会使sRNA从复合物分离的浓度的离液剂例如胍盐或尿素。在优选实施方式中,裂解缓冲液含有在0.05%(v/v)和0.2%(v/v)之间的Triton X-100,优选在0.1%(v/v)和0.2%(v/v)之间的Triton X-100,最优选0.1%(v/v)的Triton X-100。裂解缓冲液还可含有两性离子洗涤剂,例如3-[(3-胆酰胺丙基)二甲基铵]-1-丙烷磺酸盐(3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate,CHAPS),并可补充有RNase抑制剂和/或蛋白酶抑制剂。
裂解缓冲液包括或由以下组成:20mM HEPES-KOH(pH 7.9)、10-20%(v:v)甘油、1.5mM MgCl2、0.2mM EDTA、1mM DTT和100mM CH3CO2K以及0.1%Triton X-100,测量电导率为7.5-8.5mS/cm2。在优选实施方式中,裂解缓冲液的测量电导率为8mS/cm2。
在一个实施方式中,由此获得的裂解物可直接用于后续步骤,从而方便样品的处理。
在另一实施方式中,在RISC在根据本发明的裂解缓冲液中溶解后,天然裂解物从不溶性物质中澄出。这样的澄出可通过例如沉淀、过滤或短暂离心进行。在此,术语“旋转离心”和“离心”可互换使用。
在一个实施方式中,对样品施加4℃下10 000x g的离心5分钟以去除不溶性物质,例如膜碎片或植物细胞壁。将上清液转移到新鲜的管中,其构成用于根据本发明的功能性sRNA纯化的预备澄清裂解物。
在另一实施方式中,通过过滤澄出天然裂解物。例如,过滤是通过将裂解物施加于过滤柱来进行的,过滤柱保留诸如膜碎片或植物细胞壁的不溶性物质,但让溶解的RISC通过。过滤可通过使用离心或对装载裂解物的过滤柱施加真空进行。在优选实施方式中,将裂解物装载到有0.45μm醋酸纤维素滤膜如Costar Spin-X(Corning,目录号:CLS8162)的离心柱上,并在4℃下5000x g离心2分钟。
在另一实施方式中,过滤膜包括在含有树脂的柱中阴离子交换树脂的顶部,提供加入和过滤天然裂解物的第一过滤室和含有阴离子交换树脂的第二室,从而将澄出过滤和阴离子交换树脂的装载无缝地结合在一起。在优选实施方式中,第一过滤室作为可拆卸插入件提供。
澄出裂解物提供以下优点:可能堵塞柱子或阻碍RISC关联sRNA富集的细胞碎片被去除。
根据本发明的一个实施方式,当向柱中添加洗脱缓冲液时,RISC关联sRNA从柱中洗脱。洗脱缓冲液与裂解缓冲液的不同之处在于没有洗涤剂,且盐浓度较高,通过电导率测量验证。洗脱缓冲液包括或由以下组成:20mM HEPES-KOH(pH 7.9)、10%-20%(v/v)甘油、1.5mM MgCl2、0.2mM EDTA、1mM DTT,CH3CO2K浓度在400-800mM之间,以达到30至50mS/cm2的测量电导率。在优选实施方式中,洗脱缓冲液的测量电导率为40mS/cm2。
根据本发明一个实施方式的柱包括柱体和树脂。柱体可以具有任何合适的形状和体积。柱体的体积取决于所需的应用,并且可在50μl和50L之间,优选在100μl和1000μl之间,尤其优选在200μl和800μl之间。在一个实施方式中,柱体的体积可为50μl、100μl、150μl、200μl、250μl、300μl、350μl、400μl、450μl、500μl、550μl、600μl、650μl、700μl、750μl、800μl、900μl、1000μl。在优选实施方式中,柱体的体积为1000μl。在另一优选实施方式中,柱体的体积为200μl。柱体体积小使得可以以最少量的材料工作。根据本发明,柱体适于在2ml微量离心管中离心澄清样品和洗脱缓冲液,以简化程序。通常,柱体由1000μl聚丙烯管制成,但也可使用任何其他具有类似性能的材料。在优选实施方式中,MicrospinTM柱(GE healthcare,GE27-35-650,REF27356501)被用作柱体。
在一个实施方式中,柱体为96孔板。这允许使用根据本发明的方法进行自动化高通量分析。在另一个实施方式中,柱是微流体芯片,其也允许纯化过程的自动化。
根据本发明的一个实施方式,柱体包括优选地存储在存储缓冲液中的阴离子交换树脂,即,柱体用所述树脂填充。任何阴离子交换树脂均可用于本发明,只要系统保持sRNA和AGO蛋白质之间的非共价相互作用,同时允许在带正电的基质上分离所有其他不含AGO的核酸以使分离得以进行。本领域技术人员知道如何选择合适的阴离子交换树脂,以允许使用不同的缓冲液、不同的盐和不同的pH值进行sRNA纯化。在优选实施方式中,Q Sepharose HP树脂(GE healthcare GE17-5072-01)用作本发明方法中的阴离子交换树脂。
在将阴离子交换树脂装入柱体之前,可对阴离子交换树脂进行平衡。在一个优选实施方式中,阴离子交换树脂在平衡缓冲液中平衡,该平衡缓冲液包含20mM HEPES-KOH(pH 7.9)、10%-20%(v/v)甘油、1.5mM MgCl2、0.2mM EDTA、1mM DTT和100mM CH3CO2K,测量电导率为8mS/cm2。这影响阴离子交换树脂的分离性能。根据本发明,阴离子交换树脂可在填充柱子之前清洗1、2、3、4或5次,然后在适当体积的储存缓冲液中重悬并填充到柱体中。在一个优选实施方式中,使用阴离子交换树脂填充柱体的整个体积,以便在给定洗脱体积下最大化柱的分离性能。通常,在GE MicrospinTM柱中这由800μL的比例为3:5的储存缓冲液/树脂组成。
在另一个实施方式中,考虑到较低的柱容量足以用于进一步的sRNA分析,因为样品量可与树脂量成比例减少,因此柱体仅部分填充阴离子交换树脂以降低成本。在另一实施方式中,减小柱体尺寸和填充树脂的体积以允许从微量样品(例如,5000个拟南芥胚胎细胞或最多一个拟南芥花蕾)中纯化sRNA。
本发明中使用的柱和缓冲液预先分批制备,每批通过电导率测量和分子分析进行验证,随后在4℃下储存。根据本发明,储存缓冲液还可包括防止微生物污染的化合物,例如叠氮化钠(NaN3)。
在一个优选实施方式中,本发明方法的洗脱步骤重复第二次。这确保了所有RISC关联sRNA都从柱上洗掉,得到更高收率的RISC关联sRNA。在本发明的另一实施方式中,可通过应用盐浓度增加的梯度来执行洗脱步骤。
根据本发明的方法能够在最复杂的生物体、最顽固的组织和/或从最有限量的起始生物材料中稳健且一致地纯化AGO关联sRNA。
根据本发明的试剂盒是在室温下进行调节、装运和操作的,并通过为RISC关联sRNA提供高度简化、通用和单步阴离子交换纯化程序,克服了其他现有技术方法的主要问题。
根据本发明的方法可以在15分钟内在工作台上运行,对实验室要求极低,从而大大减少工作时间和成本。
根据本发明的方法完全适用于从众所周知难以处理的组织(包括含淀粉植物贮藏根或经肝素/EDTA处理的哺乳动物血液样品)中分离sRNA。
根据本发明分离的RISC关联sRNA不受样品中导致样品中全局RNA降解的苛刻条件的影响。此外,根据本发明分离的RISC关联sRNA可立即适用于Northern分析、定量RT-PCR和微阵列分析,以及使用任何内部或商业克隆方案的deep-seq和所有现有技术的测序平台。通过将分析限于AGO结合sRNA,本发明允许在deep-seq之前对sRNA库进行更高的多路复用,从而显著降低下游分析的成本。
根据本发明分离的RISC关联sRNA也特别耐受降解,并且可在分离后冷冻。
根据本发明的方法通常实现相对于污染/降解RNA的所需sRNA种类的>95%富集,从而提供前所未有的分离sRNA质量。
根据本发明的方法允许进行sRNA deep-seq,且其产量、纯度和质量至少在植物和动物中与通过凝胶上大小筛选这一金标准获得的结果相当。本发明的方法在任一生物体中均未显示基于内源性siRNA和miRNA相关性分析的测序偏差。
在优选实施方式中,本发明的方法与文库制备试剂盒(NEB,Ipswich,MA,Catalog#E7330)结合,允许完全绕过对文库进行的PCR后大小选择步骤的真正直接的sRNA克隆。
当执行本发明的方法时获得的RISC关联sRNA部分可适用于下游使用商用柱的基于硅酸盐的分离程序,完全避免RNA沉淀的需求,从而允许在不到30分钟内直接获得纯化的sRNA。在RISC纯化之后,在本发明的一个实施方式中,使用苯酚/氯仿/异戊醇执行脱蛋白步骤,然后进行基于异丙醇的沉淀以回收sRNA,以便从收集的RISC中去除蛋白质内容物。然而,这一步骤需要在低温下长时间孵育,然后高速离心,这在技术上具有挑战性且耗时。
尽管核酸沉淀是一种成本效益高、产量高的标准方法,但商用硅酸盐柱可用于脱蛋白后直接分离sRNA。在本发明的一个实施方式中,该方法需要在将蛋白质内容物去除后在醇的存在下使RNA固定在柱基质上,因此依赖于疏水相互作用。然后可以从杂质中洗出sRNA,并最终在水中洗脱。在优选实施方式中,根据制造商的说明使用ZYMO microspin IC柱(Zymo Research,德国弗莱堡,参考号C1004-50)。发明人惊奇地发现,与类似的商用系统相比,相对于洗脱体积的sRNA输出是最佳的,因此可以用于进行后续测序。
本发明还涉及用于纯化AGO关联sRNA的试剂盒,包括
a)裂解缓冲液;
b)洗脱缓冲液;和
c)包含阴离子交换树脂的柱。
根据本发明的试剂盒可以包括如上所述的缓冲液。
本发明可用于诊断与特定sRNA的存在相关的疾病或疾病亚型。例如,在人类中,Let-7miRNA水平的升高几乎总是和肺癌呈正相关,而在胶质母细胞瘤和乳腺癌中,miR-21水平通常升高。miR-15a/16a在B细胞白血病中经常缺失或显著减少,而在B细胞淋巴瘤和乳腺癌中miR-155增加。miRNA分析也可用于细化活检中肿瘤的状态或复杂成分,还可帮助确定肿瘤在转移中或在未分化肿瘤的情形中可能起源的组织。所有这些基于miRNA的读出在分析癌症及其起源方面有着巨大的前景,有效地提供了更加个性化和有针对性的疾病治疗。miRNA作为生物标志物的潜在用途并不局限于癌症,并且可以应用于细胞内稳态受到如代谢或遗传功能障碍或感染等干扰的任何情况。例如,肝细胞中miR-122升高通常是肝功能障碍和/或肝炎病毒感染的标志。已被用作生物标志物的少数循环miRNA的例子包括血浆中的miR-14,其丰度已成为明确和无创地诊断前列腺癌的主要标准;高血浆miR-141水平与结直肠癌预后不良相关。其他一些循环血浆miRNA与心血管疾病、器官功能障碍(肾脏/miR-215、肝脏/miR-122、胰腺/miR-375)或甚至与因滋养层/胎盘缺陷导致的复杂妊娠相关。
因此,本发明还涉及一种将sRNA鉴定为病理状态的生物标志物、诊断这种病理状态并最终提供关于其可能演变(预后)的信息的方法。鉴于本发明的方法甚至可以从众所周知的顽固性样本(如血浆)中产生高纯度AGO关联sRNA,其可用于鉴定循环生物标志物。这意味着从性别和年龄与患者队列相当的健康个体队列中获取血浆样本。根据本发明的方法可用于纯化血浆携带的AGO关联sRNA,然后进行deep-seq。随后可分析所得的与每个个体的循环sRNA群体相对应的数据集,以确定在患者与健康个体之间在特定sRNA组的特性或丰度(上调或下调)方面的显著差异。然后,可以就一组独立的患者验证这些标志物对诊断/预后的相关性。一旦鉴定为生物标志物的一组sRNA得到验证,本发明的方法可以在患者中与通过qRT-PCR的靶向定量联合。发明人与临床医生合作已经获得初步结果,证明了根据本发明的方法是如何可靠地用于识别患有罕见自身炎症疾病的患者。
实施方式:
(1)一种用于纯化RISC关联sRNA的方法,包括以下步骤:
a)提供来自生物标本的天然样品;
b)使用天然裂解缓冲液裂解样品;
c)选择性地从裂解物中去除非RISC关联核酸;和
d)收集包含RISC关联sRNA的RISC。
(2)根据实施方式(1)的方法,其中在步骤c)中,通过将裂解物装载到包含允许核酸固定的树脂的柱上,从裂解物中去除非RISC关联核酸。
(3)根据实施方式(1)或(2)的方法,其中所述裂解缓冲液包含或由以下组成:20mM HEPES-KOH(pH 7.9),10%-20%(v:v)甘油,1.5mM MgCl2,0.2mM EDTA,1mM DTT,100mM CH3CO2K和0.1%Triton X-100,测量电导率为7.5-8.5mS/cm2。
(4)根据实施方式(1)或(2)的方法,其中在步骤d)中通过对柱施加洗脱缓冲液来收集RISC。
(5)根据实施方式(3)的方法,其中洗脱缓冲液包含或由以下组成:20mM HEPES-KOH(pH 7.9),10%-20%(v/v)甘油,1.5mM MgCl2,0.2mM EDTA,1mM DTT以及浓度介于400-800mM之间的CH3CO2K,以达到介于30-50mS/cm2之间的测量电导率。
(6)根据实施方式中任一项的方法,其中所述树脂是阴离子交换树脂。
(7)根据实施方式中任一项的方法,其中在步骤b)中通过短暂离心或通过过滤使所述裂解物澄出。
(8)根据实施方式中任一项的方法,其中所述柱是96孔板或微流体芯片。
(9)根据实施方式中任一项的方法,其中所述标本来自人。
(10)根据实施方式中任一项的方法,其中所述生物标本是由体外、细胞内或体内RISC生产产生的含有RISC的样品。
(11)根据实施方式中任一项的方法,其还包括:
e)使用苯酚/氯仿/异戊醇提取从收集的RISC去除蛋白质内容物;或
f)通过使用蛋白酶K处理从收集的RISC去除蛋白质内容物。
(12)根据实施方式中任一项的方法,其用于诊断其诊断或预后以特定sRNA存在为特征的疾病。
(13)根据实施方式(10)的用途的方法,其中所述疾病为癌症、代谢障碍、遗传病症或传染病。
(14)一种用于纯化RISC关联sRNA的试剂盒,包括:
-天然裂解缓冲液;
-洗脱缓冲液;和
-包含存储在存储缓冲液中的阴离子交换树脂的柱。
(15)根据实施方式(12)的试剂盒,其中所述裂解缓冲液包括20mM HEPES-KOH(pH 7.9),10%-20%(v/v)甘油,1.5mM MgCl2,0.2mM EDTA,1mM DTT,100mM CH3CO2K和0.1%Triton X-100,测量电导率为7.5-8.5mS/cm2;
(16)根据实施方式(12)或(13)的试剂盒,其中所述洗脱缓冲液包含20mM HEPES-KOH(pH 7.9),10%-20%(v/v)甘油,1.5mM MgCl2,0.2mM EDTA,1mM DTT以及浓度介于400-800mM之间的CH3CO2K,以达到介于30-50mS/cm2之间的测量电导率。
(17)根据实施方式(12)到(14)的试剂盒,其中存储缓冲液包含20mM HEPES-KOH(pH 7.9),10%-20%(v/v)甘油,1.5mM MgCl2,0.2mM EDTA,1mM DTT,100mM CH3CO2K,测量电导率为7.5-8.5mS/cm2,以及2mM NaN3。
实施例
实施例1
根据本发明方法的原理
图1A所示的方案描述了根据本发明的方法的原理。以保持AGO蛋白和关联sRNA之间的非共价相互作用的方式从生物样品中产生天然裂解物。将裂解物与带正电的树脂混合,使得非AGO负载的核酸固定在树脂上,而未固定的RISC可以被洗脱。分离过程基于RISC关联RNA和其他细胞核酸之间的电荷差,生成RISC富集的级分(称为E级分)。为控制该过程,可使用高盐缓冲液将保留的游离核酸洗脱在不同的级分中(称为HS级分)。
mini-TraPR试剂盒使用的详细程序
如图1B所示的方案所示,该程序包括三个主要步骤。将样品在天然裂解缓冲液中裂解,通过快速离心澄清。然后将澄清的裂解物装载到含有带正电树脂的柱体上。将裂解物和树脂混合以助于分离,然后对柱进行离心,流过物被收集在一个新鲜的管中。然后通过向柱加入洗脱缓冲液进行洗脱步骤,接着进行短暂离心。洗脱液被收集在前一个管中。再重复一次洗脱,以确保RISC完全回收。按照此程序,样品中的RISC内容物在15分钟内被纯化,经此可从收集的级分中提取AGO关联sRNA。
缓冲液
树脂平衡缓冲液(20mM HEPES-KOH pH=7.9,10%(v/v)甘油,1.5mM MgCl2,0.2mM EDTA,1mM DTT,用指示浓度为~0.1M的醋酸钾将电导率调整在8mS.cm-2)
树脂存储缓冲液(20mM HEPES-KOH pH=7.9,10%(v/v)甘油,1.5mM MgCl2,0.2mM EDTA,1mM DTT,0.2mM叠氮化钠,用指示浓度为~0.1M的醋酸钾(KoAc)将电导率调整在8mS.cm-2)
TraPR裂解缓冲液(20mM HEPES-KOH pH=7.9,10%(v/v)甘油,1.5mM MgCl2,0.2mM EDTA,1mM DTT,0.1%(v/v)Triton X100,用指示浓度为~0.1M的KoAc将电导率调整在8mS.cm-2)
TraPR洗脱缓冲液(20mM HEPES-KOH pH=7.9,10%(v/v)甘油,1.5mM MgCl2,0.2mM EDTA,1mM DTT,用指示浓度为~0.44M的KoAc将电导率调整在40mS.cm-2)
TraPR高盐缓冲液(20mM HEPES-KOH pH=7.9,10%(v/v)甘油,1.5mM MgCl2,0.2mM EDTA,1mM DTT,用指示浓度为~4M的KoAc将电导率调整在110mS.cm-2)
1.样品裂解和澄清
在400μL TraPR裂解缓冲液中裂解速冻样品(氮预冷研钵、组织研磨器或其他方法);
将裂解物转移到新鲜的不含DNase、无RNase的1.5mL微量离心管中;
10 000x g下4℃离心5分钟以使裂解物澄清;
将300μL澄清的裂解物转移到新鲜的不含DNase、无RNase的2ml微量离心管中;
2.Mini TraPR试剂盒纯化柱准备
旋涡几秒钟使柱内树脂重悬;
将盖子松开四分之一圈,拧下底部封盖(不要丢弃底部封口);
将柱置于2ml微量离心管中;
台式微量离心机上离心15秒;
丢弃收集管,将柱置于标记为“TraPR E级分”的2mL新鲜的微量离心管上
3.点样
用底部封口将柱封闭;
打开柱盖,将300μL样品施加于树脂的顶部中心;
关闭柱盖,将样品与树脂充分倒置混合;
移除底部封口,将柱放入先前标记为“TraPR E级分”的2mL微量离心管中
4.RISC负载小RNA的洗脱
在台式微量离心机上离心15秒,保存流出液;
打开柱盖,加入300μL TraPR洗脱缓冲液;
关闭柱子并放入先前标记为“TraPR E级分”的2ml微量离心管中;
在台式微离心机上离心15秒;
重复步骤15到17一次,将洗脱液收集在相同的标记为“TraPR E级分”的2mL微量离心管中;
关闭“TraPR E级分”收集管(900μL),冰上存储供立即使用,或在-80℃下存储。这个级分包含装载有其相关sRNA的RISC蛋白(AGO)。
5.RNA提取(沉淀)
将500μL酸性PCI加到收集的级分中;
大力涡旋;
离心5分钟,全速,4℃;
将水相收集在新的1.5mL微量离心管中;
加入10%3M醋酸钠,pH 5.2和1μL糖原,均质;
加入120%冷异丙醇,均质;
-20℃孵育至少30分钟;
离心30分钟,全速,4℃;
丢弃液相;
加入400μL 80%冷乙醇;
离心5分钟,全速,4℃;
重复步骤29到30两次;
去除所有乙醇;
向团块中加入适当体积的水(或LMNb上样缓冲液1X);
上下吹打重悬(可在60℃下加热几分钟);
RNA可以在-80℃下储存,也可以立即使用。
6.RNA提取(Zymo Ic微型离心硅酸盐柱)
将500μL酸性PCI加到收集的级分中;
大力涡旋;
离心5分钟,全速,4℃;
将水相收集到2个新的1.5ml微量离心管中(将收集的相分成2个相同体积);
每管加入2个体积的RNA MAX缓冲液,搅拌均匀;
将800μL的混合物转移到Ic柱中,放入收集管中;
离心30秒,12 000g,弃掉流出液;
重复步骤41和42,直到所有混合物通过柱子;
向柱中加入400μL RNA Prep缓冲液;
离心1分钟,12 000g,弃掉流出液;
在柱中加入800μL RNA Prep缓冲液;
离心30秒,12 000g,弃掉流出液;
用400μL RNA Prep缓冲液重复步骤46和47;
将柱以12 000g离心2分钟,完全去除缓冲液;
将柱转移到一个新的1.5ml微量离心管中;
在树脂上加入6~15μL水,60℃预热;
室温孵育1分钟;
10 000g离心1分钟,洗脱RNA;
回收的RNA可以立即使用,也可以储存在-80℃。
实施例2
在根据本发明方法的RISC富集的E级分中拟南芥AGO及其关联sRNA的共纯化
拟南芥基因组编码10个旁系同源AGO基因,其中9个以蛋白质的形式表达,分为3个主要的系统发育分支,如图2A所示。对拟南芥花序中两种主要AGO蛋白(AGO1和AGO4)进行免疫印迹分析,蛋白从通过应用增加盐浓度的缓冲液而得到的柱洗脱,对缓冲液电导率进行监测(图2B,上)。提取级分中包含的RNA,在17%丙烯酰胺凝胶上进行迁移,然后用溴化乙啶染色(图2B,下)。洗脱图谱的分析显示,两种主要的拟南芥AGO蛋白在使用温和盐浓度的缓冲液(黑色箭头)之前从柱中被洗脱,而RNA则保留在树脂上,直到达到更高的盐浓度(虚化箭头)。图2B所示的结果被用来界定洗脱缓冲液中允许AGO蛋白从细胞核酸如长RNA中分离的盐浓度范围(通过在30-50mS/cm2之间的电导率监测)。
为了将该分析拓展到整个拟南芥AGO蛋白家族(9个成员),根据本发明方法,使用调整到40mS/cm2的洗脱缓冲液,在从拟南芥纯化RISC后使用抗体检测7种内源AGO蛋白进行免疫印迹蛋白分析(图2C)。为了证明抗体的特异性,对单个拟南芥ago突变系与野生型植物(凝胶左侧)进行并排分析。按照本发明方法纯化后,花序中可检测到的所有7种AGO被回收并富集在E级分,而HS级分(凝胶右侧)没有明显的信号。由于没有可识别天然AGO3和AGO7的抗体,使用在其内源性启动子下表达每个蛋白的带标签表位版本的转基因株系进行相同的实验。然后从已知表达这些特定AGO的组织即AGO3和AGO7的幼长角果(young siliques)和幼苗中提取RISC。如图2D所示,AGO3在E级分中富集,在HS级分中低于检测限。在图2E中对于AGO7观察到类似结果。结果表明,根据本发明的方法进行RISC纯化,能够在E级分中强富集所有表达的拟南芥AGO蛋白。
Deep-seq分析显示,总植物sRNA由两大主要诊断特征、离散长度组成:来自转座子和重复序列的高丰度24nt异染色质siRNA被装载在AGO4分支的AGO中,而丰富的21nt sRNA主要由装载在AGO1分支的AGO中的miRNA和装载在AGO2中的sRNA组成。为检测根据本发明方法纯化的sRNA的纯度,对所有级分(I、E和HS)均用多核苷酸激酶(PNK)进行5'端标记。图2F显示21nt和24nt sRNA物种在I和HS级分中检测不到,相反显示了异质性和不相关RNA物种的强标记。相比之下,在AGO富集的E级分中,两种物种都呈现出几乎没有任何背景的干净条带,展现了根据本发明方法的程序用于纯化RISC的能力。通过5’标记所见的纯化程度至少与通常用高特异性免疫沉淀观察到的相当。为了验证特定sRNA物种的分离,使用已知的代表性拟南芥sRNA物种的特定放射性标记寡核苷酸探针,对每个级分中包含的RNA进行Northen分析。如图2G所示,对E级分中21nt和24nt sRNA类的特定物种都观察到很强的富集。与此相反,非RISC关联U6 RNA如预期的那样在HS级分中富集。
图2所示的结果代表了通过对来自不同组织的拟南芥裂解物应用应用本发明的方法进行纯化在实验室中常规获得的模式。他们证实,该程序允许拟南芥AGO蛋白与其sRNA货物共洗脱,并有效分离大多数污染RNA及其分解产物,其反而在HS级分中洗脱。
本发明的方法定义了广泛生物体中的通用RISC纯化程序
鉴于AGO蛋白质及其与sRNA相互作用形成RISC在所有生命王国中是高度保守的特征,在一系列生物体中对根据本发明方法的纯化程序进行了测试。图3(上)所示的系统发育树示意图概述了从纤毛虫、作物、裂变酵母、蠕虫到哺乳动物的受试生物的多样性。对于每种生物体,根据本发明方法纯化RISC,并通过存在于每种级分I、E和HS中RNA的PNK放射性标记检测它们的货物,如图3(下)所示。
纤毛虫可以在同步阶段进行研究,这些同步阶段代表了积累23nt siRNA的营养阶段(WT)或积累25nt长的、独特参与DNA消除的scnRNA的有性阶段(T0)。目前还没有针对纤毛虫AGO/PIWI蛋白的商业抗体可用。尽管如此,在有性阶段细胞中的相关25nt scnRNA物种的E级分中,根据本发明方法纯化后通过PNK进行的5'-末端标记显示特异且强烈的富集。而在I和HS中几乎检测不到,如果有的话,而是显示较长RNA的强标记。
根据本发明的方法,利用从水稻叶和木薯贮藏根的裂解物中纯化的sRNA的5’-末端标记在两种关键主要作物中进行了类似的分析。与拟南芥不同,水稻的叶子有蜡状叶角质层,不利于RNA提取。水稻基因组中的转座子和重复序列也要丰富得多,使得24-nt siRNA异常丰富。根据本发明的方法进行RISC纯化后的5'-末端标记的结果完全符合这一概念,显示与I和HS级分相比,E中有较强的24-nt sRNA和更适中的21-nt sRNA富集。第二项分析涉及木薯贮藏根的裂解物,其由于淀粉含量极高,构成了提取RNA更具挑战性的组织。用于分析的木薯是非洲种植的农民首选的基因型。正如已经在拟南芥和水稻中观察到的,根据本发明方法进行RISC纯化后的5'-末端标记显示在E级分中21nt和24nt sRNA物种高度富集且几乎没有背景,但在I或HS级分中不存在。重要的是,与拟南芥不同,目前还没有针对木薯AGO蛋白的抗体,无法通过免疫沉淀分离RISC。这些结果表明,根据本发明方法的纯化允许从对众所周知难以进行RNA提取的以及没有AGO抗体可用的作物中快速且稳健地分离RISC。
在各种真菌和后生动物样本中进行了类似的分析:从单细胞酵母到更复杂的生物体如整个秀丽隐杆线虫或小鼠整个器官。在裂殖酵母中,来自着丝粒周围重复序列的异染色质siRNA构成了最大(如果不是唯一的话)的sRNA。它们的大小没有其他生物那么清晰,但仍在23nt左右。在根据本发明方法进行RISC纯化后,通过PNK进行的5'-末端标记显示,与显示较长RNA的标记的I和HS级分相比,E级分中23-nt siRNA的富集程度非常高。这一结果是非凡的,因为即使使用sRNA物种特异性放射性标记探针,Northern分析通常也无法检测到粟酒裂殖酵母(S.Pombe)的异染色质siRNA。
蠕虫拥有复杂的sRNA机制,涉及AGO/PIWI家族中超过25种的蛋白质,只有很少的商业抗体可用且可靠。如所有其他实施例所示,在根据本发明方法纯化后RNA的5'-端放射性标记显示在E级分中有强烈sRNA富集,而I或HS级分未显示。与所有其他生物中所见的一样,与其中大部分长RNA污染物或分解产物被标记的I和HS级分不同,E级分明显没有背景标记。
已经在小鼠成年大脑中测试了根据本发明方法的RISC纯化。5'-末端放射性标记显示,集中在22nt(哺乳动物Dicer产物的相关大小)的sRNA在E级分中富集,并且背景值同样较低,而I和HS级分中大部分长RNA污染物或分解产物被标记。为了归结根据本发明的方法能够普遍分离包括后生动物特异性piRNAs在内的RISC的概念,对来自小鼠睾丸(piRNAs高度表达的组织)的裂解物进行了类似的实验。如所有其他实施例所示,5'-端放射性标记显示,集中在30nt(小鼠piRNAs的相关大小)的sRNA在E级分中富集,背景值同样较低,而I和HS级分中大部分长RNA污染物或分解产物被标记。
我们从所有这些分析中得出结论,根据本发明方法的纯化可用于在所有生命王国中快速而稳健地分离RISC,包括从众所周知难以进行RNA提取的组织中。RISC与其相关货物共同纯化,可以是siRNA、miRNA、piRNA或scnRNA,它们定义了目前已知的所有沉默小RNA的全部范围。
实施例3
通过本发明方法纯化的RISC关联sRNA可直接适于基于硅酸盐的提取,绕过沉淀步骤
如上所述,根据本发明的方法可以获得RISC关联的sRNA。样品裂解后,可在15分钟内生成E级分,通常直接从中提取sRNA,但该级分也可以储存在-80℃下(AGO/PIWI结合的sRNA特别耐降解)。
sRNA通常用苯酚从RISC中提取,然后用醇沉淀,总共需要至少90分钟到一整天(图4A),实际上是sRNA可用于northern、RT-qPCR、微阵列分析或deep-seq之前下游程序中最长的步骤。为了大大减少从通过本发明对应的方法分离的RISC中提取sRNA所需的时间,对其与市售的基于硅酸盐的RNA纯化/提取试剂盒的兼容性进行了测试。这些试剂盒的原理始终依赖于在存在醇和盐(基于疏水性)的情况下RNA与硅酸盐基质结合,然后用少量不含RNA酶的水或缓冲液洗脱。
图4A提供了将硅酸盐分离接入到根据本发明的RISC关联sRNA纯化程序中的工作流程的概览。按照标准的醇基沉淀程序对三个制造的硅酸盐纯化系统(QiagenTMRNAeasy柱,ZymoTM微型柱和ZymoTMmini Ic柱)进行了测试。在该情况下使用根据本发明方法从拟南芥花序分离的RISC关联sRNA,并通过已知miRNA(miR163、miR160、miR159)的northern分析评估最终sRNA产量。如图4B所示,当根据本发明纯化的RISC关联sRNA与ZymoTMmini Ic柱一起使用时,获得了与沉淀相当的最佳产率。测试的其他柱显示显著更低的sRNA输出,可能反映了固定/洗脱步骤中材料的损失。
与ZymoTM微型柱的偶联减少了(30分钟)根据本发明方法获取RISC关联sRNA所需的时间(图4A)。此外,ZymoTM微型Ic柱由于其设计允许在小体积水中回收sRNA,非常适合于直接进行分子分析,如定量PCR前的反转录或用于deep-seq的sRNA文库制备。使用该实验装置,根据本发明方法分离的RISC关联sRNA允许在样品裂解后的规定时间内处理大量样品。
通过本发明方法纯化的RISC关联sRNA非常适合于在各种生物系统中通过RT-qPCR检测miRNA
在没有先验知识的情况下,Deep-seq仍然是在全基因组范围内识别和量化给定生物样本中的sRNA群体的金标准。尽管Deep-seq的可承受性越来越高,但对于大多数研究实验室来说,(在经济上和技术上)系统地使用Deep-seq来研究生物过程或仅用于诊断仍然是令人却步的,更别提大型sRNA数据整理/分析需要的专业知识。在大多数情况下,Deep-seq被用作下游程序,用于识别与特定过程、细胞状态或病理学相关的稳健sRNA候选者。一旦这些候选者得到验证,优选的下游方法依赖于对这些候选者进行靶向RT-qPCR定量,而不是全基因组的sRNA测序。RT-qPCR允许以适中的成本对大量样品上的多个sRNA序列进行精确定量。
基于RT-qPCR的sRNA定量的主要限制因素是反转录(RT)步骤,其中特定的sRNA序列被反转录成cDNA以实现下游PCR扩增。RNA制备的复杂性,包括感兴趣的sRNA序列的潜在低丰度,可能确实会降低RT效率,从而对定量的质量和稳健性产生负面影响。鉴于根据本发明方法的纯化显著地在E级分中富集RISC关联sRNA,使用基于内部环路的RT-qPCR程序对从拟南芥花序纯化的sRNA进行了miRNA定量的适用性测试。通过根据本发明方法的RISC关联sRNA纯化,受试miRNA(miR159,miR171)在E级分中而不是HS级分中富集。相比之下,HS级分富含拟南芥小核仁RNA snoRNA85,其未装载到任何AGO中(图4C)。在涉及富含sRNA的哺乳动物组织(如小鼠肝脏)的第二个示例中,观察到类似的模式,分别在E级分中富集两种哺乳动物miRNA,在HS级分中富集snoRNA202(图4D)。总之,这些结果表明,根据本发明方法纯化的RISC关联sRNA非常适合于在不同生物系统中通过RT-qPCR检测miRNA。
实施例4
通过本发明方法纯化的RISC关联sRNA可直接适用于一系列生物体的deep-seq
如图2中使用RNA的放射性标记所示,根据本发明方法的RISC关联sRNA纯化应用于例如拟南芥样品产生高度富集的sRNA,同时去除其他核酸污染物。在现有技术中,聚丙烯酰胺凝胶上的sRNA大小选择是用于deep-seq的sRNA文库制备的绝对先决条件。而因为担心在其他模型(如常规哺乳动物组织)中丢失样本,这一最佳输出所需的步骤很少被使用。事实上,基于凝胶的大小切除是一个长而繁琐的高技术性程序,而且需要使用放射性标记的RNA大小标尺,并且通常为下游分析(例如RT-qPCR或Deep-seq)提供低产出的sRNA材料。由于在根据本发明的方法产生的E级分中RISC关联sRNA系统性地大量富集,因此进行了一项试验,以检验在两种众所周知的困难情况下,该方法是否能够在生产能用于deep-seq的sRNA文库的过程中完全绕过凝胶选择(图5)。拟南芥和果蝇就是这样的两种生物体,在这两种生物体中,由于大量低分子量RNA污染物的存在,需要使用基于凝胶的大小切除来制备文库。
在拟南芥中的分析是在来自同一批花序的三个三次技术重复中进行的。我们比较了通过三个独立的凝胶大小选择事件、三个独立的基于TRIzol的总RNA提取或三个独立批次的根据本发明方法纯化而无凝胶大小选择的RISC关联sRNA所分离出的sRNA的deep-seq结果(图5A)。这些库是用TRUseq(Illumina)库制备试剂盒生成的。在整理和修整后,凝胶大小选择后获得的sRNA读段大小分布显示出预期的植物sRNA轮廓,在21nt(miRNA)和24nt(异染色质siRNA)处有两个峰(图5A,中间)。如预期的那样,对复杂的生物学模型(如拟南芥)中的总RNA(图5A,顶部)直接克隆与高质量的文库制备不兼容。两个sRNA峰要么几乎看不见(21nt),要么不好确定(24nt)。此外,它们被相同大小范围内的大量污染物包围,这些污染物占三个库中每个库的读段的60%以上。相比之下,使用本发明方法独立纯化的三个样品(无凝胶大小选择)获得的大小分布清楚地显示了对于拟南芥预期存在且几乎不含任何污染物的21nt和24nt峰(图5A,下)。使用根据本发明方法纯化的RISC关联sRNA获得的测序结果不仅与凝胶上的大小选择后获得的结果一致,而且它们在平行样之间表现出更少的变化,很可能体现了最低限度的样品处理要求和该方法的整体鲁棒性。
在果蝇卵巢上也进行了比较性的deep-seq分析,其中sRNA的提取和克隆在难度和繁琐性方面提供了高水平的基准。在这个复杂的组织中,发现了三类sRNA:22nt miRNA和分别被装载到AGO1和AGO2中的21nt siRNA。相比之下,后生动物特异性的piRNA(长度为23-29nt)被装载到PIWI蛋白质中。由于在相同大小范围内极高丰度的2SrRNA(30nt),piRNA测序在果蝇中得到了极大的优化。目前费力的程序需要首先以非常精确的方式在凝胶上选择长度在18到29个核苷酸之间的sRNA。在第二步中,使用商业试剂盒对纯化的sRNA进行核糖体RNA去除,然后氧化以去除不含3'甲基的RNA。后生动物siRNA和piRNAs具有这种修饰因此可以防止氧化,不像miRNA或2S rRNA那样。一个主要的问题是,在氧化后,样品不仅从主要污染物(2S)中被消耗,而且从miRNA编码的信息中被消耗,尽管如此,还是有高度价值的。
在第二组实验中,对果蝇卵巢的RNA进行了deep-seq分析。使用Brennecke实验室(IMBA,维也纳)开发的优化的内部克隆程序生成生物学重复文库,在该程序中,凝胶选择的sRNA被去除核糖体RNA,然后被氧化。将该金标准与不进行任何核糖体RNA去除和氧化的根据本发明方法纯化的RISC关联sRNA的直接克隆进行比较(图5B)。为了在广泛的输入范围内测试根据本发明的方法的性能,从2、5、10、25和50对卵巢中制备库(图5B和图6)。图5B显示了从不同策略获得的映射读段的比较尺寸概况,显示在经过凝胶选择和核糖体RNA去除但未氧化的RNA文库中存在miRNA和2S rRNA(图5B,顶部)而在经过氧化后它们显著减少(图5B,中部)。在根据本发明方法的RISC关联sRNA经直接克隆后获得的所有库中,miRNA是存在的但能观察到污染物2S rRNA的强烈消耗(图5B,下)。值得注意的是,对于纯化的RISC关联sRNA观察到几乎相同的情况,与起始材料的量无关,证明了该方法在广泛的输入量范围内的稳健性和一致性(图5B下部和图6)。
对三种类型文库的miRNA含量进行聚类分析。图7A中的热图显示,如预期的那样,由于处理引起的miRNA丢失,经凝胶选择、核糖体RNA去除和氧化的文库作为外层聚集在一起。相比之下,经凝胶选择和核糖体RNA去除的sRNA文库聚集在RISC关联sRNA文库附近。最后,使用本发明的方法从不同数量的起始材料生成的单个库聚集在一起,与所使用的起始材料的数量无关。该结果证实,本发明的方法允许在广泛的输入范围上生成健壮且可比较的RISC关联sRNA库。
使用本发明的方法所提出的一个反复而合理的问题涉及由各种程序所产生的sRNA含量的定性和定量相关性。换句话说,通过选择专一功能性的即RISC关联的sRNA,根据本发明的方法是否导致通过其他方法分离的某些sRNA物种的代表性不足或丢失?为了解决潜在的偏差,对所有果蝇文库的sRNA含量进行了相关分析(图7B)。
以核糖体RNA去除的sRNA文库为参考,在比较核糖体RNA去除的和RISC关联sRNA文库之间的miRNA和TE映射读段(piRNA和siRNA)时,观察到良好的相关性(>0.99)(图7B)。如此前分析所预期的,与在三种程序之间表现出高度相关性的siRNA和piRNA群体相比,miRNA群体在氧化的sRNA文库中丢失。总之,这些结果表明,根据本发明方法RISC关联sRNA的纯化不会引起测序文库中sRNA表现的偏差。miRNA和TE映射sRNA的不同RISC关联sRNA文库之间的显著高度相关性证实了本发明方法的稳健性和一致性,与所用起始材料的量无关。
实施例5
本发明的方法分离天然RISC并提高免疫沉淀的质量
在拟南芥中,AGO1-和AGO2-分支AGO优先与以5'尿嘧啶(U)开头的21nt长sRNA物种相关联,而AGO4分支优先与以5'腺嘌呤(A)开头的24nt物种相关联。为了确认通过本发明方法实现的天然RISC纯化适用于完整套组的拟南芥AGO蛋白,使用图5所示的sRNA测序文库进行了5'核苷酸分析。基于从经凝胶大小选择的sRNA、总RNA或根据本发明方法纯化的RISC关联sRNA产生的文库,对21nt(图8A,上)和24nt(图8A,下)物种的5'末端发现的核苷酸比例进行比较。分析显示,在凝胶选择的和RISC关联sRNA文库中,21nt物种具有强烈的5'U偏倚,24nt物种具有强烈的5'A偏倚,证实后一种程序分离出真正的、功能性的AGO-sRNA复合物。事实上,相比于凝胶上大小选择的sRNA产生的文库,相关5'A/U末端sRNA的富集在从根据本发明方法纯化的RISC关联sRNA生成的文库中似乎更为严格(图8A)。
根据本发明的方法分离功能上活跃的集合,即AGO装载的sRNA,并由此推断出天然RISC的纯化。为了确认分离的RISC的天然状态,从根据本发明方法分离的总裂解物或富含RISC的E级分的拟南芥AGO1免疫沉淀(IP)实验是并行进行的。该实验是在转基因拟南芥系的花序中进行的,该转基因拟南芥系在其内源启动子下表达带Flag标签的AGO1,该标签通过商用抗Flag抗体检测。图8B(上)所示的蛋白质印迹显示,AGO1在总裂解物和根据本发明方法的富含RISC的E级分中有效地免疫沉淀。在这两种情况下,未结合部分(Ub)中没有AGO1信号表明Flag IP是高效的。在拟南芥中,AGO1优先装载约21nt的miRNA,事实上,通过这两种程序,miR160在免疫沉淀部分中富集,证实其作为天然RISC的一部分与AGO1相互作用(图8B,中部)。每个部分中RNA的5'-末端放射性标记显示,在来自总裂解物的IP中离散的、21nt长的sRNA物种富集,尽管由于RNA污染,伴随着非特异性背景标记(图8B,下)。相比之下,在IP部分中,从根据本发明方法分离的富含RISC的E级分中免疫沉淀的sRNA标记显示出少量背景和21nt RNA物种的强烈的特异性富集,而24nt物种则保留在未结合的部分并优先装载到AGO4分支蛋白中。在AGO1 IP中发现的来自于RISC富集的E级分的低水平RNA污染物很可能通过在IP之前本发明方法内在的非RISC关联RNA的扣减来解释。总之,这些结果证实RISC是使用本发明的方法在其天然状态下纯化的,因此与下游的AGO蛋白质免疫沉淀兼容。因此,E级分中的非RISC关联RNA消减可被视为一个有价值的清理步骤,使得根据本发明的方法也可用于总体提高植物中可能还有其他生物体中的AGO IP实验的质量。
实施例6
通过本发明方法纯化的RISC关联sRNA高度耐降解
RNA是不稳定的分子,从样品采集到长期储存,在其制备和处理的任何阶段都容易降解。尽管其装载到AGO蛋白质中使调节性sRNA比其他RNA种类更稳定,但长RNA的降解产物将严重污染通过总RNA或者sRNA凝胶大小选择从质量不佳的样品中制得的sRNA文库。由于本发明的方法分离其中sRNA与其相关AGO效应子结合的RISC,因此使用本发明方法预期能够强有力地筛选掉质量次优的样品中存在的长RNA降解产物,因而即使从高度降解的RNA制备物中也有可能制备出高质量的sRNA deep-seq文库。为了验证这一想法,用RNase T1处理来自小鼠肝脏的未澄清的裂解物,并在室温下培养30分钟,然后根据本发明的方法进行sRNA纯化。使用完整或经RNAse处理的样本,从输入(总RNA)和RISC关联sRNA制备deep-seq文库(三个生物学重复)。在进行deep-seq之前,进行低分子量RNA印迹分析(图9A)。其表明,尽管其他RNA发生强烈降解,但根据本发明方法分离的AGO结合的sRNA,例如Let7a或肝细胞特异性miR-122,在完整的或RNAse T1处理的样品中都能被轻易检测到。此降解通过在转移尼龙膜之前丙烯酰胺凝胶的溴化乙锭染色得到证明,并且在通用U6 RNA杂交后也可见这种降解,在所有RNA酶T1处理的样品中表现为“阶梯”。注意,从U6-和溴化乙锭染色的较长RNA种类中产生的这些丰富的降解产物在沉默sRNA的大小范围内,因此,可能污染通过总RNA提取或甚至凝胶上的大小选择制备的sRNA文库。
通过对标准Truseq(Illumina)程序制备的sRNA文库的测序结果进行检查,确实证实了这一想法。图9B中的注释信息揭示了RNA降解(RNase T1处理)对直接从总RNA中制备的sRNA文库质量有严重负面影响:此类文库始终受到高达80%的tRNA衍生片段的高度污染,与从完整肝脏样本制备的文库相比,测序miRNA的数量减少了几乎一个数量级。相比之下,根据本发明制备的RISC关联sRNA文库仅受到降解的适度影响,并且显示出与未经RNase-T1处理制备的文库相当的质量。在两个RISC关联sRNA文库中,基因组图谱读段主要是以22nt为中心的miRNA,与样品的降解状态无关。相比之下,该峰值在总sRNA提取后的降解样品中几乎无法检测到,因为大量峰值在32nt的tRNA污染,其可能影响沉默sRNA的克隆。
为了测试根据本发明的方法在应用于降解样品时的潜在偏差,对从各种库中测序的sRNA进行了相关分析。图9C中所示的结果表明,与从完整样品中分离的或在未经RNase T1处理的总RNA制备后分离的miRNA池(相关度=0.954)相比,应用于经RNase T1处理的样品的根据本发明的方法分离的miRNA池保持无偏(相关度=0.984)。因此,本发明的方法通过在库制备之前纯化AGO负载的sRNA使得能够独立于样品各自的降解状态进行高度准确的样品比较。
这一结果对于使用不稳定样品、在不同时间间隔采集的样品和/或在不同条件下储存的样品(包括一些不能防止RNA降解的样品)进行的工作是高度相关的。因此,本发明的方法特别适合于研究大量患者来源的活组织检查或生物液体,这些活组织检查或生物液体易于降解,并且有时经过多年(例如>10年)收集。这通常会强烈限制通过deep-seq对样品中包含的sRNA群组进行稳健比较,但根据本发明的方法能够通过其RISC内容物实现sRNA库的标准化。
实施例7
根据本发明的方法能够以高度可再现性和鲁棒性从哺乳动物血浆中分离sRNA
到目前为止,哺乳动物血浆的复杂性、RNA降解倾向和非常低的sRNA含量严重阻碍了临床研究中对该体液及其他体液中sRNA生物标志物的有力探索。这同样也阻碍了使用RT-qPCR可靠地检测已经确定的循环生物标志物以进行诊断/预后。因此,我们使用小鼠血浆测试了根据本发明的方法的性能,以评估该程序是否能够弥补这些主要的、往往无法克服的负担。从四只单独小鼠采集血浆样品。每个样品都经过了从150μL血浆中提取总RNA,或根据本发明方法从相同体积中纯化RISC关联sRNA。在所有条件下,采用Lexogen生产的小RNA文库制备试剂盒克隆RNA。
图10A显示了对各血浆sRNA文库获得的测序读段比例(见注释)。正如预期的那样,总RNA文库包含高达80%的tRNA污染物。相比之下,由根据本发明方法纯化的RISC关联sRNA制备的文库在miRNA中高度富集,占其内容物的90%以上,几乎没有任何污染或降解产物的痕迹。这些结果通过使读段映射到小鼠基因组的长度分布得到了证实(图10B)。事实上,来自总RNA的文库仅显示出一个以22nt为中心的小峰(miRNA),相反,以30nt为中心显示出一个tRNA主峰,反映了严重的污染。相比之下,根据本发明方法纯化的RISC关联sRNA文库显示出以22nt为中心的独特、尖锐的miRNA信号,并占据了所有测序读段。对单独总RNA文库(图10C,上)中存在的miRNA群体和根据本发明方法纯化的RISC关联sRNA产生的文库(图10C,下)进行相关性分析。该分析显示,与总RNA文库相比,在RISC关联sRNA纯化后产生的文库中的miRNA群体具有显著更高的个体内相关性。此外,如图10D所示,对每四分位miRNA丰度显示的miRNA离散度的分析显示,与总RNA文库相比,RISC关联sRNA文库中低丰度miRNA的变化显著较小。该结果表明,根据本发明的方法对于deep-seq文库中的低丰度sRNA序列产生显著更具可比性的结果。
对代表组织或细胞类型特异性状态或病理病症的sRNA的探索,通常通过deep-seq实现。一旦确定了该状态/病症的稳健sRNA标记,就可以有针对性地将其用作所述状态/病症的定量指标。图11A中呈现的结果显示,与输入(总RNA)相比,通过RT-qPCR对两种此类miRNA的靶向检测在根据本发明方法纯化的小鼠血浆RISC关联sRNAE级分中富集了两个数量级。
在对根据本发明的方法作为在复杂样本(拟南芥和果蝇)中产生高质量功能性sRNA测序文库的稳健利器进行验证的期间,应用了不同的克隆策略。对植物样品采用标准化TRUseq(Illumina)程序(图5)。另一方面,对果蝇样品(图5和图6)采用Brennecke实验室(IMBA,维也纳)专门针对果蝇卵巢制定的定制方案。在这两种情况下,使用本发明的方法大大提高了deep-seq结果的质量和鲁棒性,也表明样品完整性是sRNA文库制备中的一个关键参数。
我们的目的是通过比较另一种商业克隆试剂盒(由Lexogen生产)和Illumina TRUseq来确认这一假设。图11B中显示的结果显示,如果按照本发明的方法处理样本,则与总RNA文库(0.879)相比,两个方案之间的miRNA群体具有更好的相关性(0.903)。我们得出结论,本发明的方法适用于使用从商业到定制设计等大批的文库制备方案进行sRNA克隆。更一般地说,本发明证实了获得用于deep-seq的稳健且高质量的sRNA文库的关键步骤是sRNA样品制备,对此,本发明的方法在每个方面(时间、复杂性、技术性、可承受性等)都优于目前采用的所有方法。
我们得出结论,本发明的方法适用于适于RT-qPCR定量和deep-seq分析(主要包括miRNA)的高质量sRNA制备。因此,本发明的方法在再现性和深度方面为改进诊断/预后打开了巨大的前景,在对包括哺乳动物血浆在内的多种样品中复杂的定性和定量变化的一致和稳健检测方面提供了保证。
- 还没有人留言评论。精彩留言会获得点赞!